

Abstract
Synapse formation in the CNS is a complex process that involves the dynamic interplay of numerous signals exchanged between pre- and postsynaptic neurons as well as perisynaptic glia. Members of the neurotrophin family, which are widely expressed in the developing and mature CNS and are well-known for their roles in promoting neuronal survival and differentiation, have emerged as key synaptic modulators. However, the mechanisms by which neurotrophins modulate synapse formation and function are poorly understood. Here, we summarize our work on the role of neurotrophins in synaptogenesis in the CNS, in particular the role of these signaling molecules and their receptors, the Trks, in the development of excitatory and inhibitory hippocampal synapses. We discuss our results that demonstrate that postsynaptic TrkB signaling plays an important role in modulating the formation and maintenance of NMDA and GABAA receptor clusters at central synapses, and suggest that neurotrophin signaling coordinately modulates these receptors as part of mechanism that promotes the balance between excitation and inhibition in developing circuits. We also discuss our results that demonstrate that astrocytes promote the formation of GABAergic synapses in vitro by differentially regulating the development of inhibitory presynaptic terminals and postsynaptic GABAA receptor clusters, and suggest that glial modulation of inhibitory synaptogenesis is mediated by neurotrophin-dependent and -independent signaling. Together, these findings extend our understanding of how neuron–glia communication modulates synapse formation, maintenance and function, and set the stage for defining the cellular and molecular mechanisms by which neurotrophins and other cell–cell signals direct synaptogenesis in the developing brain.
Keywords: neurotrophin, presynaptic, postsynaptic, astrocyte, synaptogenesis
INTRODUCTION
The assembly of CNS synapses is a complex and highly dynamic process, requiring the coordinated exchange of anterograde and retrograde signals between pre- and postsynaptic neurons and surrounding glia (reviewed in Garner et al., 2002). Although synapses can be formed in the absence of glia, recent work demonstrates that activity-dependent and -independent interactions between neurons and glia modulate synapse formation and function (Pfrieger and Barres, 1997; Haydon, 2001; Ullian et al., 2001; Cohen-Cory, 2002; Zhang et al., 2003; Ullian et al., 2004).
Several lines of evidence indicate that astrocytes coordinately modulate pre- and postsynaptic development of excitatory synapses by soluble and contact-mediated factors. Glial-derived soluble factors enhance neurite outgrowth, synapse formation and function (Aoyagi et al., 1994; Pfrieger and Barres, 1997; Ullian et al., 2001; Ullian et al., 2004). In purified retinal ganglion cell and spinal motor neuron cultures, glia-conditioned medium dramatically increases the number of presynaptic contacts made between neurons, the quantal size and the efficacy of transmitter release (Pfrieger and Barres, 1997; Nagler et al., 2001; Ullian et al., 2001; Ullian et al., 2004). Mauch and colleagues have shown that astrocyte-derived cholesterol complexed to ApoE is both necessary and sufficient to increase the number of functional presynaptic terminals (Mauch et al., 2001). Treatment with glia-conditioned media also increases the number of postsynaptic AMPA receptor clusters and their responsiveness to glutamate. Once mature synapses have formed, astrocytes continue to provide soluble factors that regulate synaptic transmission. In hippocampal slices, perisynaptic astrocytes release glutamate in response to GABAB-receptor activation, which potentiates GABA release from interneurons and inhibitory transmission (Kang et al., 1998). Recent work has demonstrated that thrombospondin-1 and -2, expressed by immature astrocytes, can induce the formation of immature synapses between retinal ganglion cells in vitro and in vivo. These immature synapses are active presynaptically but silent postsynaptically, indicating that multiple additional, as yet unknown, factors coordinate pre-and postsynaptic maturation (Christopherson et al., 2005). Glial contact with neurons also enhances synapse formation. In hippocampal neurons grown in vitro, integrin-mediated contact between astrocytes and pyramidal neurons induces neuron-wide activation of protein kinase C (PKC) signaling. PKC activation, in turn, promotes the maturation of excitatory presynaptic terminals, but has no effect on postsynaptic AMPA receptor cluster number and localization (Hama et al., 2004). As synapses mature, astrocytes continue to modulate synaptic function by potentiating and suppressing activity at presynaptic glutamatergic terminals via the release of glutamate and ATP, respectively (Zhang et al., 2003; Fiacco and McCarthy, 2004). Ephrin–Eph signaling between neurons and glial cells has also been suggested to modulate postsynaptic spine morphology. EphrinA3, localized to astrocytic processes that envelop spines, interacts with EphA4 receptors in dendritic spines of pyramidal neurons, providing repulsive cues that refine spine morphology (Murai et al., 2003). Moreover, presynaptic ephrinB1 ligands and postsynaptic EphB2 receptors modulate the clustering of NMDA receptors in cortical and hippocampal neurons in vitro (Dalva et al., 2000). These studies underscore the complexity of glial regulation of synapse formation and function by mechanisms that include soluble and contact-mediated factors.
