

Abstract
The Arabidopsis NPR1 gene is essential in activating systemic, inducible plant defense responses. To gain a better understanding of NPR1 function, we conducted a yeast two-hybrid screening procedure and identified a differential interaction between NPR1 and all known members of the Arabidopsis TGA family of basic leucine zipper transcription factors. In the electrophoretic mobility shift assay, NPR1 substantially increased the binding of TGA2 to its cognate promoter element (as-1) as well as to a positive salicylic acid–inducible element (LS7) and a negative element (LS5) in the promoter of the pathogenesis-related PR-1 gene. Proteins encoded by npr1 mutants interacted poorly with TGA2 and did not substantially increase TGA2 binding to the as-1, LS5, or LS7 elements, thus establishing a link between the loss of disease resistance and the loss of TGA2 interaction and NPR1-enhanced DNA binding. Coupled with observations that the DNA binding activity of TGA factors is deregulated in npr1 plants, the results suggest that NPR1-mediated DNA binding of TGA2 is critical for activation of defense genes.
INTRODUCTION
Localized exposure of plants to certain microbes can induce subsequent resistance to a broad range of otherwise virulent pathogens in distant, noninfected tissues. To date, two types of such induced, or acquired, systemic disease resistance have been described: systemic acquired resistance (SAR) (Ryals et al., 1996) and induced systemic resistance (ISR) (van Loon et al., 1998). The former is triggered by pathogens that cause cell death at the site of infection and is associated with the activation of pathogenesis-related (PR) genes and the accumulation of PR proteins (Ward et al., 1991). Salicylic acid (SA) is a key metabolite in the deployment of SAR. Transgenic plants expressing the bacterial nahG gene encoding a SA-degrading enzyme are unable to mount SAR and are impaired in PR gene expression (Delaney et al., 1994). ISR is induced by certain nonpathogenic, root-colonizing rhizobacteria (van Loon et al., 1998). In contrast to SAR, ISR is not associated with the activation of PR genes and is presumed to rely on the activation of genes yet to be identified (Pieterse et al., 1998). ISR is independent of SA but requires intact jasmonate and ethylene response pathways (Pieterse et al., 1998).
Despite their differences, SAR and ISR both require the activity of the Arabidopsis NPR1 gene (Cao et al., 1994; Pieterse et al., 1998), also known as NIM1 (Ryals et al., 1997). Plants containing mutations at the NPR1 locus are compromised in their ability to mount effective SAR and ISR and are more susceptible to normally incompatible pathogens (Cao et al., 1994; Delaney et al., 1995; Pieterse et al., 1998). Transgenic plants expressing lesser amounts of NPR1 are also more susceptible to infection from compatible pathogens, whereas lines overexpressing NPR1 display enhanced resistance against bacterial and fungal pathogens (Cao et al., 1998). Genetic analyses have established that NPR1 acts downstream of SA in the SAR signal transduction pathway (Cao et al., 1994; Delaney et al., 1995) and downstream of jasmonate and ethylene in the ISR signaling pathway (Pieterse et al., 1998).
The NPR1 gene has recently been isolated and predicted to encode a protein containing ankyrin-like repeats (Cao et al., 1997; Ryals et al., 1997), which is a motif known to be responsible for mediating protein–protein interactions (Sedgwick and Smerdon, 1999). It has been proposed that NPR1 possesses extensive sequence conservation with the mammalian protein IκBα and that it may represent the plant homolog of IκBα (Ryals et al., 1997). IκB proteins bind to and control the transcriptional activity of NF-κB, a member of the Rel family of transcription factors and a critical regulator of several cellular events, such as response to stress and to pathogens (Baldwin, 1996).
To gain a better understanding of NPR1 function, we and others have conducted yeast two-hybrid screening procedures to identify proteins capable of interacting with NPR1. Recently, one of these groups (Zhang et al., 1999) screened a tomato cDNA library with a putative tomato homolog of NPR1 (TomNPR1) and reported that it interacted with a basic leucine zipper (bZIP) transcription factor called NIF1 (for NPR1 Interacting Factor1). NIF1 belongs to a subclass of bZIP proteins that includes the Arabidopsis TGA factors. Subsequently, these authors showed by using directed yeast two-hybrid tests that Arabidopsis NPR1 interacts with three members of the Arabidopsis TGA family: AHBP-1b/TGA2, OBF5/TGA5, and TGA6. Mutant derivatives of NPR1 that abolish SAR in the plant failed to interact in the yeast two-hybrid system with the two TGA factors tested; these were TGA2 and TGA6 (Zhang et al., 1999). Furthermore, it was shown that TGA2 binds to sequences in the PR-1 promoter. Although it has been reported that TGA2 binds to sequences required for SA induction of PR-1 (Zhang et al., 1999), the probe used in the electrophoretic mobility shift assay (EMSA) contained two cis-acting elements previously identified by linker scanning mutagenesis (Lebel et al., 1998). These elements, linker scan7 (LS7) and LS5, contain the sequence ACGTCA, which is complementary to TGACGT, the sequence reported to be the preferred substrate for TGA factors (Schindler et al., 1992). LS7 represents a positive regulatory element involved in 2,6-dichloroisonicotinic acid and SA responsiveness, whereas LS5 is a negative regulatory element (Lebel et al., 1998). To demonstrate that TGA2 binds the PR-1 promoter sequence required for SA induction (LS7), Zhang et al. (1999) used a mutant competitor in which both the LS5 and LS7 sequences were mutated. Therefore, it is still not clear whether TGA factors bind to the SA response element, the negative regulatory element, or both. Nevertheless, the results they obtained suggest that NPR1 may regulate the expression of genes involved in SAR by interacting with a subclass of bZIP transcription factors (Zhang et al., 1999).
Here, we report on our yeast two-hybrid screen, using Arabidopsis NPR1 as bait. Our results confirm those of Zhang et al. (1999) and expand upon them to show the following points. (1) NPR1 interacts with two additional members of the Arabidopsis TGA family, TGA3 and a newly identified member, TGA7, but does not interact with TGA1 and TGA4. (2) TGA factors bind to both a positive (LS7) and a negative (LS5) regulatory element found within the PR-1 promoter. (3) NPR1 enhances the binding of an interacting TGA factor to a well-characterized cis-acting element (as-1 from the cauliflower mosaic virus 35S promoter) as well as to the PR-1 LS5 and LS7 elements, but it has little or no effect on the DNA binding properties of a noninteracting TGA factor. (4) NPR1 fractionates to both the cytoplasmic and nuclear fractions in unstimulated Arabidopsis tissues. And (5) the DNA binding activity of TGA factors is deregulated in npr1 plants. These results suggest that the activity of TGA2, and possibly of other members of the TGA family, is regulated by their interaction with NPR1 and that disruption of the interaction between these proteins can compromise systemic induced disease resistance.
RESULTS
NPR1 Differentially Interacts with bZIP Transcription Factors Belonging to the TGA Family
To investigate whether NPR1 interacts with other proteins, we screened a yeast two-hybrid library using a full-length NPR1–GAL4 DNA binding domain fusion as bait. From ∼107 transformants, eight colonies producing blue color on the X-gal filter test and capable of growth on medium lacking tryptophan, leucine, and histidine and supplemented with 5 mM 3-amino-1′,2′,4′-triazole (His−/3-AT medium) were identified. Sequence analysis of the GAL4 transcriptional activation domain library inserts that were present in the individual colonies expressing the lacZ gene and capable of growth on His−/3-AT medium revealed that all but one encoded bZIP transcription factors belonging to the TGA family. All bZIP-interacting clones contained truncated cDNA copies lacking the basic domain and leucine zipper coding regions. Three of the interactors had been previously identified as OBF5/TGA5 (Zhang et al., 1993) or TGA6 (Xiang et al., 1995) and shown to interact with NPR1 (Zhang et al., 1999). Two interactors corresponded to TGA3 (Miao et al., 1994), whereas the remaining two represent a new member of the Arabidopsis family, which we have named TGA7. Detailed characterization of TGA7 will appear elsewhere (C. Després and P.R. Fobert, manuscript in preparation). Thus, our screen identified two new interactors of NPR1 (TGA3 and TGA7) but failed to recover TGA2, which has previously been shown to interact with NPR1 (Zhang et al., 1999).
