

Abstract
The genes required for γ-polyglutamic acid (PGA) production were cloned from Bacillus subtilis IFO16449, a strain isolated from fermented soybeans. There were four open reading frames in the cloned 4.2-kb DNA fragment, and they were almost identical to those in the ywsC and ywtABC genes of B. subtlis 168. Northern blot analysis showed that the four genes constitute an operon. Three genes, ywsC, ywtA, and ywtB, were disrupted to determine which gene plays a central role in PGA biosynthesis. No PGA was produced in ΔywsC and ΔywtA strains, indicating that both of these genes are essential for PGA production. To clarify the function of the YwsC protein, histidine-tagged YwsC (YwsC-His) was produced in the ΔywsC strain and purified from the lysozyme-treated lysate of the transformant by Ni-nitrilotriacetic acid affinity chromatography. Western blot analysis revealed that the YwsC-His protein consists of two subunits, the 44-kDa and 33-kDa proteins, which are encoded by in-phase overlapping in the ywsC gene. 14C-labeled PGA was synthesized by the purified proteins from l-[14C]-glutamate in the presence of ATP and MnCl2, through an acylphosphate intermediate, indicating that the ywsC gene encodes PGA synthetase (EC 6.3.2), a crucial enzyme in PGA biosynthesis.
Some Bacillus strains produce γ-polyglutamic acid (PGA), an amino acid polymer that consists of only d-glutamic acid or d- and l-glutamic acid polymerized through γ-glutamyl bonds, as a capsular or an extracellular viscous material (6). PGA was first discovered as a component of the capsule of Bacillus anthracis (19) and Bacillus mesentericus (18) and was isolated from the culture medium of Bacillus subtilis (7). Since then, a number of bacteria producing PGA, including B. subtilis (8, 17, 21, 22), Bacillus licheniformis (9, 42), and Bacillus megaterium (13, 41), have been reported. PGA is a main constituent of the sticky material in natto, a Japanese traditional food made from soybeans that have been steamed and then fermented by B. subtilis (14).
Concerning PGA biosynthesis, Makino et al. reported cloning of three genes, capBCA, responsible for capsular PGA biosynthesis from B. anthracis, and the gene products occurred together as membrane-associated proteins in the Escherichia coli transformant (26, 27, 43). The complete genome sequence of B. subtilis 168, in which ywsC and ywtAB were found to be highly homologous to the capBCA genes of B. anthracis, has been made available in databases (31). Recently, pgsBCA genes for PGA biosynthesis were also cloned from B. subtilis IFO3336, and their sequences were found to be the same as those of the ywsC and ywtAB genes of B. subtilis 168 (2). These three genes seem to be involved in PGA production; however, little is known about the function of each gene product in PGA biosynthesis.
In this paper, we describe the cloning and gene disruption of the ywsC and ywtAB genes, which are responsible for PGA production in B. subtilis IFO16449, a strain isolated from natto, and we also describe the characterization of the YwsC 44-kDa and 33-kDa proteins, which catalyze the biosynthesis of PGA from l-glutamate, a crucial enzyme in PGA production.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Bacillus subtilis IFO16449, a strain isolated from commercial fermented soybeans (29), was used in this study. E. coli JM109 and plasmids pUC19 and pHY300PLK were purchased from Takara Shuzo Co. (Kyoto, Japan). Plasmids pMutin4MCS (44) and pC194 (15) were used for gene disruption. B. subtilis and E. coli were routinely grown at 37°C overnight in Luria-Bertani (LB) medium (35). If necessary, ampicillin, tetracycline, and chloramphenicol were added to final concentrations of 50, 20, and 5 μg/ml, respectively.
For PGA production of B. subtilis IFO16449 and its derivatives, we used a chemically defined medium (PGA medium) consisting of 2% glucose, 2% sodium l-glutamate, 1% (NH4)2SO4, 0.1% Na2HPO4, 0.1% KH2PO4, 0.05% MgSO4 · 7H2O, 0.002% MnCl2 · 4H2O, 0.005% FeCl3 · 6H2O, and 0.5 μg of biotin (pH 7.5) per ml (22). Spizizen minimal medium (1) was also used for cell growth. Cells were grown in a 200-ml conical flask containing 30 ml of the medium at 37°C on a rotary shaker. The E. coli transformant was cultured in LB medium supplemented with 2% sodium l-glutamate and 0.05% MnCl2 · 4H2O for PGA production.
General DNA manipulation.
Restriction enzymes, T4 DNA ligase, and other DNA-modifying enzymes were purchased from Takara Shuzo (Kyoto, Japan). Genomic DNA of B. subtilis IFO16449 was isolated by the method of Saito and Miura (34). DNA sequencing was performed by the dideoxy chain termination method (36) using a Thermo Sequence premixed cycle sequencing kit (Amersham) on a Hitachi SQ-5500 DNA sequencer. Nucleotide sequences were analyzed using the Genetyx Mac computer program version 10 (Software Development Co., Ltd., Tokyo, Japan).
Cloning of ywsC and ywtABC genes.