Based on these, and other, studies it is clear that astrocytes actively modulate synapse formation by multiple mechanisms. Neurotrophins are potent morphogenetic modulators that are sensitive to activity-dependent regulation and, as discussed below, induce the formation of new synaptic contacts and modify the structure of existing synapses by pre- and post-synaptic mechanisms. Astrocytes express a subset of tropomyosin-related kinase (Trk) receptors and both release and recycle neurotrophins (Alderson et al., 2000). Here, we discuss how neurotrophins and Trks modulate synapse formation and function, and discuss the evidence that suggests that neurotrophin signaling is one of several mechanisms by which astrocytes and other glial cells influence synaptic circuitry in the developing and mature nervous system.
Neurotrophins, Trks and p75 receptors
The neurotrophins are a family of neuronal growth factors that include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT3) and neurotrophin-4/5 (NT4/5). Neurotrophins bind with high-affinity (Kd ~10−9 M) to members of the Trk-receptor family. NGF binds preferentially to TrkA, BDNF and NT4/5 bind to TrkB and NT3 binds to TrkC. Neurotrophin binding results in dimerization and autophosphorylation of Trk receptors, which triggers tyrosine-kinase activation and initiation of one or more signal transduction cascades, including mitogen-activated protein kinase (MAPK), phosphoinositol 3-kinase and phospholipase C-γ signaling pathways (reviewed in Huang and Reichardt, 2001; Patapoutian and Reichardt, 2001; Chao, 2003). These signals either induce rapid changes in the structure and function of local proteins or are propagated to the nucleus where they activate transcription factors that alter gene expression.
In addition to full-length receptors, TrkB and TrkC have differentially spliced variants known as truncated (t) Trk isoforms, which bind neurotrophin with the same affinity as full-length receptor but lack the intracellular kinase domain. Truncated receptors are expressed by neurons and by glial cells, and have traditionally been thought to function only through their ability to sequester neurotrophins or to dominant-negatively inhibit activation of Trk kinases by forming non-signaling heterodimers (reviewed in Huang and Reichardt, 2001). Recent work has shown that TrkB.t1 mediates BDNF-induced Ca2+ signaling in astrocytes (Rose et al., 2003). Thus, the pleiotropic effects of neurotrophin signaling are caused, in part to the activation of diverse Trk receptors.
All neurotrophins also bind with low affinity to a structurally distinct receptor of the tumor necrosis factor receptor superfamily, the p75 neurotrophin receptor (p75NR). The p75 receptor serves numerous roles in the developing nervous system including increasing the affinity and specificity of Trk–neurotrophin interactions when binding neurotrophins as a co-receptor and inducing apoptosis in neurons and other cell types in the absence of Trk activation. p75 can also act as a high-affinity receptor for uncleaved peptide precursors of the neurotrophins, known as proneurotrophins (Lee et al., 2001). Proneurotrophins are secreted during neural injury and can stimulate apoptosis in oligodendrocytes and vascular smooth muscle cells by selectively binding to and activating p75 receptors (Lee et al., 2001; Beattie et al., 2002). Neurotrophin activation of p75-mediated apoptosis has also been associated with the elimination of neurons that do not make appropriate connections during development (Lee et al., 2001). In addition, recent work indicates that p75 plays a role in modulating peripheral nerve myelination via interactions with neurotrophins and with myelin-associated glycoprotein and the NOGO receptor complex (Cosgaya et al., 2002; Wang et al., 2002). Although p75-mediated signaling appears to play several roles in the developing and mature nervous system, this signaling has not yet been implicated in either synapse formation or function and remains to be carefully addressed.
Activity-dependent neurotrophin synthesis, transport and release
At developing and mature synapses, neurotrophins are exchanged between pre- and postsynaptic neurons via anterograde, retrograde and trans-synaptic transport. Neurotrophins are synthesized and packaged into vesicles in neuronal cell bodies and transported subsequently to presynaptic axon terminals and postsynaptic dendrites for local secretion (reviewed in Poo, 2001). The synthesis, transport and release of neurotrophins are tightly regulated by neural activity. In neurons in culture, neuronal depolarization produced by application of glutamate, elevated extracellular K+ and high-frequency stimulation increase levels of BDNF mRNA and subsequent BDNF secretion (Zafra et al., 1991; Lindholm et al., 1994). In hippocampal slices, normal physiological activity capable of potentiating glutamatergic synapses is sufficient to increase BDNF mRNA (Patterson et al., 1992). By contrast, GABA-mediated inhibition of neuronal activity decreases BDNF mRNA in hippocampal neurons (Berninger et al., 1995). In studies in vivo, blockade of visual input induces a down-regulation in BDNF mRNA in rat visual cortex (Castren et al., 1992). Consistent with activity-dependent changes in expression, BDNF and TrkB mRNA transcripts are targeted to dendrites in an activity-dependent fashion where they have been proposed to undergo local translation (Tongiorgi et al., 1997). Trans-synaptic neurotrophin transport is also modulated by activity. Kohara and colleagues showed that GFP-tagged BDNF is transferred from presynaptic terminals to postsynaptic sites in hippocampal neurons in culture and that BDNF transfer declines as neuronal activity decreases (Kohara et al., 2001).