The full-length coding region of TGA3 and other known members of the TGA family that were not recovered during our two-hybrid screenings, namely, TGA1 (Schindler et al., 1992), TGA2 (Kawata et al., 1992), and OBF4/TGA4 (Zhang et al., 1993), were isolated and fused to the transcriptional activation domain of GAL4 to study their interaction with NPR1. Table 1 shows that only cells containing the NPR1–GAL4 DNA binding domain fusion with either the TGA2–GAL4 transcriptional activation domain fusion or the TGA3–GAL4 transcriptional activation domain fusion were capable of growth on His−/3-AT medium and activation of the lacZ reporter gene. Cells containing the NPR1–GAL4 DNA binding domain fusion with either the TGA1–GAL4 transcriptional activation domain fusion or the TGA4–GAL4 transcriptional activation domain fusion did not activate either reporter gene (Table 1). In Figure 1, results of an immunoblot analysis show that TGA1 and TGA4 were expressed at levels similar to TGA2 in the yeast cells. Together, these results indicate that NPR1 specifically interacts with five of the seven known members of the Arabidopsis TGA family.
Table 1.
Differential Interaction between NPR1 and TGA Factors
| DB Hybrida | TA Hybridb | Growthc | Color d |
|---|---|---|---|
| NPR1 | TGA1 | − | White |
| NPR1 | TGA2 | + | Blue |
| NPR1 | TGA3 | + | Blue |
| NPR1 | TGA4 | − | White |
| NPR1 | — | − | White |
| — | TGA3 | − | White |
DB hybrid indicates a GAL4 DNA binding domain fusion protein. The dash indicates that the yeast cells contained the empty GAL4 DB vector.
TA hybrid indicates a GAL4 transcriptional activation domain fusion protein. The dash indicates that the yeast cells contained the empty GAL4 DB vector.
Growth on medium lacking leucine, tryptophan, and histidine but containing 3-AT monitors the activation of the HIS3 reporter gene by interacting GAL4 fusions. The plus signs indicate growth; the minus signs indicate no growth.
Activation of lacZ reporter gene monitored by X-gal filter assay from colonies grown on medium lacking leucine and tryptophan. Blue color was detected after ∼1 hr at 30°C; white color was observed after 1 and 16 hrs at 30°C.
Figure 1.
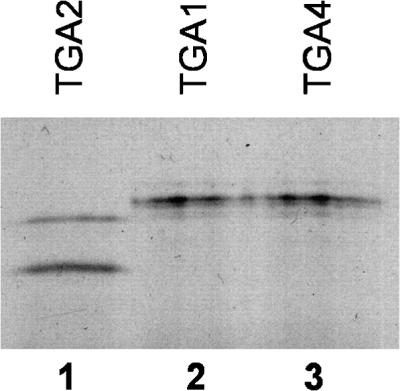
Expression of TGA Proteins in the Yeast Two-Hybrid System.
Immunoblot analysis of yeast cells demonstrating that noninteracting TGA–GAL4 transcriptional activation domain fusions are expressed at levels similar to those of interacting TGA–GAL4 transcriptional activation domain fusions. Yeast cells containing the NPR1–GAL4 DNA binding domain fusion together with the TGA2–GAL4 transcriptional activation domain (lane 1), the TGA1–GAL4 transcriptional activation domain (lane 2), or the TGA4–GAL4 transcriptional activation domain (lane 3) fusions were grown on synthetic dextrose medium lacking leucine and tryptophan only. Proteins were extracted, fractionated on 8% SDS–polyacrylamide gels, and immunoblotted with an anti–GAL4 transcriptional activation domain antibody.
In control experiments, yeast cells transformed with either the NPR1–GAL4 DNA binding domain fusion together with the GAL4 transcriptional activation domain vector alone or the TGA3–GAL4 transcriptional activation domain fusion together with the GAL4 DNA binding domain vector alone were unable to grow on His−/3-AT medium or activate the lacZ reporter gene (Table 1). This indicates that the result observed with the NPR1–GAL4 DNA binding domain fusion and the TGA3–GAL4 transcriptional activation domain fusion is due to a specific interaction between the two proteins. Identical results were obtained with all of the TGA factors (data not shown).
NPR1 Mutant Proteins That Fail to Mount SAR in the Plant Fail to Interact with TGA2 and TGA3 in the Yeast Two-Hybrid System
Several NPR1 mutant alleles have now been characterized (Glazebrook et al., 1996; Cao et al., 1997; Ryals et al., 1997; Shah et al., 1997). Some contain single base substitutions that translate into single amino acid changes, whereas others harbor premature stop codons or frameshift mutations. We selected three NPR1 mutants, npr1-1, nim1-2, and npr1-2, that harbor single amino acid replacements and are unambiguously compromised in their ability to mount SAR (Cao et al., 1997; Ryals et al., 1997), and we tested, using the yeast two-hybrid system, their capacity to interact with TGA2 and TGA3. One of these factors, TGA2, has already been shown not to interact with NPR1-1 and NPR1-2 (Zhang et al., 1999). NPR1-1 and NIM1-2 each contain a histidine-to-tyrosine replacement in one of the putative ankyrin-like repeats at positions 334 and 300, respectively, whereas NPR1-2 contains a cysteine-to-tyrosine replacement at position 150. Table 2 shows that in contrast to the wild-type NPR1, none of the mutants was able to interact significantly with either TGA2 or TGA3, as determined by the lack of growth on His−/3-AT medium and failure to activate the lacZ reporter gene. Figure 2 shows an immunoblot analysis of yeast cells harboring the mutant NPR1 fusions, confirming that these were expressed at similar amounts as the wild-type NPR1 fusion. The fact that NPR1 mutants that fail to mount systemic resistance have also lost the ability to interact with TGA2 and TGA3 suggests that these transcription factors, and possibly other TGA family members, are involved in the NPR1 signal transduction pathway.
Table 2.
NPR1 Mutants Do Not Interact with TGA3 in Yeast
| DB Hybrida | TA Hybridb | Growthc | Color d |
|---|---|---|---|
| NPR1 | TGA2 | + | Blue |
| NPR1-1 | TGA2 | − | White |
| NPR1-2 | TGA2 | − | White |
| NIM1-2 | TGA2 | − | White |
| NPR1 | TGA3 | + | Blue |
| NPR1-1 | TGA3 | − | White |
| NPR1-2 | TGA3 | − | White |
| NIM1-2 | TGA3 | − | White |
DB hybrid indicates a GAL4 DNA binding domain fusion protein.
TA hybrid indicates a GAL4 transcriptional activation domain fusion protein.
Growth on medium lacking leucine, tryptophan, and histidine but containing 3-AT monitors the activation of the HIS3 reporter gene by interacting GAL4 fusions. The plus signs indicate growth; the minus signs indicate no growth.
Activation of the lacZ reporter gene monitored by X-gal filter assay from colonies grown on medium lacking leucine and tryptophan. Blue color was detected after ∼1 hr at 30°C; white color was observed after 1 and 16 hrs at 30°C.
Figure 2.

Expression of Wild-Type and Mutant NPR1 Proteins in the Yeast Two-Hybrid System.
Immunoblot analysis of yeast cells demonstrating similar expression levels of wild-type and mutant NPR1 proteins. Yeast cells containing the NPR1–GAL4 DNA binding domain fusion (lane 1), the NPR1-1–GAL4 DNA binding domain fusion (lane 2), the NPR1-2–GAL4 DNA binding domain fusion (lane 3), the NIM1-2–GAL4 DNA binding domain fusion (lane 4), or the empty GAL4 DNA binding domain vector (lane 5), together with a full-length TGA3–GAL4 transcriptional activation domain fusion (lanes 1 to 5), were grown on synthetic dextrose medium lacking leucine and tryptophan only. Proteins were extracted, fractionated on 8% SDS–polyacrylamide gels, and immunoblotted with an anti-NPR1 antibody.