A sense primer, 5′-GTGTGACTATACGTCAGAAAGG-3′, and an antisense primer, 5′-TGCGAATTGCTGTGTGCCGATCG-3′, were designed on the basis of the sequence of the ywsC gene of B. subtilis 168 (31). A DNA fragment was amplified by PCR, and the amplified fragment was inserted into the SmaI site of pUC19 to analyze the nucleotide sequence. When used as a probe, the fragment was labeled with horseradish peroxidase using an ECL labeling kit (Amersham) according to the protocol of the supplier. The genomic DNA of B. subtilis IFO16449 was digested with BglII, and 4- to 5-kb fragments were collected and inserted into the BamHI site of pUC19. The ligation mixture was transformed into E. coli JM109, and the transformants were screened by colony hybridization using the PCR-amplified fragment as a probe. Hybridization was carried out overnight in an ECL hybridization buffer (Amersham), and the plasmid isolated from a positive clone was named pBF1 (Fig. 1). Transformation of E. coli JM109 was carried out by the method of Inoue et al. (16).
FIG. 1.

Physical map of the insert of pBF1. Construction of the plasmid is described in Materials and Methods.
Disruption of ywsC and ywtAB genes.
Plasmid pUC19-CS with a cat-spac cassette was constructed by the following procedure. A 0.6-kb SmaI fragment containing a spac promoter was excised from pMutin4MCS and inserted into the SmaI site of pUC19, generating pUC19-S. A 1.0-kb NaeI-XhoII fragment containig the cat gene was cut from pC194 and inserted into SacI of pUC19-S. The constructed plasmid, in which the cat gene was located upstream of the spac promoter, was named pUC19-CS. Plasmids for disruption of the ywsC, ywtA, and ywtB genes were constructed as follows. The 1.6-kb cat-spac cassette of pUC19-CS was excised with EcoRI and inserted into the BalI, NaeI, and Van91I sites of pBF1 (Fig. 1). The constructed plasmids, named pBCD, pBAD, and pBBD, respectively, were transformed into B. subtilis IFO16449 for construction of ΔywsC, ΔywtA, and ΔywtB strains by the method of Cutting and Horn (10).
Construction of a plasmid containing ywsC with a histidine-tagged codon.
Plasmid pYWSC containing ywsC with a histidine-tagged codon was constructed by the following procedure. The 3′-terminal region of the ywsC gene was amplified by PCR with pBF1 DNA as a template and the following two primers: a sense primer, 5′-AAGAAATCGGTTACCCACC-3′ (underlining indicating the BstPI site), and an antisense primer, 5′-ATCGAGGATCCCTAATGATGATGATGATGATGGCTTACGAGCTGCTT-3′ (underlining and italic letters indicate the BamHI site and histidine-tagged codon, respectively). The amplified fragment was digested with BstPI and BamHI, and a 300-bp BstPI-BamHI fragment was recovered from agarose gel. The DNA fragment including the 34-bp palindromic transcriptional terminator was amplified by PCR with pBF1 DNA as a template and the following two primers: a sense primer, 5′-ATCGAGGATCCTTCAAAAAAGAGAGTGTC-3′ (underlining indicating the BamHI site), and an antisense primer, 5′-CTTCTTGAGCTCGCCAGTGTGTTCACT-3′ (underlining indicating the SacI site). The amplified fragment was cut with BamHI and SacI, and a 150-bp BamHI-SacI fragment was collected. These two fragments were then inserted in order of the restriction sites into the BstPI-SacI fragment (4.7 kb) cut from pBF1, generating pYHT. A 2.0-kb fragment containing ywsC with a histidine-tagged codon and the transcriptional terminator region was excised with SphI and SacI from pYHT and then inserted into the SmaI site of pHY300PLK. The resulting plasmid, named pYWSC, was transformed into the ΔywsC strain by the method of Cutting and Horn (10).
Deletion of the ywsC gene.
The plasmid pYHT mentioned above was digested with SacII, and then both of the termini were blunted by T4 DNA polymerase. The 2-bp short linear plasmid was circularized by the ligase reaction, and the circular plasmid was cut with SphI and SacI to excise the 2.0-kb fragment containing ywsC with a histidine-tagged codon. The fragment was then inserted into the SmaI site of pHY300PLK, generating pYWSC-Sac, which was transformed into the ΔywsC strain in the manner described above.
Northern blot analysis.
Cells were grown in PGA medium as described above at 37°C for 4 h with shaking. Total RNA was extracted from the cells with Isogen (Nippon Gene) according to the manual of the supplier. After denaturation, 10 μg of the total RNA was subjected to electrophoresis on a 1% agarose gel and blotted onto a Hybond N+ membrane (Amersham). Hybridization was performed using a digoxigenin-labeled DNA probe according to the protocol of the supplier (Rosh Diagnostics). The SacII-BstPI fragment (0.7 kb) excised from pBF1(Fig. 1) was used as a probe specific for ywsC mRNA.
Western blot analysis.
Histidine-tagged proteins were separated by sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride membrane (Bio-Rad). The blotted membrane was incubated with nickel-nitrilotriacetic acid (NTA)-alkaline phosphatase (AP) conjugate (Qiagen), and the proteins were visualized with 5-bromo-4-chloro-3-indolylphosphate (Sigma) and nitro blue tetrazolium (Sigma) according to the manual of the supplier.
Production and purification of histidine-tagged YwsC.