TrkB expression, membrane incorporation and accumulation at synapses are also modulated by synaptic activity (Tongiorgi et al., 1997; Meyer-Franke et al., 1998; Du et al., 2000). Depolarization and elevated cAMP levels induce the rapid recruitment of TrkB from internal stores to the plasma membrane of hippocampal neurons (Meyer-Franke et al., 1998; Du et al., 2000). On binding ligand, neurotrophin–Trk complexes are internalized in endocytic vesicles that might continue transducing signal and/or might eventually be re-released at nerve terminals or postsynaptic dendrites to synaptic partners in an activity-dependent manner (reviewed in Poo, 2001).
Neurotrophins as synaptic modulators: presynaptic terminal formation and function
Neurotrophins, in particular BDNF, have profound modulatory effects on the growth, remodeling and stability of dendrites and axons in hippocampal, cortical and cerebellar neurons (Cohen-Cory and Fraser, 1995; McAllister et al., 1996; McAllister et al., 1997; Alsina et al., 2001). By increasing the complexity of axonal and dendritic arbors, neurotrophins increase the number of potential contact sites between pre-and postsynaptic neurons and thereby modulate the density of synaptic innervation. Following the establishment of these nascent connections, BDNF and TrkB signaling actively promote the maturation and stabilization of CNS synapses via pre- and postsynaptic mechanisms.
During synaptogenesis, retrograde Trk signaling modulates the development of presynaptic terminals, in part by regulating the expression and/or distribution of synaptic vesicle proteins (reviewed in Vicario-Abejon et al., 2002). BDNF and NT3 treatment in E16 dissociated and P7 slice hippocampal cultures results in an increase in the number of synaptic vesicles docked at active zones (Collin et al., 2001; Tyler and Pozzo-Miller, 2001). Consistent with these observations, several groups report that BDNF treatment of hippocampal and cortical neuronal cultures increase synaptobrevin expression (Takei et al., 1997; Tartaglia et al., 2001; Yamada et al., 2002). In accordance with the effects of exogenous neurotrophins, mice that are deficient in either BDNF or TrkB exhibit marked reductions in the total number of docked vesicles at hippocampal synapses and a redistribution of docked vesicles to areas far from the active zone in the cerebellar synapses (Martinez et al., 1998; Pozzo-Miller et al., 1999; Carter et al., 2002). Moreover, hippocampal sections from TrkB-deficient mice exhibit an overall reduction in syntaxin 1 and SNAP25 immunoreactivity (Martinez et al., 1998). Consistent with reduced exocytotic machinery, TrkB-deficient mice exhibit impaired Ca2+-evoked glutamate and GABA release in cortical synaptosomal preparations from postnatal mice (Carmona et al., 2003). Despite these reports, the mechanisms by which neurotrophins modulate synaptic vesicle machinery remains controversial because several groups report that expression of synaptobrevin, syntaxin 1 and SNAP25 is not altered following BDNF treatment in culture (Vicario-Abejon et al., 1998; Vicario-Abejon et al., 2002) or in BDNF-deficient mice (Pozzo-Miller et al., 1999). Thus, although neurotrophins modulate the formation of presynaptic axon terminals, the precise cellular and molecular mechanisms remain to be determined.
Once nascent synaptic contacts have formed, BDNF signaling promotes the maturation of presynaptic terminals and modulates ongoing neural transmission at excitatory and inhibitory synapses. In dissociated cell and slice cultures from the hippocampus and cortex, BDNF treatment increases mEPSC frequency and FM1-43 turnover at excitatory terminals (Rutherford et al., 1998; Collin et al., 2001; Tyler and Pozzo-Miller, 2001). Reducing BDNF signaling by scavenging BDNF with TrkB–IgG fusion proteins decreases mEPSC frequency in hippocampal neurons (Cabelli et al., 1997). Consistent with these findings, BDNF enhances glutamatergic transmission at mature hippocampal synapses, in part by increases in presynaptic glutamate release (Kang and Schuman, 1996; Carmignoto et al., 1997; Li et al., 1998). At hippocampal and cortical synapses in vivo and in vitro, BDNF facilitates long-term potentiation, attenuates long-term depression and promotes homeostatic and competitive interactions that refine neural circuitry (Korte et al., 1995; Akaneya et al., 1996; Cabelli et al., 1996; Huang et al., 1999; Turrigiano and Nelson, 2000; Turrigiano and Nelson, 2004). At hippocampal and cerebellar synapses in BDNF-deficient mice, high-frequency stimulation and paired-pulse facilitation are impaired, which is consistent with compromised presynaptic release (Pozzo-Miller et al., 1999; Carter and Regehr, 2002). These studies demonstrate that BDNF acts presynaptically to modulate the dynamics of glutamate release at developing and mature synapses.