TGA2 but Not TGA4 Physically Interacts with Wild-Type NPR1 in Vitro
To confirm the results from the yeast two-hybrid analysis and to study the physical interaction between TGA factors and NPR1, we incubated purified biotinylated wild-type NPR1 bound to streptavidin–agarose beads with in vitro–translated radiolabeled TGA2. This member of the TGA family was chosen for further studies because it accounts for ∼50% of the TGA binding activity in Arabidopsis leaves (Lam and Lam, 1995). Analysis of the bound fraction by SDS-PAGE, as presented in Figure 3A, revealed the presence of a signal on the autoradiogram, demonstrating the existence of an in vitro interaction between these two proteins (lane 2). When the protein–protein interaction assay was performed with biotinylated NIM1-2, the signal either was not observed or was greatly reduced (Figure 3A, lane 3). Immunoblot analysis revealed that similar amounts of biotinylated NPR1 and NIM1-2 were coupled to the streptavidin–agarose beads (Figure 3B), indicating that the weaker signal was due to a greatly reduced interaction between TGA2 and this mutant form of NPR1. These results are in agreement with those obtained with the yeast two-hybrid system (Tables 1 and 2), thus confirming, in vitro, the authenticity of the lack of interaction between mutant forms of NPR1 and TGA factors.
Figure 3.

TGA2 Interacts with NPR1 but Not with the NIM1-2 Mutant Protein in Vitro.
(A) Biotinylated NPR1 (lanes 2 and 5) or NIM1-2 (lane 3) was produced in Escherichia coli and coupled to streptavidin–agarose beads. The beads were mixed and incubated for 1 hr with in vitro–translated radiolabeled TGA2 (lanes 2 and 3) or TGA4 (lane 5). After washing the beads, the bound proteins were eluted by boiling in SDS sample buffer, resolved by 8% SDS-PAGE, and visualized by autoradiography. Lanes 1 and 4 contain 20% of the input radiolabeled TGA2 and TGA4, respectively.
(B) Immunoblot analysis of NPR1 and NIM1-2 biotinylated resins, demonstrating that they contain similar amounts of coupled proteins. Biotinylated NPR1 and NIM1-2 were produced in E. coli and coupled to streptavidin–agarose beads. The NPR1 (lane 1) and the NIM1-2 (lane 2) beads were boiled in SDS sample buffer, resolved by 8% SDS-PAGE, and immunoblotted with an anti-NPR1 antibody. The position of the intact NPR1 fusion protein is marked by an arrow.
We also tested the in vitro protein–protein interaction between NPR1 and TGA4, a member of the TGA family that did not interact with NPR1 in the yeast two-hybrid system (Table 1). Using an amount of TGA4 comparable with that of TGA2 as shown in Figure 3A, lane 1, we observed a noticeably reduced signal (Figure 3A, lane 5). Therefore, NPR1 and TGA4 appear to interact weakly, even though this interaction was not apparent in the yeast two-hybrid system.
NPR1 Enhances the Binding of TGA2 to Its Cognate as-1 DNA Element
It is well documented that IκBα can inhibit the binding of NF-κB to the κB element as well as displace preformed NF-κB–DNA complexes (Zabel and Baeuerle, 1990). To test whether NPR1 behaves similarly to IκBα, we investigated the effect of NPR1 on the binding of TGA factors to the as-1 promoter element. This element is one of the best-characterized DNA sequences recognized by the TGA family of transcription factors (Lam et al., 1989).
Figure 4 shows results from EMSA experiments performed in the presence or absence of in vitro–translated TGA2, NPR1, or both. As anticipated, a retarded band was observed when TGA2 was present in the binding mixture (Figure 4B, lane 2). This complex is most likely formed by the specific binding of TGA2 to the as-1 element because incubation of the probe with the reticulocyte extract alone or after transcription and translation of a plasmid containing TGA2 with a mutated version of the as-1 element as probe did not yield a retarded band (Figure 4A, lanes 1 and 3). The addition of NPR1 to the TGA2–as-1 binding mixture substantially increased the amount of complex formed in an EMSA reaction containing only TGA2–as-1 (Figure 4B, cf. lanes 2 and 3). The observations that neither NPR1 nor NIM1-2 bound to the as-1 probe in the absence of TGA2 (Figure 4A, lanes 4 and 5) and that NPR1 did not alter the mobility of the TGA2–as-1 complex (Figure 4B, cf. lanes 2 and 3) suggest that NPR1 enhances the DNA binding activity of TGA2 without binding stably to the complex. This is reminiscent of the human viral protein TAX, which stimulates DNA binding of bZIP proteins (Wagner and Green, 1993) and of the Arabidopsis OBP1 protein, which stimulates DNA binding of TGA4 and TGA5 to the as-1 element (Zhang et al., 1995). Both TAX and OBP1 have been shown to stimulate the DNA binding activity of their interacting partner without altering the mobility of the complex in the EMSA.
Figure 4.

Binding of TGA2 to the as-1 Element Is Enhanced by NPR1 but Not by the NIM1-2 Mutant Protein.
(A) EMSA using in vitro–translated TGA2 (lanes 2 and 3), NPR1 (lane 4), NIM1-2 (lane 5), or the reticulocyte (Retic.) extract alone (lane 1), together with the wild type (lanes 1, 2, 4, and 5) or a mutated version of the as-1 element (lane 3) as the probe. An equal amount of reticulocyte lysate extract was used in all lanes.
(B) EMSA using in vitro–translated TGA2 (lanes 2 to 4) or TGA4 (lanes 5 and 6) and the as-1 DNA element as a probe (lanes 1 to 6). NPR1 (lane 3) or NIM1-2 (lane 4) was added to the TGA2–as-1 EMSA reaction. NPR1 (lane 6) was added to the TGA4–as-1 EMSA reaction. The open arrow indicates the position of the TGA2 complex, the black arrow indicates the position of the TGA4 complex, the asterisk indicates the position of the slower migrating band detected in reactions containing NPR1 proteins, and the bracket indicates the position of the free probe. An equal amount of reticulocyte lysate extract was used in all lanes.
(C) Autoradiography of 4 μL of reticulocyte lysates after separation on 8% SDS–polyacrylamide gels, demonstrating the presence of similar amounts of NPR1 and NIM1-2 used in the EMSAs. The arrow marked 66 kD denotes the apparent molecular mass of NPR1 and NIM1-2.
It is noteworthy that upon addition of NPR1, a slower migrating band became apparent (Figure 4B, lane 3). Although the exact nature of this complex is not known, it could represent a higher order complex composed of NPR1–TGA2–DNA. However, attempts to supershift this band with an anti-NPR1 antibody were unsuccessful (data not shown), suggesting that the complex with lower mobility could be nonspecific. The addition of a mutant NPR1 protein, NIM1-2, that fails to mount SAR (Ryals et al., 1997), that did not interact with TGA2 in the yeast two-hybrid system (Table 2), and that interacted weakly in the in vitro assay (Figure 3) did not substantially increase the amount of TGA2–as-1 complex formed (Figure 4B, lane 4).
The addition of NPR1 resulted in only a slight increase in the binding of TGA4 to the as-1 element (Figure 4B, lane 6). This is consistent with the weak interaction between NPR1 and TGA4 observed in the in vitro assay (Figure 3, lane 5). Similar amounts of in vitro–translated NPR1 and NIM1-2 were used in these experiments, as shown in Figure 4C.
NPR1 Enhances the Binding of TGA2 to a Positive (LS7) and a Negative (LS5) Regulatory Element of the Arabidopsis PR-1 Promoter
As detailed in the Introduction, two functional cis-acting elements in the Arabidopsis PR-1 promoter (LS5 and LS7), previously identified by linker scanning mutagenesis (Lebel et al., 1998), contain the preferred target sequence for TGA factors. Therefore, we tested whether these elements might represent binding sites for TGA2 and whether NPR1 could enhance the binding of TGA2 to these elements.