The ΔywsC strain harboring pYWSC was grown at 37°C for 10 h with shaking in PGA medium containing 20 μg of tetracycline and 5 μg of chloramphenicol per ml. Cells were harvested from 1,000 ml of culture by centrifugation and resuspended in 100 ml of 10 mM imidazole buffer (10 mM imidazole, 20 mM Tris-HCl, 0.3 M NaCl, 20% glycerol, pH 8.0). Lysozyme (Sigma) and DNase I (Sigma) were added to final concentrations of 1.0 and 0.1 mg/ml, respectively. After incubation for 30 min at 37°C, 4 ml of 5% Triton X-100 was added to the lysate, and the incubation was continued for 1 h at 4°C with shaking. The lysate was centrifuged at 100,000 × g for 30 min, and the supernatant was applied to an Ni-NTA-agarose column (2 ml; Qiagen) equilibrated with 10 mM imidazole buffer. The column was washed with 10 mM imidazole buffer, and then histidine-tagged proteins were eluted with imidazole buffer (20 mM Tris-HCl, 0.3 M NaCl, 20% glycerol, pH 8.0) containing a stepwise gradient of imidazole from 100 to 500 mM.
Assay for PGA synthetase.
PGA synthetase activity was measured by the method of Troy (42) with slight modifications. The assay mixture contained 50 mM Tris-HCl buffer (pH 8.0), 22.2 μM l-[U-14C]glutamate (9.25 GBq/mmol; Moravec Biochemicals, Inc.), 20 mM ATP, 4 mM MnCl2, and enzyme solution (30 μg of protein), in a final volume of 50 μl. The incubation was carried out for 2 h at 30°C, and 10 μl of the mixture was spotted onto Whatman no. 3MM paper and chromatographed using the solvent system of 1-butanol-acetic acid-water (40:8.8:20, by volume). The chromatogram was dried, the 14C-labeled PGA at the origin was cut into a vial, and the radioactivity in a toluene scintillation solution was measured. The synthesized [14C]PGA was also detected by SDS-PAGE after the mixture had been treated with proteinase K (Sigma) and then concentrated by a centrifugal filter (Millipore). Protein was determined by the method of Lowry et al. (25) with bovine serum albumin as a standard.
Detection of the product of ATP cleavage.
AMP, ADP, and ATP were separated on a phosphoethyleneimine-cellulose thin-layer plate by the method of Randerath and Randerath (32). Incubation was carried out at 30°C for 2 h in a reaction mixture (50 μl) containing 50 mM Tris-HCl buffer (pH 8.0), 20 mM l-glutamate, 89 μM [8-14C]ATP (2.07 GBq/mmol; New England Nuclear), 10 mM ATP, 4 mM MnCl2, and enzyme solution (30 μg of protein).
SDS-PAGE and PGA determination.
SDS-PAGE of proteins was performed using a 10% polyacrylamide gel at pH 7.0 by the method of Laemmli (23). The gel was stained with Coomassie brilliant blue R-250. The molecular weight of proteins was estimated by comparison with protein markers (Daiichi Pure Chemistry Co.). SDS-PAGE of PGA was also done using a 10% polyacrylamide gel by the method of Yamaguchi et al. (45). The amount of PGA was determined by an amino acid analyzer after acid hydrolysis according to the method of Ogawa et al. (30).
Nucleotide sequence accession number.
The sequences of the ywsC and ywtABC genes of B. subtilis IFO16449 have been submitted to the GenBank, EMBL, and DDBJ databases and can be accessed under accession number AB046355.
RESULTS
Cloning of the genes required for PGA production.
A 4.2-kb DNA fragment containing the ywsC and ywtABC genes was cloned from B. subtilis IFO16449. The ywsC and ywtAB genes were almost identical to the ywsC-ywtAB and pgsBCA genes of B. subtilis 168 (31) and B. subtilis IFO3336 (2), respectively. The E. coli transformant harboring pBF1 was grown in LB medium containing 2% l-glutamate and 0.05% MnCl2, and PGA productivity was examined by SDS-PAGE. As shown in Fig. 2, PGA was produced by the transformant, indicating that the cloned 4.2-kb DNA fragment possesses genes required for PGA production. Northern blot analysis showed that one transcript hybridized with a probe specific for ywsC mRNA is approximately 3.0 kb, suggesting that the ywsC and ywtABC genes consist of an operon (data not shown).
FIG. 2.
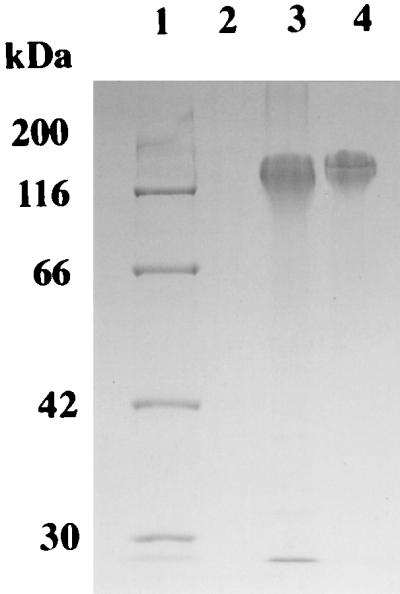
SDS-PAGE analysis of PGA production of E. coli transformants. PGA was purified by the ethanol precipitation method (2). Lane 1, protein markers; lane 2, E. coli JM109/pUC19; lane 3, E. coli JM109/pBF1; lane 4, purified PGA.