In addition to modulating excitatory presynaptic terminals, BDNF and TrkB signaling also influence the construction and function of inhibitory synapses. BDNF promotion of inhibitory axon outgrowth and synapse formation is well characterized in hippocampus, cortex and, in particular, cerebellum in vitro and in vivo (Huang et al., 1999; Marty et al., 2000; Seil and Drake-Baumann, 2000; Yamada et al., 2002). Accordingly, fewer GABAergic synapses are observed in cerebellar slices following scavenging of endogenous BDNF (Seil and Drake-Baumann, 2000) and in conditional TrkB deletion mutants (Rico et al., 2002). Far less is known about how TrkB signaling modulates the formation of presynaptic inhibitory terminals. Presynaptic activation of TrkB increases glutamic acid decarboxylase (GAD) expression in interneurons, and enhances presynaptic GABA release and uptake at inhibitory terminals (Vicario-Abejon et al., 1998; Marty et al., 2000; Yamada et al., 2002). Consistent with these findings, increased miniature inhibitory postsynaptic current (mIPSC) frequency and enhanced quantal content of GABA containing vesicles have been observed in hippocampal and cerebellar neurons following treatment with BDNF (Bao et al., 1999; Bolton et al., 2000). These studies underscore the role of the retrograde actions of BDNF and TrkB in regulating the formation and the function of inhibitory CNS synapses.
Ongoing work in our lab indicates that astrocytes modulate the formation and function of inhibitory presynaptic terminals via soluble factors as well as contact-mediated factors that are not neurotrophins. Preliminary results indicate that inhibitory axon outgrowth and the number of inhibitory synaptic contacts increase when neuronal cultures are treated with astrocyte-conditioned medium (ACM), and that this astrocyte effect is activity and neurotrophin independent (S.B. Elmariah and R. Balice-Gordon, unpublished). Thus, astrocytes release soluble factors that modulate interneuron maturation and the formation of inhibitory contacts in the absence of either neuronal activity or neurotrophin signaling. ACM and contact with astrocytes increases the number of presynaptic terminals that are immunopositive for synaptophysin and the vesicular GABA transporter (VGAT). However, an increase in the number of functional inhibitory synaptic terminals, assayed by AM1-43 labeling, is only observed when neurons are plated in direct contact with astrocytes (S.B. Elmariah, E. Hughes and R. Balice-Gordon, unpublished). Together, these results indicate that astrocytes modulate the formation and function of inhibitory presynaptic terminals, at least in vitro, by both secreted and contact-mediated factors.
The mechanisms by which neurotrophins enhance the formation and function of presynaptic terminals remain unclear. Neurotrophins might modulate either the assembly or the stabilization of synaptic vesicles through post-translation modifications of SNARE complex proteins (Vicario-Abejon et al., 1998; Pozzo-Miller et al., 1999; Tartaglia et al., 2001). Consistent with this hypothesis, BDNF-deficient mice exhibit decreased synaptosomal levels of the vesicular proteins synaptobrevin and synaptophysin, although levels form whole hippocampal extracts appear to be similar to controls (Pozzo-Miller et al., 1999). Thus, by altering the associations between SNARE proteins and other vesicular proteins, neurotrophins might regulate vesicle distribution and properties of vesicular release. Recent work demonstrates that Synapsin 1 is phosphorylated following BDNF-mediated activation of MAPK, which promotes the mobilization of synaptic vesicles and enhances neurotransmitter release (Jovanovic et al., 2004). Long-term and acute neurotrophin signaling may also influence, and be influenced by, Ca2+ levels in presynaptic terminals. BDNF and NT3 potentiate neurotransmitter release by increasing presynaptic Ca2+ concentrations at developing neuromuscular synapses of Xenopus (Lohof et al., 1993; Stoop and Poo, 1995; Stoop and Poo, 1996). BDNF-induced MAPK activation increases the number of P/Q-type Ca2+ channels expressed in hippocampal neurons (Levine et al., 1995; Baldelli et al., 2000). These studies show that BDNF and TrkB signaling modulate the formation, maturation and function of presynaptic terminals at CNS synapses, but the mechanisms by which such modulation occurs remain to be determined.