Figure 5A depicts the PR-1 promoter region surrounding the LS7 element and the oligonucleotides used in the EMSA. Figure 5B shows that TGA2 can bind to the LS7 element (lane 3). Binding of TGA2 to the as-1 element is also shown (Figure 5B, lane 2). Incubation of the LS7 probe with the reticulocyte extract alone or after transcription and translation of a plasmid containing the NPR1 or nim1-2 coding sequence did not result in a retarded band (Figure 5B, lanes 1, 5, and 6), indicating that the complex observed in the EMSA was due to binding of TGA2 and that NPR1 does not bind to the LS7 element. Furthermore, binding of TGA2 to the LS7 element was specific, because no complex formation was observed when a mutated version of the LS7 element (LS7mut) was used as a probe (Figure 5B, lane 4). The addition of in vitro–translated NPR1 to the binding mixture substantially increased the amount of complex formed on the LS7 element when compared with an EMSA reaction containing only TGA2 (Figure 5C, cf. lanes 3 and 2). The addition of NIM1-2 did not substantially increase the amount of TGA2–LS7 complex formed (Figure 5C, lane 4). These results are similar to those observed with the as-1 probe (Figure 4), indicating that NPR1 can enhance the binding of TGA2 to at least two different promoter elements. TGA4 was also able to bind to the LS7 element, and, as observed with the as-1 element (Figure 4), addition of NPR1 did not result in a substantial increase in DNA binding activity (data not shown).
Figure 5.

Binding of TGA2 to the LS7 SA Response Element of the Arabidopsis PR-1 Promoter Is Enhanced by NPR1 but Not by the NIM1-2 Mutant Protein.
(A) Nucleotide sequence of the PR-1 promoter surrounding the LS7 element (boxed). The numbers indicate the position of the element relative to the PR-1 RNA start site. LS7 and LS7mut indicate the sequence of the wild-type and mutant oligonucleotides, respectively, used as probes in EMSAs. Note that the LS7 oligonucleotide contains mutated LS6 and LS8 sites. Dashes indicate nucleotides identical to the top strand of the wild-type sequence.
(B) EMSA using in vitro–translated TGA2 (lanes 2 to 4), NPR1 (lane 5), NIM1-2 (lane 6), or the reticulocyte (Retic.) extract alone (lane 1), together with the wild-type as-1 probe (lane 2), the wild-type LS7 (lanes 1, 3, 5, and 6), or a mutated version of the LS7 element (lane 4) as the probe.
(C) EMSA using in vitro–translated TGA2 (lanes 2 to 4) and the LS7 DNA element as a probe (lanes 1 to 4). NPR1 (lane 3) or NIM1-2 (lane 4) was added to the TGA2–LS7 EMSA reaction. Equal amounts of NPR1 and NIM1-2 were used in these experiments (see Figure 4C). For (B) and (C), an equal amount of reticulocyte lysate extract was used in all lanes. The black arrow indicates the position of the TGA2 complex. Retic. indicates that these EMSA reactions contain only protein from the reticulocyte lysate extract.
Figure 6A depicts the PR-1 promoter region surrounding the LS5 element and the oligonucleotides used in the EMSA as competitors. Figure 6B shows the results of an EMSA demonstrating that TGA2 can bind to the LS5 element (lane 4). Binding of TGA2 to the as-1 element is also shown (Figure 6B, lane 3). Incubation of the LS5 probe with NPR1 or NIM1-2 produced from the reticulocyte did not result in a retarded band (Figure 5B, lanes 1 and 2), indicating that the complex observed in the EMSA was due to binding of TGA2 and that NPR1 does not bind to the LS5 element. The results of competition experiments using the LS5 probe and a 10-fold excess of various mutant oligonucleotides are shown in lanes 5 through 10. All of the oligonucleotides were able to compete with the LS5 probe for TGA2 binding, with the exception of mutant LS5mut, indicating that on the fragment tested, only the region corresponding to LS5 is able to bind TGA2. Figure 6C illustrates that the addition of NPR1 in the EMSA reaction increased the binding of TGA2 to the LS5 probe (lane 2), whereas mutant NIM1-2 did not (lane 3). Together, these results indicate that TGA2 can bind both the positive (LS7) and the negative (LS5) elements of the PR-1 promoter and that NPR1 enhances this binding.
Figure 6.

Binding of TGA2 to the LS5 Negative Regulatory Element of the Arabidopsis PR-1 Promoter Is Enhanced by NPR1 but Not by the NIM1-2 Mutant Protein.
(A) Nucleotide sequence of the PR-1 promoter surrounding the LS5 element (boxed). The numbers indicate the position of the element relative to the PR-1 RNA start site. LS5 indicates the sequence of the wild-type oligonucleotide used as a probe in EMSA. The sequences of mutant oligonucleotide LS3mut, LS4mut, LS5mut, and LS6mut used as competitors in EMSAs are also shown. Dashes indicate nucleotides identical to the top strand of wild-type LS5.
(B) EMSA using in vitro–translated TGA2 (lanes 3 to 10), NPR1 (lane 1), or NIM1-2 (lane 2), together with the wild-type as-1 probe (lane 3) or the wild-type LS5 probe (lanes 1, 2, and 4 to 10). The as-1 (lane 5), LS5 (lane 6), LS3mut (lane 7), LS4mut (lane 8), LS5mut (lane 9), and LS6mut (lane 10) denote double-stranded oligonucleotides added in the EMSA reactions as competitors. The black arrow indicates the position of the TGA2 complex. An equal amount of reticulocyte lysate extract was used in all lanes.
(C) EMSA using in vitro–translated TGA2 (lanes 1 to 3) and the LS5 DNA element as a probe (lanes 1 to 3). NPR1 (lane 2) or NIM1-2 (lane 3) was added to the TGA2–LS5 EMSA reaction. Equal amounts of NPR1 and NIM1-2 were used in these experiments (see Figure 4C). The black arrow indicates the position of the TGA2 complex; the asterisk indicates the position of the slower migrating band detected in reactions containing NPR1 proteins. An equal amount of reticulocyte lysate extract was used in all lanes.
NPR1 Localizes to the Nucleus
The in vitro stimulation of TGA2 DNA binding activity by NPR1 suggested to us that the in vivo role of NPR1 could be exerted, at least in part, in the nucleus. To test whether endogenous NPR1 can localize to the nucleus, we isolated Percoll-purified nuclei and subjected nuclear proteins to immunoblot analysis using anti-NPR1 antibodies. Microscopic evaluation of the purified nuclei indicated that the preparations were free of visible contamination and that the nuclei were intact (data not shown). The quality of the fractionation was also demonstrated by monitoring the activity of localization-specific marker enzymes, as described in Methods.
The quality of the anti-NPR1 antiserum was tested for specific recognition of NPR1 in immunoblot analysis. Figure 7A indicates that the anti-NPR1 antiserum reacted against a crude lysate from an E. coli strain overexpressing NPR1 (lanes 2 and 3) but not against a lysate from a strain expressing an empty vector (lane 1) or an unrelated His6-tagged protein (lane 4) (His6-ICK2, for interactor of cyclin-dependent kinase2; Lui et al., 2000). In addition, when we attempted to deplete the anti-NPR1 antibody with streptavidin–agarose beads bound by an unrelated biotinylated protein (the 13-kD peptide expressed from the Pinpoint Xa-1 vector), a 66-kD band was detected in the Arabidopsis protein extract even after a 1-min exposure (Figure 7B, lane 1). However, when the antiserum was depleted of the anti-NPR1 antibody with the streptavidin–agarose beads bound by biotinylated NPR1, no signal could be detected from the Arabidopsis protein extract, even after a 10-min exposure (lane 2). This result indicates that the 66-kD band detected in the plant extracts is related to NPR1. The results of the immunoblot analysis after fractionation of the Arabidopsis proteins into a cytoplasmic and a nuclear fraction is presented in Figure 7C. The results indicate that in unstimulated Arabidopsis leaves, NPR1 was detected in the cytoplasmic fraction (lane 1) as well as in the purified nuclear fraction (lane 2).