Disruption of ywsC, ywtA, and ywtB genes.
To determine which gene plays a central role in PGA production in B. subtilis, we constructed three gene disruptants defective in ywsC, ywtA, and ywtB as described in Materials and Methods. Disruption of each gene was verified by Southern blot analysis (data not shown). The ΔywsC, ΔywtA, and ΔywtB strains were grown in PGA medium, and cell growth was monitored to investigate the physiological effect of gene disruption, but all three disruptants grew nearly two times faster than the parent strain, indicating that these genes are not required for cell growth in this strain.
PGA productivity of the three strains was examined by SDS-PAGE analysis (Fig. 3). PGA was not detected from the supernatant of the ΔywsC or ΔywtA strain. On the other hand, the ΔywtB strain produced PGA, but PGA productivity was greatly reduced. These results indicate that the ywsC and ywtAB genes are required and that YwsC and YwtA are essential for PGA production. YwtB seems to be required for maximum PGA production.
FIG. 3.

Construction and PGA productivity of gene disruptants. (A) Structures of ywsC, ywtA, and ywtB gene disruptants. The cat gene and spac promoter are indicated by a solid box and a solid arrow, respectively. The position of the putative hairpin structure is indicated by an open circle. (B) SDS-PAGE analysis of PGA production. Cells were grown in PGA medium, and the culture supernatant was subjected to SDS-PAGE. Lane 1, protein markers; lane 2, wild-type strain; lane 3, ΔywsC strain; lane 4, ΔywtA strain; lane 5, ΔywtB strain.
Expression of YwsC-His proteins.
The results described above suggested that YwsC or YwtA is a PGA synthetase in B. subtilis IFO16449. We therefore constructed pYWSC containing a histidine-tagged codon at the 3′ end of the ywsC gene and produced histidine-tagged YwsC under the control of the promoter of the ywsC gene as described in Materials and Methods. Plasmid pYWSC was transformed into the B. subtilis IFO16449 ΔywsC strain, and PGA productivity of this complemented strain was investigated. As shown in Fig. 4A, the ΔywsC strain harboring pYWSC produced PGA, indicating that the histidine-tagged ywsC gene is expressed in this transformant.
FIG. 4.

Production of YwsC-His in B. subtilis ΔywsC strain. (A) SDS-PAGE analysis of PGA production by B. subtilis IFO16449 and transformants. Cells were grown in PGA medium, and the culture supernatant was subjected to SDS-PAGE. (B) Western blot analysis of YwsC-His. Proteins prepared from the wild-type strain and transformants were separated by SDS-PAGE, and YwsC-His was detected with an Ni-NTA-AP conjugate. Arrows indicate the positions of subunits of YwsC-His. Lane 1, protein markers; lane 2, wild strain; lane 3, ΔywsC strain; lane 4, ΔywsC/pYWSC strain; lane 5, ΔywsC/pYWSC-Sac strain. The positions of the 33- and 44-kDa proteins are shown (arrows).
Cells harboring pYWSC were grown in PGA medium. Crude extract was prepared, and the histidine-tagged YwsC protein was analyzed by Western blotting using an Ni-NTA-AP conjugate, which can be used for detection of recombinant protein with an accessible histidine tag. As shown in Fig. 4B, two major signals of 44 and 33 kDa were detected. The molecular mass of the 44-kDa protein was in good agreement with the molecular weight of 44,070 deduced from the ywsC gene. Since the control experiments using the crude extracts from the wild-type and ΔywsC strains gave no signal, the 33-kDa protein is also the product of the ywsC gene as a histidine-tagged protein.
It is possible that the 33-kDa protein is derived from the 44-kDa protein by posttranslational modification. To test this possibility, we constructed a 2-bp deletion mutant at the SacII site in the ywsC gene for generating a stop codon 42 codons downstream from the SacII site. This plasmid was transformed into the B. subtilis IFO16449 ΔywsC strain, and histidine-tagged YwsC protein was analyzed by Western blotting (Fig. 4B). The strain harboring the deletion plasmid could not produce the 44-kDa protein but produced the 33-kDa protein, indicating that the 33-kDa protein is not derived from the 44-kDa protein but rather is translated independently. Based on this result and the molecular mass of the 33-kDa protein, we propose that the initiation codon of the 33-kDa protein is at ATG 287 bases downstream from the initiation codon of the 44-kDa protein (Fig. 5).
FIG. 5.

Nucleotide sequence of the ywsC gene. The possible promoter sequence (−35, TTGAGA; −10, TATACT), similar to the consensus sequences of B. subtilis ςA (20), is indicated as −35 and −10. SD, Shine-Dalgarno sequence. Possible translation initiation codons are enclosed in small boxes.
Purification and properties of YwsC-His proteins.
YwsC-His proteins were purified from the solubilized membrane fraction by nickel affinity chromatography, and the purified proteins were analyzed by SDS-PAGE (Fig. 6). Two bands corresponding to 44 and 33 kDa, which were identical to the proteins detected by Western blot analysis, were detected by Coomassie brilliant blue staining. On the other hand, gel filtration using Sephacryl S-200 HR showed that the molecular weight of purified YwsC-His is 150,000, suggesting that the 44-kDa and 33-kDa proteins constitute the YwsC protein.