Neurotrophins as synaptic modulators: postsynaptic neurotransmitter receptor clusters
In addition to their roles in establishing presynaptic terminals and regulating neurotransmitter release, neurotrophins are emerging as key modulators of postsynaptic specializations. Prolonged signaling by neurotrophins promotes quantal scaling of excitatory and inhibitory synapses in postnatal neural networks by modulating postsynaptic receptors (Rutherford et al., 1997; Rutherford et al., 1998). At glutamatergic synapses, BDNF promotes maturation of silent synapses by increasing the translocation of the GluR2 AMPA-receptor subunit to the neuronal surface in neocortical neurons (Narisawa-Saito et al., 1999; Narisawa-Saito et al., 2002). In hippocampal neurons, BDNF rapidly increases the phosphorylation and potentiates the conductance of NMDA receptors (Levine et al., 1996; Levine et al., 1998). At inhibitory synapses in hippocampal cultures, BDNF treatment increases the expression of GABAA-receptor subunits, differentially modulates the number of postsynaptic GABAA-receptor clusters, regulates GABAA receptor recruitment to the neuronal surface, and modulates GABAA-channel conductance (Brunig et al., 2001; Kilman et al., 2002; Yamada et al., 2002; Jovanovic et al., 2004). Examination of current density in hippocampal cultures at times before many synapses have formed indicates that astrocytes contribute to the maintenance of GABAA receptors at the neuronal cell surface (Liu et al., 1996; Liu et al., 1997). Although upregulation of GABA current density requires Ca2+ elevation in astrocytes, this effect does not depend on direct contact with astrocytes because ACM treatment mimicks the effects observed in co-cultures (Liu et al., 1996; Liu et al., 1997). Thus, soluble factors released by astrocytes modulate the distribution of GABAA receptors prior to synapse formation. These studies demonstrate that neurotrophins might modulate synaptic integrity and strength by regulating the expression, distribution and kinetics of postsynaptic neurotransmitter receptors at excitatory and inhibitory synapses.
Recently, we have showed that full-length and truncated TrkB are distributed diffusely throughout the dendrites and soma of rat hippocampal neurons grown in vitro and that some TrkB is localized to some, but not all, postsynaptic specializations (Fig. 1A) (Elmariah et al., 2004). Manipulation of TrkB-mediated signaling results in dramatic changes in the number and synaptic localization of postsynaptic NMDA-receptor (NMDAR) and GABAA-receptor (GABAAR) clusters (Fig. 1B,C). BDNF treatment increases in the number of NMDAR and GABAAR clusters and increases the proportion of clusters that are apposed to presynaptic terminals. Down-regulation of TrkB signaling decreases the number of receptor clusters and their synaptic localization (Fig. 1B). Examination of the time course of the effect of BDNF on receptor clusters shows that the increase in GABAAR clusters precedes the increase in NMDAR clusters by >12 hours (Fig. 1C). Moreover, the TrkB-mediated effects on NMDAR clusters are dependent on GABAAR activation. Although treatment with TTX, APV and 6-cyano-7-nitro-quinoxaline-2, 3-dione (CNQX) have no effect, blockade of GABAARs with bicuculline abolishes the BDNF-mediated increase in NMDAR cluster number and synaptic localization (Fig. 1D). By contrast, application of exogenous GABA prevents the decrease in NMDAR clusters induced by BDNF scavenging (Fig. 1E). Taken together, these results indicate that TrkB-mediated signaling modulates the clustering of postsynaptic GABAARs whose activity is required for a subsequent upregulation of NMDAR clusters (Elmariah et al., 2004).
Fig. 1. TrkB signaling modulates neurotransmitter receptor cluster formation and synaptic localization in hippocampal neurons in vitoe.

Rat hippocampal neurons at 10 days in vitro (div) were immunostained and analyzed with confocal microscopy. (A) Immunostaining of permeabilized neurons with an antibody against all isoforms of TrkB (red) and MAP2 (green) shows that TrkB is distributed diffusely throughout dendrites. Scale bar, 10 μm. Area in white box is shown below at higher magnification (scale bar, 2 μm). (B) Compared with no-treatment controls (top left), treatment with 50 ng ml−1 BDNF (bottom left) for 48 hours increases the number and synaptic localization of NMDAR clusters in pyramidal neurons. In contrast, neither treatment with 2.0 μg ml−1 TrkB-IgG (top right) to scavenge endogenous BDNF nor infection with an adenovirus encoding truncated TrkB (AdTrkB.t1ha; bottom right) to dominant-negatively downregulate TrkB signaling decreases NMDAR cluster number and synaptic localization. Hippocampal neurons were immunostained with an antibody against NMDAR1 (red) and synaptophysin (SP, green) to visualize presynaptic terminals. Scale bar, 2 μm. (C) Time-course of the effects of BDNF on GABAAR and NMDAR clusters. Top panels: treatment with 50 ng ml−1 BDNF for 36 hours and 48 hours increases the number of GABAAR clusters compared with untreated neurons. GABAAR clusters visualized with antibody against GABAA β2/3 subunits (top); overlay of GABAAR (red) and SP (green) is shown below. Bottom panels: an increase in NMDAR clusters is observed only after 48 hours. NMDAR clusters visualized with antibody against NMDAR1 (top); overlay of NMDAR (red) and SP (green) is shown below. Scale bar, 2 μm. Graphs: quantification of the fold change in GABAAR and NMDAR clusters per 20 μm-dendrite segment in BDNF-treated compared with untreated neurons at 24, 36 and 48 hours. Bars represent the ratio of the number of clusters per segment in BDNF-treated neurons compared with untreated controls. Graph right: quantification of the fold change in the proportion of synaptically localized GABAAR and NMDAR clusters after BDNF for 24, 36 and 48 hours. Points represent the ratio of the percentage of synaptically-localized clusters in BDNF-treated neurons compared with untreated controls. * (GABAARs) and ** (NMDARs) indicates significant change from 0 hours treatment (P<0.005, chi-square and other tests). (D,E) Hippocampal neurons treated with 50 ng ml−1 BDNF and 10 μM bicuculline, a GABAA receptor antagonist, for 48 hours (D) and 2 μg ml−1 TrkB-IgG and 100 μM GABA for 48 hours (E) were immunostained at 10 div with an antibody against NMDAR1. Treatment with BDNF + bicuculline decreased NMDAR-cluster number compared with neurons treated with BDNF alone whereas treatment with TrkB-IgG + 100 μM GABA increased NMDAR-cluster number compared with neurons treated with TrkB-IgG alone. Scale bars, 10 μm. Areas in white boxes are shown below at higher magnification (scale bars, 2 μm).