Figure 7.

Immunoblot Analysis of NPR1 Protein in Wild-Type Unstimulated Arabidopsis.
(A) Specificity of the NPR1 antibody. Aliquots (5 μg) of total E. coli protein extracts expressed from an empty pBluescript SK+ vector (Stratagene) (lane 1), a Pinpoint–NPR1 fusion (lanes 2 and 3), or a pRSETB–ICK2 fusion (lane 4) were fractionated on 8% SDS–polyacrylamide gels and immunoblotted with an anti-NPR1 antibody. Lane 2 contains one-tenth (0.5 μg) the amount of proteins loaded in the other lanes.
(B) Specificity of the anti-NPR1 antibody. Arabidopsis proteins from the cytoplasmic (240 μg) and the nuclear (20 μg) fractions were pooled and then fractionated on 8% SDS–polyacrylamide gels (lanes 1 and 2). Proteins in lane 1 were immunoblotted with the anti-NPR1 antibody that had been depleted by treatment with streptavidin–agarose beads bound by the protein moiety produced from the Pinpoint vector. Proteins in lane 2 were immunoblotted with the anti-NPR1 antibody that was depleted on streptavidin–agarose beads bound by the NPR1 fusion protein produced from the Pinpoint–NPR1 vector. Exposure of the films was 1 min for lane 1 and 10 min for lane 2.
(C) Localization of NPR1 in Arabidopsis extracts. Tissues were ground in a nuclei-stabilizing buffer. Nuclei were pelleted, and the supernatant was kept as the cytoplasmic fraction (lane 1). Nuclei were further purified on a Percoll gradient and lysed, and the recovered proteins were kept as the nuclear fraction (lane 2). The proteins were immunoblotted with an anti-NPR1 antibody.
Numbers in (B) and (C) refer to the molecular mass standards expressed in kilodaltons. The cytoplasmic (240 μg) and the nuclear (20 μg) fractions were separated on 3- and 1.5-mm-thick SDS–polyacrylamide gels, respectively. The running conditions were otherwise identical. The position of the NPR1 bands relative to one another was determined by superimposing the molecular mass standards run with each gel. In (A) to (C), arrows denote the position of the NPR1 protein.
NPR1 Controls the TGA (as-1 Binding Factors) Binding Activity in Vivo
It has been reported that TGA factors represent a significant component of the Arabidopsis as-1 binding activity (Lam and Lam, 1995). Therefore, as a first step to investigate the in vivo role of the NPR1–TGA interaction, we conducted EMSAs with the as-1 probe and protein extracts from wild-type and npr1 mutant Arabidopsis plants. Figure 8A shows the results of an EMSA in which as-1 binding activity was determined in leaf extracts of wild-type and npr1 Arabidopsis plants after treatment with water or SA. For comparison, binding of TGA3 produced in vitro is shown in Figure 8A, lane 2. In extracts from water-treated wild-type plants, as-1 binding activity was weak (Figure 8A, lane 3). However, after a 1-hr treatment with 0.5 mM SA, binding to the as-1 probe was substantially increased (Figure 8A, lane 4). To demonstrate binding specificity, a 10-fold excess of cold wild-type or mutant as-1 oligonucleotide was added to the EMSA reaction as competitor. As shown in Figure 8B, the as-1–specific complex was greatly reduced by the addition of cold as-1 (lane 3) when compared with an EMSA reaction in which no competitor was added (lane 1). However, the as-1–specific complex was not dramatically reduced by the addition of cold mutant as-1 (lane 2) when compared with an EMSA reaction in which no competitor was added (lane 1).
Figure 8.

TGA Binding Activity Is Deregulated in npr1 Mutant Plants.
(A) Extracts from wild-type (Wt; lanes 3 and 4) or npr1 mutant (lanes 5 and 6) Arabidopsis plants treated for 1 hr with water (lanes 3 and 5) or with a solution of 0.5 mM SA (lanes 4 and 6) were subjected to an EMSA, using the as-1 element as a probe (lanes 1 to 6). The EMSA reaction in lane 1 contained no protein (−Prot.), whereas the one in lane 2 contained in vitro–translated TGA3.
(B) Extracts from SA-treated Arabidopsis plants were subjected to an EMSA using the as-1 element as a probe (lanes 1 to 3). A 10-fold molar excess of mutant as-1 (lane 2) or wild-type as-1 (lane 3) DNA was added as competitor. In lane 1, the dash denotes a reaction in which no competitor was added.
In (A) and (B), the brackets indicate the position of the specific complexes; asterisks indicate the positions of a nonspecific complex.
In contrast to the wild type, extracts from water-treated npr1 plants displayed a substantial amount of as-1 binding activity (Figure 8A, cf. lanes 5 and 3), and the addition of SA did not increase this binding activity (cf. lanes 6 and 5). These results indicate that in vivo, the binding activity of the TGA factors is regulated by NPR1, although they do not address whether this is necessarily by a direct physical interaction.
DISCUSSION
Using a combination of in vivo and in vitro assays, we have identified a differential physical interaction between a key positive regulator of acquired and induced systemic resistance (SAR/ISR), NPR1, and members of the Arabidopsis TGA family of bZIP transcription factors. Although the precise in vivo function of these transcription factors is unclear, they are known to bind to promoter elements, such as the cauliflower mosaic virus 35S as-1 element, that are inducible by phytohormones implicated in SAR and ISR signaling (Xiang et al., 1996; van Loon et al., 1998). As described in this study, at least one TGA factor (TGA2) is also capable of binding in the EMSA to the SA response element LS7 of the PR-1 gene promoter, which is a gene that is known to be induced during SAR (Ward et al., 1991) and whose expression is abolished in npr1 mutants (Cao et al., 1994; Delaney et al., 1995). Furthermore, TGA factors, such as tobacco TGA1a, act as genuine transcription factors in heterologous and homologous in vitro transcription systems (Katagiri et al., 1990; Yamazaki et al., 1990) and can transcriptionally activate as-1–containing promoters in plants (Lam et al., 1989) and yeast (Rüth et al., 1994). Taken together, these results suggest that a subgroup of the TGA factors acts immediately downstream of NPR1 in the SAR and ISR signaling pathways and that ultimately the expression of genes involved in SAR and ISR could be controlled by the NPR1-regulated binding of TGA factors to cis-acting promoter elements. In support of this last statement, we present two lines of evidence suggesting that NPR1 is capable of regulating TGA factor activity.
First, in vitro–synthesized NPR1 enhanced the binding of an NPR1-interacting TGA factor (TGA2) to its cognate promoter elements in the EMSA (Figures 4 to 6). This enhancement of binding was specific, because it was not observed with a mutant version of NPR1 (NIM1-2) that fails to mount SAR (Ryals et al., 1997), that did not interact with TGA factors in the yeast two-hybrid system (Figure 2), and that bound weakly with TGA2 in the in vitro assay (Figure 3). Also, this enhancement was not observed when a member of the TGA family (TGA4) that failed to interact with NPR1 in the yeast two-hybrid system and interacted only weakly with NPR1 in the in vitro binding assay was tested. These results suggest that interaction with NPR1 in the plant cell could stimulate the binding of specific TGA factors to cognate promoter elements found in genes involved in SAR and ISR, resulting in their activation. Accordingly, the inability of npr1 mutants to establish SAR and/or ISR would be attributed to the production of NPR1 proteins with greatly reduced ability to interact with TGA factors (Table 2 and Figures 3 to 6). In the absence of wild-type NPR1, TGA factors that normally interact with this regulator would be unable to productively bind cognate promoter elements found in genes involved in SAR and ISR, thus failing to activate gene transcription. Enhanced disease resistance and PR gene expression observed in plants overexpressing NPR1 (Cao et al., 1998) could be explained by enhanced TGA binding to promoter elements stimulated by the increased availability of NPR1 protein.