FIG. 6.

SDS-PAGE of purified YwsC-His. Cells were grown in PGA medium (lanes 2 and 4) or Spizizen minimal medium containing 2% l-glutamate (lane 3). Purification of YwsC-His is described in Materials and Methods. Lane 1, protein markers; lanes 2 and 3, ΔywsC/pYWSC strain; lane 4, ΔywsC/pYWSC-Sac strain.
In vitro PGA synthesis by purified YwsC-His was carried out by the incorporation of [14C]glutamate into PGA. The reaction mixture contained 50 mM Tris-HCl (pH 8.0), 22.2 μM l-[U-14C]glutamate (9.25 GBq/mmol), 20 mM ATP, 4 mM MnCl2 and enzyme solution. 14C-labeled PGA synthesized in the reaction mixture was detected by SDS-PAGE, indicating that the molecular weight was between 20,000 and 140,000 (data not shown). Table 1 shows the radioactivity of the synthesized [14C]PGA, in which a certain amount of the incorporated radioactivity was found under the conditions mentioned above. The activity of this enzyme was dependent on ATP and Mn2+ ions and was decreased in the absence of ATP or Mn2+ ions. When d-[14C]glutamate was used instead of l-[14C]glutamate, little incorporation of the radioactivity was found, indicating that this enzyme incorporates only l-isomer.
TABLE 1.
Effect of incubation conditions on γ-polyglutamate synthetase activity
| Assay mixturea | Relative activity (% of control) |
|---|---|
| Complete (control) | 100 |
| −ATP | 4 |
| −MnCl2 | 4 |
| +d-[14C]-glutamateb | 3 |
| +CoCl2 | 6 |
| +CaCl2 | 6 |
| +MgCl2 | 0 |
| +GTP | 33 |
| +CTP | 4 |
| +UTP | 0 |
The complete reaction mixture, containing 30 μg of purified YwsC-His, is described in Materials and Methods. In these conditions, 3,865 cpm of [14C]PGA was detected in 50 μl of reaction mixture. Divalent cations were added at 4 mM. Nucleotides were added at 20 mM.
72.7 μM d-[1-14C]-glutamate (2.04 GBq/mmol) was added to the reaction mixture.
When CTP, GTP, and UTP were used as substrates in this reaction, GTP exhibited lower activity. Of the divalent ions examined, Mn2+ ions stimulated the synthetic reaction, but neither Mg2+, Ca2+, nor Co2+ ions stimulated the reaction. Considering the properties of the enzyme, the synthetic enzyme may be called γ-polyglutamic acid synthetase (EC class 6.3.2) according to the recommended nomenclature (28).
Amide bond ligases are divided into two subgroups depending on the mode of ATP activation (37). One subgroup activates the γ-carboxyl group via adenylate-substrate intermediates, and the other activates the γ-carboxyl group via acylphosphate intermediates. Figure 7 shows the hydrolysis product of 14C-labeled ATP. Only ADP was detected as a nucleotide released from ATP in the enzymatically synthetic reaction of polyglutamate from l-glutamate. This result indicates that ADP + Pi is the product of the reaction and that YwsC is an ADP-forming amide bond ligase.
FIG. 7.

Detection of the products of ATP cleavage. The polyglutamate synthetase reaction was carried out as described in Materials and Methods. At the times indicated, 10 μl of the reaction mixture was chromatographed on a polyethyleneimine-cellulose thin-layer plate and developed in sodium formate. Nucleotides were detected by their UV absorption, and radioactivities of labeled ATP (Rf 0.31), ADP (Rf 0.64), and AMP (Rf 0.83) were determined using a liquid scintillation counter. Symbols: •, ADP; ○, AMP.
Requirement of 44-kDa and 33-kDa YwsC proteins for PGA biosynthesis.
When the ΔywsC strain harboring pYWSC was grown in Spizizen minimal medium containing 2% l-glutamate, neither PGA nor the 33-kDa protein was produced (data not shown). Also, no PGA was produced from the ΔywsC strain harboring pYWSC-Sac (Fig. 4A, lane 5). These results suggested that both the 44-kDa and 33-kDa proteins are required for PGA biosynthesis in B. subtilis IFO16449.
We then purified the 44-kDa protein from the ΔywsC strain harboring pYWSC grown in Spizizen minimal medium containing 2% l-glutamate, and the 33-kDa protein was purified from the ΔywsC strain harboring pYWSC-Sac (Fig. 6, lane 3 and lane 4, respectively). PGA synthetase activity was assayed using the purified enzyme solution. Incorporation of l-[14C]glutamate into PGA was not observed in the reaction mixture containing only the 44-kDa or 33-kDa protein (data not shown). This result indicates that both subunits of the 44-kDa and 33-kDa proteins are required for PGA synthesis.