These results support a model in which TrkB-mediated signaling directly increases inhibitory interneuron synapses onto pyramidal neurons, which leads to a homeostatic increase in NMDAR clusters and their synaptic localization (Fig. 2). One important question raised by this simple model is what mechanism limits homeostatic increases in excitatory and inhibitory synapses, preventing synapse number from constantly increasing. One possibility is that activity-dependent mechanisms underlying synaptic competition and loss might counterbalance these effects as circuits develop.
Fig. 2. Neurotrophin signaling modulates GABAA- and NMDA-receptor clustering at hippocampal synapses.
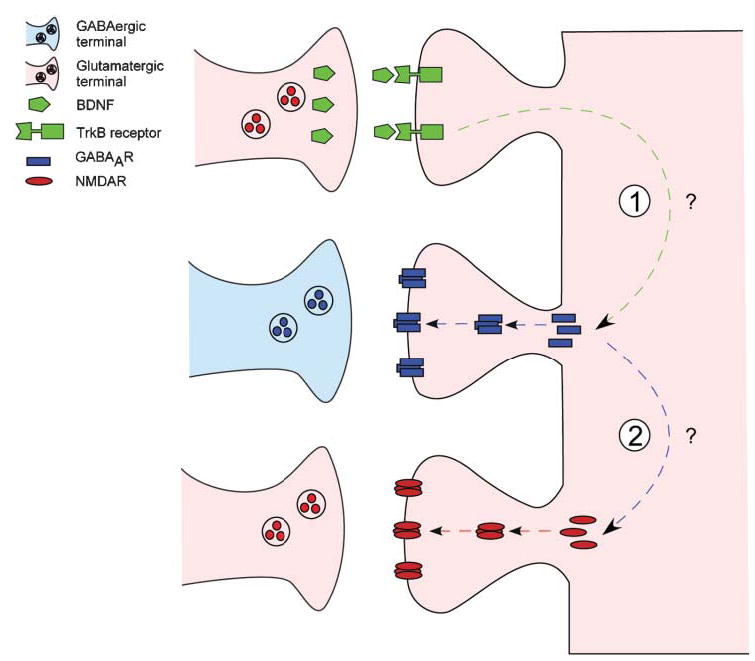
TrkB-mediated signaling might be part of a homeostatic mechanism that balances excitatory and inhibitory synaptic activity in developing neural circuits. (1) Activation of postsynaptic TrkB signaling by BDNF (top) increases the number and synaptic localization of GABAA receptor clusters (middle) in pyramidal neurons. Upregulation of functional GABAergic synapses causes a relative imbalance in inhibition and excitation, thereby reducing excitability in target pyramidal neurons. (2) In turn, this leads to a compensatory increase in the number of NMDA-receptor clusters (bottom) and synaptic localization in pyramidal neurons. Adapted from Elmariah et al. (2004).
We have also studied the postsynaptic effects of neurotrophin signaling at neuromuscular synapses, where bidirectional neurotrophin signaling influences postsynaptic development via direct and indirect mechanisms. Retrograde signaling by BDNF, NT3 and NT4 released by muscle cells regulates neuregulin expression in presynaptic nerve terminals, which, in turn, stimulates synthesis of acetylcholine receptors (AChRs) in postsynaptic muscle fibers (Loeb and Fischbach, 1997; Loeb et al., 2002). Previous work from our lab has highlighted a role for anterograde neurotrophin signaling at neuromuscular synapses, with the observation that dominant-negative disruption of postsynaptic TrkB signaling results in the disassembly of existing AChR clusters in muscle fibers in vivo and in vitro (Gonzalez et al., 1999), suggesting a role for TrkB signaling in receptor cluster maintenance. Taken together, this work at neuromuscular synapses in vivo and at CNS synapses in vitro indicates that TrkB-dependent signaling plays an important role in modulating postsynaptic neurotransmitter clusters. In the CNS as least, this signaling might be part of a mechanism that balances inhibitory and excitatory synaptic transmission in developing neural circuits.