The observation that the DNA binding activity of TGA4 is not greatly affected by NPR1 suggests that the activity of members of the TGA family that interact poorly with NPR1 may not be regulated by NPR1 in vivo. Such TGA factors could be involved in regulating the expression of genes in an NPR1-independent manner. Thus, differential interaction with NPR1 may be one mechanism by which the in vivo specificity and activity of TGA transcription factors, which bind to the same target sequences in vitro, are regulated.
The observation that NPR1 enhanced TGA2 interaction to a negative regulatory element (LS5) of the PR1 promoter (Figure 6) suggests that in addition to a stimulatory role, the NPR1-regulated DNA binding activity of the TGA factors could be required for the transcriptional repression of genes. There are numerous reports of mammalian and Drosophila transcription factors acting as repressors. One example is provided by the transcription factor Dorsal, the Drosophila homolog of NF-κB, which activates the twist gene but represses the zen gene (Lehming et al., 1994). The specific promoter context surrounding NPR1-regulated TGA binding sites and the identity of the TGA factors bound to NPR1 are two variables that could influence whether TGA factor binding results in transcriptional activation or repression.
The second line of evidence suggesting that NPR1 is important for TGA function comes from a comparison of as-1 binding activity in wild-type and npr1 plants. We observed that as-1 binding in wild-type Arabidopsis leaves is inducible by SA (Figure 8). To our knowledge, this phenomenon has not been reported for Arabidopsis, although the as-1 element is known to confer SA-inducible gene expression in this species (Xiang et al., 1996). Our results are reminiscent of those obtained in tobacco, in which SA was reported to stimulate the as-1 binding of a factor called SA response protein that is immunologically related to TGA1a (Jupin and Chua, 1996). The tobacco results, together with previous reports that TGA factors, in particular TGA2, represent a significant component of nuclear as-1 binding in Arabidopsis (Lam and Lam, 1995), suggest that the SA-inducible as-1 binding we observed in wild-type Arabidopsis is mediated by TGA factors.
In extracts from water-treated npr1 mutant plants, as-1 binding activity was substantially higher than that observed in similarly treated wild-type plants (Figure 8). Furthermore, this binding activity did not increase after treatment with SA. These results demonstrate that TGA factor activity is altered in the absence of wild-type NPR1 function and, consequently, that NPR1 is an important regulator of TGA factors in plants. Although this line of research does not provide information as to how NPR1 may regulate TGA factor activity in vivo, the observations that these proteins interact with each other in the yeast two-hybrid system and in vitro suggest that it is likely through a direct physical interaction.
The observation that extracts from water-treated npr1 plants possessed higher as-1 binding activity than did wild-type plants suggests that the role of NPR1 in regulating gene expression is complex and likely to extend beyond simply enhancing the DNA binding activity of the TGA factors. Indeed, any model of NPR1 function during SA-activated PR-1 gene expression must consider the NPR1-dependent DNA binding enhancement of TGA factors, the cytoplasmic and nuclear localization of NPR1, the SA inducibility of as-1 binding activity in wild-type Arabidopsis, as well as the constitutive as-1 binding activity in npr1 plants.
A rise in the levels of SA concentrations could result in the modification of NPR1, TGA factors, or both partners, altering one or more of their functions and/or properties. For example, it could modify the transactivation activity of TGA factors, the ability of NPR1 and TGA factors to interact with each other or with additional partners, and/or the abundance or availability of either protein at the PR-1 promoter. These effects, in turn, could be mediated by a number of different mechanisms, including (de)phosphorylation, altered protein turnover, changes in subcellular localization, and/or sequestration. The importance of these processes in regulating the activity of transcription factors in other systems has been abundantly documented (Calkhoven and Ab, 1996).
Although it is too early to advocate any one particular mechanism, it has been previously proposed that the TGA factors, or at least the related SA response protein, is sequestered under noninductive conditions by an inhibitor protein called SAI (Jupin and Chua, 1996). After SA treatment, the SA response protein is released from SAI in a process that involves phosphorylation (Jupin and Chua, 1996). Based on our results of TGA binding in npr1 plants, it appears possible that NPR1 could be SAI or could control SAI activity.
Clearly, additional research is required to resolve the possible functions of NPR1 implied from our current work. Future research should include analysis of the subcellular localization of NPR1 before and after activation of SAR and of PR-1 gene expression in plants containing dominant-negative or knockout versions of TGA factors.
METHODS
Plasmid Constructions
All polymerase chain reaction amplifications were performed using cloned Pfu polymerase under conditions recommended by the manufacturer (Stratagene, La Jolla, CA), using either Arabidopsis thaliana genomic DNA or the GAL4 transcriptional activation domain library DNA (Kohalmi et al., 1997) as template. Polymerase chain reaction primers were designed to incorporate a SalI restriction site before the initiation codon and a NotI restriction site after the termination codon. The npr1 mutants were created by using the Quick-Change site-directed mutagenesis kit (Stratagene), according to the manufacturer's instructions. For yeast two-hybrid assays, N-terminal protein fusions to either the yeast GAL4 DNA binding domain (amino acids 1 to 147 of the intact GAL4 protein) or the GAL4 transcriptional activation domain (amino acids 768 to 881 of GAL4) were created by ligating full-length coding regions of appropriate genes into the SalI-NotI sites of plasmids pBI880 and pBI881 (Kohalmi et al., 1997), respectively. For in vitro transcription and translation, genes were cloned in pBC SK+ (Stratagene) as SalI-NotI inserts. For the in vitro protein–protein interaction assays, the full-length NPR1 or nim1-2 coding sequences were cloned in-frame into the EcoRV site of plasmid Pinpoint Xa-1 (Promega, Madison, WI) after end-filling the SalI site of the insert by using T4 DNA polymerase. All plasmid constructs were verified by sequencing.
Yeast Two-Hybrid System
All methods for two-hybrid screening and assays were as described (Kohalmi et al., 1997). Strain YPB2 of Saccharomyces cerevisiae was used throughout this study. Yeast cells were transformed using a lithium acetate–based protocol and grown on synthetic dextrose media. For library screening, YPB2 cells containing the full-length NPR1 fused to the GAL4 DNA binding domain were transformed with a library of Arabidopsis cDNA fused to the GAL4 transcriptional activation domain prepared as previously described (Kohalmi et al., 1997). For two-hybrid assays, cells were cotransformed with two plasmids. The first, a pBI880 derivative, carried a GAL4 DNA binding domain fusion and a leucine prototrophic marker. The second, a pBI881 derivative, carried a GAL4 transcriptional activation domain fusion and a tryptophan prototrophic marker. The ability to drive the HIS3 reporter gene was assessed by growing transformants on selective medium lacking tryptophan, leucine, and histidine and supplemented with 5 mM 3-amino-1′,2′,4′-triazole (3-AT). The activity of the lacZ reporter gene was monitored visually by using the X-gal filter assay.
In Vitro Transcription and Translation of Proteins
Radiolabeled proteins were synthesized by using the TnT T7 quick-coupled transcription/translation system (Promega) with 35S-methionine (Amersham) as the radiolabel.