DISCUSSION
Troy et al. reported that the poly-γ-d-glutamyl capsule of B. licheniformis is synthesized by a membrane-associated enzyme reaction in which the synthetic complex catalyzes the activation, racemization, and polymerization of l-glutamate to form poly-γ-d-glutamate; however, the membrane enzymes catalyzing the reaction have not yet been purified (12, 42). A similar experiment using the membrane fraction of the same strain has been carried out by other researchers, but still no purified enzyme has been obtained (24). On the other hand, the capBCA genes responsible for biosynthesis of the poly-γ-glutamyl capsule of B. anthracis have been cloned in an E. coli transformant, but there is no information on the function of each gene product (27).
In this study, we purified histidine-tagged YwsC, which consists of two subunits (44-kDa and 33-kDa proteins), and we demonstrated that the purified proteins catalyze the biosynthesis of high-molecular-weight poly-γ-glutamate from l-glutamate in the presence of ATP and Mn2+ ions, indicating that the YwsC proteins may be called poly-γ-glutamate synthetase and belong to the ATP-dependent amide ligase superfamily (4, 11).
ADP was only detected as a nucleotide released from ATP in the enzymatically synthetic reaction of polyglutamate from l-glutamate (Fig. 7). This result clearly indicates that YwsC proteins catalyze PGA polymerization in an ADP-forming manner, like enzymes belonging to the family of ADP-forming amide bond ligases, such as murein ligases involved in peptidoglycan biosynthesis (5) and folyl-γ-polyglutamate ligases (38). In contrast, Troy et al. reported that a membrane-bound enzyme preparation from B. licheniformis catalyzes the biosynthesis of poly-γ-glutamate through an adenyl-l-glutamate intermediate, on the basis of detection of AMP as a major product from ATP (12), which differs from our result. Another difference between the enzyme activities is the requirement for a divalent cation; the membrane-associated enzyme of B. licheniformis requires Mg2+ ions, but the purified YwsC proteins require Mn2+ ions.
Makino et al. reported that gene products of capBCA from B. anthracis consisted of four proteins (CapB, CapB′, CapC, and CapA) as membrane-associated proteins in recombinant cells of E. coli, indicating that the capB gene is an overlapping gene for the CapB and B′ proteins (27). Western blot and SDS-PAGE analysis of the YwsC-His protein indicated that the gene product is comprised of 44-kDa and 33-kDa proteins (Fig. 4 and 5). Furthermore, the results of deletion of the gene and estimation of the molecular weight by SDS-PAGE suggest that the initiation codon of the 33-kDa protein may be the ATG 287 bases downstream from the original one and upstream of which a putative Shine-Dalgarno sequence (GGAGA) exists (Fig. 5), indicating that ywsC is also an overlapping gene for the 44-kDa and 33-kDa proteins. It is conceivable that the YwsC 33-kDa protein may be expressed in the same manner as the CapB′ protein of B. anthracis, but the molecular weight of the former is greater than that of the latter, while the molecular weight of the YwsC 44-kDa protein is nearly the same as that of the CapB protein.
YwtA is a hydrophobic protein similar to CapC, which has been suggested to exist as a membrane-integrated protein in the outer surface in B. anthracis (27). On the other hand, a homology search revealed that YwtA has similarity with SmpA of Staphylococcus epidermidis, which functions as a transport protein for erythromycin (33). These facts indicate that YwtA plays a role in transportation of γ-polyglutamate synthesized inside the membrane of B. subtilis. Actually, the ΔywtA strain failed to secrete PGA into the medium (Fig. 3), but purified YwsC was able to synthesize a certain amount of PGA (Table 1).
Eveland et al. reported that there are four common homologous regions in enzymes belonging to the family of ADP-forming amide bond ligases, including a large number of murein and folyl-γ-polyglutamate ligases, the four regions of which were also found to exist in the CapB protein of B. anthracis (11). More recently, Sheng et al. demonstrated high degrees of similarity in the structure and function of UDP-N-acetylmuramoyl-l-alanine-d-glutamate and folyl-γ-polyglutamate ligases, although the degree of similarity of the amino acid sequences is not so high (37).
The fact that there are four common regions in the YwsC 44-kDa and CapB proteins and the fact that ADP is only released as a product of ATP in the biosynthetic reaction of γ-polyglutamate from l-glutamate by the 44-kDa and 33-kDa proteins suggest that the YwsC 44-kDa protein belongs to the family of ADP-forming amide bond ligases. However, it seems likely that the YwsC protein, working as a γ-polyglutamate synthetase, differs in structure from other synthetic enzymes of the ADP-forming ligase family, since enzymes such as murein ligases and folyl-γ-polyglutamate ligase function as monomers, while γ-polyglutamate synthesis is catalyzed by two subunits, the YwsC 44-kDa and 33-kDa proteins.
Acknowledgments
This work was partly supported by a grant-in-aid for scientific research from the Ministry of Education, Science, and Culture of Japan (grant 11660081) and by a grant from the Takano Life Science Research Foundation (to Y.T.).