Neurotrophin signaling at tripartite synapses
A role for neurotrophin signaling in the glial modulation of synapse formation and function, and vice-versa, has begun to emerge. Several groups have reported that primary astrocytes in culture produce NGF, BDNF and NT3, and that the expression of these neurotrophins and their receptors in astrocytes is modulated by cAMP signaling (Furukawa et al., 1987; Houlgatte et al., 1989; Rudge et al., 1992; Condorelli et al., 1994). Thus, astrocytes might release neurotrophins that affect neurons that, in turn, enhance synapse formation and function. Similarly, neurons release neurotrophins, which might affect astrocytes that, in turn, signal back to neurons. Furthermore, glia might regulate the release of neurotrophins from neurons. For example, in hippocampal-cell cultures, stimulated astrocytes release activity-dependent neurotrophic factor, which induces NT3 release from postsynaptic neurons. Retrograde NT3 then acts on presynaptic pyramidal neurons to increase glutamatergic-synapse formation (Blondel et al., 2000). Finally, astrocytes might modulate the expression of neuronal Trk receptors indirectly by influencing neuronal activity. Because the expression and distribution of neurotrophin and Trk mRNA and protein are highly sensitive to neuronal activity levels, glial modulation of neuronal excitation by, for example, the release either glutamate or ATP might regulate Trk signaling between neurons. These and other cellular and molecular mechanisms by which neurotrophin and other signaling pathways affect and are affected by glia remain to be explored.
We have examined the development of inhibitory synapses in dissociated cultures of embryonic rat hippocampal neurons grown in the presence and absence of either astrocytes or ACM. We showed that astrocytes enhance inhibitory synaptogenesis by promoting the formation and postsynaptic localization of GABAAR clusters via neurotrophin and Trk-mediated signaling (Elmariah et al., 2005). We found that astrocytes increase the number and synaptic localization of GABAAR clusters during the first week in vitro (Fig. 3A). Scavenging endogenously released BDNF prevents the astrocyte-induced increase in postsynaptic GABAAR clusters but has no effect on the number of presynaptic GABAergic terminals (Fig. 3B,C). Moreover, TrkB-deficient astrocytes increase postsynaptic GABAAR clusters in wild-type neurons. However, wild-type astrocytes failed to induce an increase in the synaptic localization of GABAAR clusters in either TrkB- or BDNF-deficient neurons (Fig. 3D,E). We also observe that NT3-mediated signaling decreases the synaptic localization of GABAAR clusters in the presence of astrocytes. Together, these results indicate that neurotrophin and Trk signaling are not required in astrocytes, but are required in neurons, to increase postsynaptic GABAAR clusters (Elmariah et al., 2005).
Fig. 3. Astrocytes promote inhibitory-synapse formation in hippocampal neurons via TrkB-mediated signaling.

Hippocampal neurons cultured in either the presence or the absence of astrocytes and ACM and immunostained with antibodies against GABAAR-β2/3 (green), SP (red) and VGAT (blue) to visualize inhibitory presynaptic terminals. (A) Co-culture of neurons with astrocytes increases the number of SP+ and VGAT+ presynaptic terminals and GABAAR clusters at 10 div. The proportion of GABAAR clusters apposed to presynaptic terminals also increases in neuron–astrocyte co-cultures. Scale bar, 10 μm. Areas in white boxes are shown below at higher magnification (scale bar, 2 μm). (B) Treating neuron–astrocyte co-cultures with 2.0 μg ml−1 TrkB-IgG (bottom) to scavenge endogenous BDNF reduced the number of postsynaptic GABAAR clusters at 10 div but did not alter the number of SP+ boutons. GABAAR clusters visualized with antibody against GABAA β2/3 subunits (top), overlay of GABAAR (green) and SP (red) below. Scale bar, 2 μm. (C) Left: the effect of TrkB-IgG on the number of GABAAR clusters on dendrite segments at 4, 7 and 10 div. * P<0.001 compared with no treatment in pure neuronal cultures; ** P<0.001 compared to no treatment in neuron–astrocyte co-cultures. Right: the effect of TrkB-IgG on the synaptic localization of GABAAR clusters. * P<0.001 compared with no treatment in pure neuronal cultures; ** P<0.001 decrease compared with no treatment in neuron–astrocyte co-cultures. (D) Left: hippocampal neurons from BDNF−/− mice and wild-type (WT) littermates cultured in the presence of WT ACM. ACM increased the number and synaptic localization of GABAAR clusters in BDNF+/+ neurons (left) but not in BDNF−/− neurons (right). Right: ACM from BDNF+/+ (left) and BDNF−/− (right) astrocytes caused similar increases in GABAAR-cluster number and synaptic localization in WT neurons. GABAAR clusters visualized with antibody against GABAA β2/3 subunits (top); overlay of GABAAR (green) and SP (red) is below. Scale bar, 2 μm. (E) Left: hippocampal neurons from TrkB−/− mice and WT littermates were cultured in the presence of WT ACM. ACM increased the number and synaptic localization of GABAAR clusters in TrkB +/+ neurons (left), but in TrkB−/− neurons (right). Right: ACM from TrkB+/+ (left) and TrkB−/− (right) astrocytes caused similar increases in GABAAR-cluster number and synaptic localization in WT neurons. GABAAR clusters visualized with an antibody against GABAA β2/3 subunits (top); overlay of GABAAR (green) and SP (red) below. Scale bar, 2 μm.