In Vitro Protein–Protein Interaction Assays
Escherichia coli XL1 Blue cells harboring the Pinpoint Xa-1 fusion proteins were grown at 37°C to mid-log phase and induced with 100 μM isopropyl β-d-thiogalactopyranoside for 4 hr, according to the manufacturer's instructions (Promega). Cells were lysed by sonication in lysis buffer (20 mM Hepes-KOH, pH 7.9, 150 mM KCl, 2 mM DTT, 20% glycerol, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 0.5 mM phenylmethylsulfonyl fluoride, and 1 mM benzamidine). The biotinylated proteins were affinity-purified on Tetralink streptavidin–agarose beads (Promega). Twenty microliters of beads was incubated in 500 μL of binding buffer (lysis buffer containing 0.1% Triton X-100 and 0.5% nonfat dry milk) and 5 μL of in vitro–synthesized 35S-methionine–radiolabeled protein. After a 1-hr incubation at 4°C, the beads were washed twice with binding buffer and once with the same buffer lacking milk. The bound proteins were eluted by boiling in 2 × SDS-PAGE loading buffer, separated on 8% SDS–polyacrylamide gels, and visualized by autoradiography.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assays (EMSAs) were essentially performed as previously described (Després et al., 1995), using as-1 (5′-TCGAGCTGACGTAAGGGATGACGCACG-3′ annealed to 5′-CGTGCGTCATCCCTTACGTCAGCTCGA-3′) and mutant as-1 (5′-TCGAGCTGCTGTAAGGGATCTCGCACG-3′ annealed to 5′-CGTGCG-AGATCCCTTACAGCAGCTCGA-3′). The sequences of the other oligonucleotides used in this study are presented in Figures 5 and 6. One hundred nanograms of double-stranded synthetic oligonucleotides was labeled by using T4 polynucleotide kinase. The specific activity ranged from 105 cpm/ng to 1.1 × 105 cpm/ng. EMSA reactions were performed at 22°C for 15 min in 40 μL of EMSA buffer (20 mM Hepes-KOH, pH 7.9, 100 mM KCl, 2 mM DTT, and 20% glycerol) containing 1 μL (20,000 cpm) of the labeled oligonucleotide, 100 ng of the respective noncoding strand (to favor double-stranded DNA formation), and either 0.4 μL of in vitro–synthesized 35S-methionine–radiolabeled TGA2 or TGA4 protein with 4 μL of reticulocyte extract or 2 μg of extract from wild-type (ecotype Columbia) Arabidopsis or the npr1 mutant (from the Arabidopsis Biological Resource Center, Ohio State University, Columbus). In reactions in which NPR1 or NIM1-2 was used, 4 μL of in vitro–synthesized 35S-methionine–radiolabeled NPR1 or NIM1-2 replaced the 4 μL of reticulocyte extract. The EMSA reactions were loaded onto 5.4% polyacrylamide gels (29.2:0.8 acrylamide-bisacrylamide in 100 mM Tris, 100 mM borate, and 1 mM EDTA) and run at 8 V/cm for 90 min. After electrophoresis, gels were blotted on Whatman (Maidstone, UK) 3MM paper and autoradiographed at −80°C on Kodak (Rochester, NY) XAR films.
Bacterial Protein Expression, Purification, and Antibody Production
A partial NPR1 (amino acids 354 to 593) lacking the ankyrin-like repeats was fused to an N-terminal histidine tag and purified from E. coli by Ni+ chelate chromatography, according to the supplier's instructions (Qiagen, Chatsworth, CA). The purity of NPR1 was assessed by SDS-PAGE followed by Coomassie Brilliant Blue R 250 staining. Rabbit polyclonal anti-NPR1 antibody was made by immunizing rabbits with the His6–NPR1 fusion protein. The titer and specificity of the antibody were tested by immunoblot analysis.
Preparation of Cytoplasmic and Nuclear Fractions
Percoll-purified nuclei were prepared essentially as previously described (Després et al., 1995) from 2 g of Arabidopsis (ecotype Columbia) leaves. The supernatant was saved as the cytoplasmic fraction; the nuclear pellet was further purified on a 45 to 88% Percoll step gradient. The absence of cytosol or cytosol/plastid contamination of nuclei was confirmed by assaying for the cytosol-specific marker alcohol dehydrogenase (MacDonald and Rees, 1983) and the cytosol/plastid–specific marker glucose-6-phosphate dehydrogenase (Doehlert et al., 1988). Reduction of NAD and NADP was monitored spectrophotometrically at A340. We ensured that the activities were linear for at least 10 min and were proportional to the amount of protein added to the assay. The activity of alcohol dehydrogenase was 30 nmol min−1 mg−1 protein in the cytoplasmic fraction and was undetectable in the nuclear fraction. The activity of glucose-6-phosphate dehydrogenase was 100 nmol min−1 mg−1 protein in the cytoplasmic fraction and 0.5 nmol min−1 mg−1 protein in the Percoll-purified nuclear fraction. To verify that the low activity present in the nuclear fraction was not due to the presence of inhibitors, we added commercially available glucose-6-phosphate dehydrogenase to the nuclear fraction, and the resulting activity was monitored. The activity of the commercial enzyme assayed in the nuclear fraction was 96% of that in the control buffer. Protein concentration was quantified by the Bradford assay, according to the manufacturer's instructions (Bio-Rad), with BSA as a standard.
Immunoblot Analysis of NPR1 and GAL4 Transcriptional Activation Domain Fusion Proteins
Two hundred forty micrograms of protein from the plant cytoplasmic fraction and 20 μg from the nuclear fraction were resolved on 8% SDS–polyacrylamide gels by electrophoresis and blotted onto nitrocellulose membranes. Similarly, to monitor fusion protein production in yeast, 20 μg of total protein was electrophoresed on 8% SDS–polyacrylamide gels and blotted onto nitrocellulose membranes. The blots were stained with Ponceau S and blocked as previously described (Subramaniam et al., 1997). The anti-NPR1 antiserum was used at a dilution of 1:1000. The anti–GAL4 transcriptional activation domain (sc-429; Santa Cruz Biotechnology, Santa Cruz, CA) antibody was used as recommended by the supplier. The blots were developed with an enhanced chemiluminescence detection system, according to the manufacturer's instructions (Amersham).
NOTE ADDED IN PROOF
A paper on this topic (Zhou, J.-M., Trifa, Y., Silva, H., Pontier, D., Lam, E., Shah, J., and Klessig, D.F. [2000]. NPR1 differentially interacts with members of the TGA/OBF family of transcription factors that bind an element of the PR-1 gene required for induction by salicylic acid. Mol. Plant-Microbe Interact. 13, 191–202) is appearing in the February issue of Molecular Plant-Microbe Interactions.
Acknowledgments
We are very grateful to Dr. William L. Crosby (Plant Biotechnology Institute) and his group for their help with the yeast two-hybrid system and their generous gift of the cDNA library; to the DNA Technology Unit, Plant Biotechnology Institute, for sequence analysis and oligonucleotide synthesis; and to Catherine Evans for help with the construction of the npr1-2 site-directed mutant. This research was supported by a grant from the Canada–Saskatchewan Agri-Food Innovation Fund and the National Research Council–Plant Biotechnology Institute core program. This is NRCC publication No. 42629.
References
- Baldwin, A.S., Jr. (1996). The NF-κB and IκB proteins: New discoveries and insights. Annu. Rev. Immunol. 14 649–683. [DOI] [PubMed] [Google Scholar]
- Calkhoven, C.F., and Ab, G. (1996). Multiple steps in the regulation of transcription factor level and activity. Biochem. J. 317 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, H., Bowling, S.A., Gordon, S., and Dong, X. (1994). Characterization of an Arabidopsis mutant that is nonresponsive to inducers of systemic acquired resistance. Plant Cell 6 1583–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, H., Glazebrook, J., Clarke, J.D., Volko, S., and Dong, X. (1997). The Arabidopsis NPR1 gene that controls systemic acquired resistance encodes a novel protein containing ankyrin repeats. Cell 88 57–63. [DOI] [PubMed] [Google Scholar]
- Cao, H., Li, X., and Dong, X. (1998). Generation of broad-spectrum disease resistance by overexpression of an essential regulatory gene in systemic acquired resistance. Proc. Natl. Acad. Sci. USA 95 6531–6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney, T.P., Uknes, S., Vernooij, B., Friedrich, L., Weymann, K., Negrotto, D., Gaffney, T., Gutrella, M., Kessmann, H., Ward, E., and Ryals, J. (1994). A central role of salicylic acid in plant disease resistance. Science 266 1247–1250. [DOI] [PubMed] [Google Scholar]
- Delaney, T.P., Friedrich, L., and Ryals, J.A. (1995). Arabidopsis signal transduction mutant defective in chemically and biologically induced disease resistance. Proc. Natl. Acad. Sci. USA 92 6602–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Després, C., Subramaniam, R., Matton, D.P., and Brisson, N. (1995). The activation of the potato PR-10a gene requires the phosphorylation of the nuclear factor PBF-1. Plant Cell 7 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehlert, D.C., Kuo, T.M., and Felker, F.C. (1988). Enzymes of sucrose and hexose metabolism in developing kernels of two inbred maize lines. Plant Physiol. 86 1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazebrook, J., Rogers, E.E., and Ausubel, F.M. (1996). Isolation of Arabidopsis mutants with enhanced disease susceptibility by direct screening. Genetics 143 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jupin, I., and Chua, N.-H. (1996). Activation of the CaMV as-1 cis-element by salicylic acid: Differential DNA-binding of a factor related to TGA1a. EMBO J. 15 5679–5689. [PMC free article] [PubMed] [Google Scholar]
- Katagiri, F., Yamazaki, K.-I., Horikoshi, M., Roeder, R.G., and Chua, N.-H. (1990). A plant DNA-binding protein increases the number of active preinitiation complexes in a human in vitro transcription system. Genes Dev. 4 1899–1909. [DOI] [PubMed] [Google Scholar]
- Kawata, T., Imada, T., Shiraishi, H., Okada, K., Shimura, Y., and Iwabuchi, M. (1992). A cDNA clone encoding HBP-1b homologue in Arabidopsis thaliana. Nucleic Acids Res. 20 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohalmi, S.E., Nowak, J., and Crosby, W.L. (1997). The yeast two-hybrid system. In Differentially Expressed Genes in Plants: A Bench Manual, E. Hansen and G. Harper, eds (London: Taylor and Francis), pp. 63–82.