REFERENCES
- 1.Anagnostopoulos, C., and J. Spizizen. 1960. Requirements for transformation in Bacillus subtilis. J. Biol. Chem. 81:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashiuchi, M., K. Soda, and H. Misono. 1999. A poly-γ-glutamate synthetic system of Bacillus subtilis IFO3336: gene cloning and biochemical analysis of poly-γ-glutamate produced by Escherichia coli clone cells. Biochem. Biophys. Res. Commun. 263:6–12. [DOI] [PubMed] [Google Scholar]
- 3.Avakyan, A. A., L. N. Katz, K. N. Levina, and I. B. Pavlova. 1965. Structure and composition of the Bacillus anthracis capsule. J. Bacteriol. 90:1082–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertrand, J. A., E. Fanchon, L. Martin, L. Chantalat, G. Auger, D. Blanot, J. van Heijenoort, and O. Dideberg. 2000. “Open” structures of MurD: domain movements and structural similarities with folylpolyglutamate synthetase. J. Mol. Biol. 301:1257–1266. [DOI] [PubMed] [Google Scholar]
- 5.Bertrand, J. A., G. Auger, E. Fanchon, L. Martin, D. Blanot, J. van Heijienoort, and O. Dideberg. 1997. Crystal structure of UDP-N-acetylmuramoyl-l-alanine:d-glutamate ligase from Escherichia coli. EMBO J. 16:3416–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birrer, G. A., A. Crowmick, and R. A. Gross. 1994. γ-Poly(glutamic acid) formation by Bacillus licheniformis 9945A: physiological and biochemical studies. Int. J. Biol. Macromol. 16:265–275. [DOI] [PubMed] [Google Scholar]
- 7.Bovarnick, M. 1942. The formation of extracellular d(-)glutamic acid polypeptide by Bacillus subtilis. J. Biol. Chem. 145:415–424. [Google Scholar]
- 8.Cheng, C., Y. Asada, and T. Aida. 1989. Production of γ-polyglutamic acid by Bacillus licheniformis A35 under denitrifying conditions. Agric. Biol. Chem. 53:2369–2375. [Google Scholar]
- 9.Cromwick, A., and R. A. Gross. 1995. Effects of manganese(II) on Bacillus licheniformis ATCC 9945A physiology and γ-poly(glutamic acid) formation. Int. J. Biol. Macromol. 17:259–267. [DOI] [PubMed] [Google Scholar]
- 10.Cutting, S. M., and P. B. Vander Horn. 1990. Genetic analysis, p. 27–74. In C. R. Harwood and S. M. Cutting (ed.), Molecular biological methods for Bacillus. John Wiley & Sons Ltd., Chichester, United Kingdom.
- 11.Eveland, S. S., D. L. Pompliano, and M. S. Anderson. 1997. Conditionally lethal Escherichia coli murein mutants contain point defects that map to regions conserved among murein and folyl poly-γ-glutamate ligases: identification of a ligase superfamily. Biochemistry 36:6223–6229. [DOI] [PubMed] [Google Scholar]
- 12.Gardner, J. M., and F. A. Troy. 1979. Chemistry and biosynthesis of the poly(γ-d-glutamyl) capsule in Bacillus licheniformis: activation, racemization, and polymerization of glutamic acid by a membranous polyglutamyl synthetase complex. J. Biol. Chem. 254:6262–6269. [PubMed] [Google Scholar]
- 13.Guex-Holzer, S., and J. Tomcsik. 1956. The isolation and chemical nature of capsular and cell-wall haptens in a Bacillus species. J. Gen. Microbiol. 14:14–25. [DOI] [PubMed] [Google Scholar]
- 14.Hara, T., Y. Fujio, and S. Ueda. 1982. Polyglutamate production by Bacillus subtilis (natto). J. Appl. Biochem. 4:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horinouchi, S., and B. Weisblum. 1982. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 150:815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue, H., H. Nojima, and H. Okayama. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 9:23–28. [DOI] [PubMed] [Google Scholar]
- 17.Ito, Y., T. Tanaka, T. Ohmachi, and Y. Asada. 1996. Glutamic acid independent production of poly(γ-glutamic acid) by Bacillus subtilis TAM-4. Biosci. Biotechnol. Biochem. 60:1239–1242. [Google Scholar]
- 18.Ivanovics, G., and V. Bruckner. 1937. Chemishe und immunologiche Studien uber den Mechanismus der milzbrandinfektin und Immunitat; die chemische Struktur der Kapselsubstanz des Milzbrandbazillus und der serologisch identischen spezifischen Subtanz des Bazillus mesentericus. Z. Immunitatsforsch. 90:304–318. [Google Scholar]
- 19.Ivanovics, G., and L. Erdos. 1937. Ein Beitrag zum Wesen der Kapselsubstnz des Milizbrandbazillus. Z. Immunitatsforsch. 90:5–19. [Google Scholar]
- 20.Johnson, W. C., C. P. Moran, and R. Losick. 1983. Two RNA polymerase sigma factors from Bacillus subtilis discriminate between overlapping promoters for a developmentally regulated gene. Nature 302:800–804. [DOI] [PubMed] [Google Scholar]
- 21.Kubota, H., T. Matsunobu, K. Uotani, H. Takabe, A. Satoh, T. Tanaka, and M. Taniguchi. 1993. Production of poly (γ-glutamic acid) by Bacillus subtilis F-2-01. Biosci. Biotechnol. Biochem. 57:1212–1213. [DOI] [PubMed] [Google Scholar]
- 22.Kunioka, M., and A. Goto. 1994. Biosynthesis of poly(γ-glutamic acid) from L-glutamic acid, citric acid and ammonium sulfate in Bacillus subtilis IFO3335. Appl. Microbiol. Biotechnol. 40:867–872. [Google Scholar]
- 23.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. [DOI] [PubMed] [Google Scholar]
- 24.Leonard, C. G., and R. D. Housewright. 1963. Polyglutamic acid synthesis by cell-free extracts of Bacillus licheniformis. Biochim. Biophys. Acta 73:530–532. [DOI] [PubMed] [Google Scholar]
- 25.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275. [PubMed] [Google Scholar]
- 26.Makino, S., C. Sasakawa, I. Uchida, N. Terakado, and M. Yoshikawa. 1988. Cloning and CO2-dependent expression of the genetic region for encapsulation from Bacillus anthracis. Mol. Microbiol. 2:371–376. [DOI] [PubMed] [Google Scholar]
- 27.Makino, S., I. Uchida, N. Terakado, C. Sasakawa, and M. Yoshikawa. 1989. Molecular characterization and protein analysis of the cap region, which is essential for encapsulation in Bacillus anthracis. J. Bacteriol. 171:722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nomenclature Committee of the International Union of Biochemistry and Molecular Biology. 1992. Enzyme nomenclature. Academic Press Inc., New York, N.Y.