- Lam, E., and Lam, Y.K.P. (1995). Binding site requirements and differential representation of TGA factors in nuclear ASF-1 activity. Nucleic Acids Res. 23 3778–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, E., Benfey, P.N., Gilmartin, P.M., Fang, R.-X., and Chua, N.-H. (1989). Site-specific mutations alter in vitro factor binding and change promoter expression pattern in transgenic plants. Proc. Natl. Acad. Sci. USA 86 7890–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel, E., Heifetz, P., Thorne, L., Uknes, S., Ryals, J., and Ward, E. (1998). Functional analysis of regulatory sequences controlling PR-1 gene expression in Arabidopsis. Plant J. 16 223–233. [DOI] [PubMed] [Google Scholar]
- Lehming, N., Thanos, D., Brickman, J.M., Ma, J., Maniatis, T., and Ptashne, M. (1994). An HMG-like protein that can switch a transcriptional activator to a repressor. Nature 371 175–179. [DOI] [PubMed] [Google Scholar]
- Lui, H., Wang, H., DeLong, C., Fowke, L.C., Crosby, W.L., and Fobert, P.R. (2000). The Arabidopsis Cdc2a-interacting protein ICK2 is structurally related to ICK1 and is a potent inhibitor of cyclin-dependent kinase activity in vitro. Plant J., in press. [DOI] [PubMed]
- MacDonald, F.D., and Rees, T. (1983). Enzymic properties of amyloplasts from suspension cultures of soybean. Biochim. Biophys. Acta 755 81–89. [Google Scholar]
- Miao, Z.-H., Liu, X., and Lam, E. (1994). TGA3 is a distinct member of the TGA family of bZIP transcription factors in Arabidopsis thaliana. Plant Mol. Biol. 25 1–11. [DOI] [PubMed] [Google Scholar]
- Pieterse, C.M.J., van Wees, S.C.M., van Pelt, J.A., Knoester, M., Laan, R., Gerrits, H., Weisbeek, P.J., and van Loon, L.C. (1998). A novel signaling pathway controlling induced systemic resistance in Arabidopsis. Plant Cell 10 1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüth, J., Schweyen, R.J., and Hirt, H. (1994). The plant transcription factor TGA1 stimulates expression of the CaMV 35S promoter in Saccharomyces cerevisiae. Plant Mol. Biol. 25 323–328. [DOI] [PubMed] [Google Scholar]
- Ryals, J.A., Neuenschwander, U.H., Willits, M.G., Molina, A., Steiner, H.Y., and Hunt, M.D. (1996). Systemic acquired resistance. Plant Cell 8 1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryals, J.A., Weymann, K., Lawton, K., Friedrich, L., Ellis, D., Steiner, H.Y., Johnson, J., Delaney, T.P., Jesse, T., Vos, P., and Uknes, S. (1997). The Arabidopsis NIM1 protein shows homology to the mammalian transcription factor inhibitor IκB. Plant Cell 9 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler, U., Beckmann, H., and Cashmore, A.R. (1992). TGA1 and G-box binding factors: Two distinct classes of Arabidopsis leucine zipper proteins compete for the G-box-like element TGACGTGG. Plant Cell 4 1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick, S.G., and Smerdon, S.J. (1999). The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 24 311–316. [DOI] [PubMed] [Google Scholar]
- Shah, J., Tsui, F., and Klessig, D.F. (1997). Characterization of a salicylic acid–insensitive mutant (sai) of Arabidopsis thaliana identified in a selective screen utilizing the SA-inducible expression of the tms2 gene. Mol. Plant Microbe Interact. 10 69–78. [DOI] [PubMed] [Google Scholar]
- Subramaniam, R., Després, C., and Brisson, N. (1997). A functional homolog of mammalian protein kinase C participates in the elicitor-induced defense response in potato. Plant Cell 9 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loon, L.C., Bakker, P.A.H.M., and Pieterse, C.M.J. (1998). Systemic resistance induced by rhizosphere bacteria. Annu. Rev. Phytopathol. 36 453–483. [DOI] [PubMed] [Google Scholar]
- Wagner, S., and Green, M. (1993). HTLV TAX protein stimulation of DNA binding of bZIP proteins by enhancing dimerization. Science 262 395–399. [DOI] [PubMed] [Google Scholar]
- Ward, E., Uknes, S., Williams, S.C., Dincher, S.S., Wiederhold, D.L., Alexander, D.C., Ahl-Goy, P., Métraux, J.P., and Ryals, J. (1991). Coordinate gene activity in response to agents that induce systemic acquired resistance. Plant Cell 3 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, C., Miao, Z., and Lam, E. (1995). Isolation of TGA6, a new member of the TGA family of bZIP transcription factors in Arabidopsis thaliana. Plant Physiol. 109 721–722.7480354 [Google Scholar]
- Xiang, C., Miao, Z.-H., and Lam, E. (1996). Coordinated activation of as-1–type elements and a tobacco glutathione S-transferase gene by auxins, salicylic acid, methyl-jasmonate and hydrogen peroxide. Plant Mol. Biol. 32 415–426. [DOI] [PubMed] [Google Scholar]
- Yamazaki, K.-I., Katagiri, F., Imaseki, H., and Chua, N.-H. (1990). TGA1a, a tobacco DNA-binding protein, increases the rate of initiation in a plant in vitro transcription system. Proc. Natl. Acad. Sci. USA 87 7035–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel, U., and Baeuerle, P.A. (1990). Purified human IκB can rapidly dissociate the complex of the NF-κB transcription factor with its cognate DNA. Cell 61 255–265. [DOI] [PubMed] [Google Scholar]
- Zhang, B., Foley, R.C., and Singh, K.B. (1993). Isolation and characterization of two related Arabidopsis ocs-element bZIP binding proteins. Plant J. 4 711–716. [DOI] [PubMed] [Google Scholar]
- Zhang, B., Chen, W., Foley, R.C., Buttner, M., and Singh, K.B. (1995). Interactions between distinct types of DNA binding proteins enhance binding to ocs element promoter sequence. Plant Cell 7 2241–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., Fan, W., Kinkema, M., Li, X., and Dong, X. (1999). Interaction of NPR1 with basic leucine zipper protein transcription factors that bind sequences required for salicylic acid induction of the PR-1 gene. Proc. Natl. Acad. Sci. USA 96 6523–6528. [DOI] [PMC free article] [PubMed] [Google Scholar]