- 29.Ogawa, Y., H. Hosoyama, M. Hamano, and H. Motai. 1991. Purification and properties of γ-glutamyltranspeptidase from Bacillus subtilis (natto). Agric. Biol. Chem. 55:2971–2977. [PubMed] [Google Scholar]
- 30.Ogawa, Y., F. Yamaguchi, K. Yuasa, and Y. Tahara. 1997. Efficient production of γ-polyglutamic acid by Bacillus subtilis (natto) in jar fermenters. Biosci. Biotechnol. Biochem. 61:1684–1687. [DOI] [PubMed] [Google Scholar]
- 31.Presecan, E., I. Moszer, L. Boursier, R. H. C. Ramos, V. de la Fuente, M.-F. Hullo, C. Lelong, S. Schleich, A. Sekowska, B. H. Song, G. Villani, F. Kunst, A. Danchin, and P. Glaser. 1997. The Bacillus subtilis genome from gerBC (311 degrees) to licR (334 degrees). Microbiology 143:3313–3328. [DOI] [PubMed] [Google Scholar]
- 32.Randerath, K., and E. Randerath. 1967. Thin-layer separation methods for nucleic acid derivatives. Methods Enzymol. 12A:323–350. [Google Scholar]
- 33.Ross, J. I., E. A. Eady, J. H. Cove, S. Baumberg. 1995. Identification of a chromosomally encoded ABC-transport system with which the staphylococcal erythromycin exporter MsrA may interact. Gene 153:93–98. [DOI] [PubMed] [Google Scholar]
- 34.Saito, H., and K. Miura. 1963. Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim. Biophys. Acta 72:619–629. [PubMed] [Google Scholar]
- 35.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring, Harbor, N.Y.
- 36.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheng, Y., X. Sun, Y. Shen, A. L. Bognar, E. N. Baker, and C. A. Smith. 2000. Structural and functional similarities in the ADP-forming amide bond ligase superfamily: implications for a substrate-induced conformational change in folylpolyglutamate synthetase. J. Mol. Biol. 302:427–440. [DOI] [PubMed] [Google Scholar]
- 38.Sun, X., A. L. Bognar, E. N. Baker, and C. A. Smith. 1998. Structural homologies with ATP-and folate-binding enzymes in the crystal structure of folylpolyglutamate synthetase. Proc. Natl. Acad. Sci. USA 95:6647–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorne, C. B., C. G. Gomez, G. R. Blind, and R. D. Housewright. 1953. Synthesis of glutamic acid and glutamyl polypeptide by Bacillus anthracis. J. Bacteriol. 65:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thorne, C. B., and C. G. Leonard. 1958. Isolation of D-and L-glutamyl polypeptides from culture filtrates of Bacillus subtilis. J. Biol. Chem. 233:1109–1112. [PubMed] [Google Scholar]
- 41.Torii, M., O. Kurimura, S. Utsumi, H. Nozu, and T. Amano. 1959. Decapsulation of Bacillus megaterium. Biken J. 2:265–276. [Google Scholar]
- 42.Troy, F. A. 1973. Chemistry and biosynthesis of the poly(γ-D-glutamyl) capsule in Bacillus licheniformis: properties of the membrane-modiated biosynthetic reaction. J. Biol. Chem. 248:305–315. [PubMed] [Google Scholar]
- 43.Uchida. I., K. Hashimoto, S. Makino, C. Sasakawa, M. Yoshikawa, and N. Terakado. 1987. Restriction map of a capsule plasmid of Bacillus anthracis. Plasmid 18:178–181. [DOI] [PubMed] [Google Scholar]
- 44.Vagner, V., E. Dervyn, and S. D. Ehrlich. 1998. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144:3097–3104. [DOI] [PubMed] [Google Scholar]
- 45.Yamaguchi, F., Y. Ogawa, M. Kikuchi, K. Yuasa, and H. Motai,. 1996. Detection of γ-polyglutamic acid (γ-PGA) by SDS-PAGE. Biosci. Biotechnol. Biochem. 60:255–258. [DOI] [PubMed] [Google Scholar]