One hypothesis generated by our observations is that astrocytes upregulate activity-independent release of BDNF from pre- or postsynaptic neurons, which acts in either paracrine or autocrine fashion to upregulate GABAAR clusters (Fig. 4). Once mature networks have formed, activity-dependent BDNF and TrkB signaling provides ongoing modulation of postsynaptic GABAAR clusters. Taken together, this work indicates that astrocytes regulate the formation of inhibitory synapses by modulating the number of postsynaptic GABAAR clusters, and that these effects are mediated, in part, by neurotrophin and Trk signaling in neurons, which is enhanced by astrocytes.
Fig. 4. Astrocytes regulate BDNF and TrkB-mediated modulation of inhibitory synaptogenesis.

Astrocytes coordinately modulate the pre- and postsynaptic development of inhibitory connections. Soluble factors released by astrocytes promote the formation of inhibitory presynaptic terminals via neutrotrophin- and TrkB-independent signaling. In contrast, astrocytes enhance BDNF and TrkB signaling between neurons and, thereby, promote the formation and postsynaptic localization of GABAAR clusters. Our data indicate that neurons, not astrocytes, are the source of BDNF and the site of TrkB activation for postsynaptic GABAAR modulation. Astrocytes might increase TrkB-mediated modulation of postsynaptic GABAAR clusters by enhancing neuronal BDNF release and/or TrkB activation in neurons via unknown signals.
Conclusions and future directions
There are several areas in which our understanding of how neuronal and glial signals coordinately modulate synapse assembly, maturation and function is currently lacking. First, our understanding of the cellular and molecular mechanisms that underlie neurotrophins and other cell–cell signals that modulate synaptogenesis is in its infancy. Second, although neurotrophin and glial modulation of synaptic morphology and activity has been studied extensively in dissociated cell and slice cultures, comparatively less is known about how neuron–glia signaling contributes to the construction and modulation of CNS synapses in vivo. The combination of imaging approaches to observe development in vivo, and molecular approaches to manipulate neuronal and glial signaling using targeted genetic approaches is crucial to our understanding of how different cells and different molecules, including the neurotrophins, interact to shape synaptic connectivity. Addressing these issues is necessary to understanding how synaptic connections are formed and function in the developing and mature brain.
Acknowledgments
We thank M. Maronski, M.O. Scott, and H.Y. Zhou for technical assistance, and Drs M. Dichter, S. Gibbs, P. Haydon, S. Moss, J. Panzer, S. Potluri, and D. Hess, Y. Song and R. Wyatt for helpful discussions. This work was supported by grants from the NIH (NS/AR40763) and NSF (0130822) to R.B.-G. and an NIH NRSA (NS43821) to S.B.E.
References
- Akaneya Y, Tsumoto T, Hatanaka H. Brain-derived neurotrophic factor blocks long-term depression in rat visual cortex. Journal of Neurophysiology. 1996;76:4198–4201. doi: 10.1152/jn.1996.76.6.4198. [DOI] [PubMed] [Google Scholar]
- Alderson RF, Curtis R, Alterman AL, Lindsay RM, DiStefano PS. Truncated TrkB mediates the endocytosis and release of BDNF and neurotrophin-4/5 by rat astrocytes and Schwann cells in vitro. Brain Research. 2000;871:210–222. doi: 10.1016/s0006-8993(00)02428-8. [DOI] [PubMed] [Google Scholar]
- Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nature Neuroscience. 2001;4:1093–1101. doi: 10.1038/nn735. [DOI] [PubMed] [Google Scholar]
- Aoyagi A, Nishikawa K, Saito H, Abe K. Characterization of basic fibroblast growth factor-mediated acceleration of axonal branching in cultured rat hippocampal neurons. Brain Research. 1994;661:117–126. doi: 10.1016/0006-8993(94)91188-6. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Forni PE, Carbone E. BDNF, NT-3 and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons. European Journal of Neuroscience. 2000;12:4017–4032. doi: 10.1046/j.1460-9568.2000.00305.x. [DOI] [PubMed] [Google Scholar]
- Bao S, Chen L, Qiao X, Thompson RF. Transgenic brain-derived neurotrophic factor modulates a developing cerebellar inhibitory synapse. Learning and Memory. 1999;6:276–283. [PMC free article] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo, et al. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36:375–386. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger B, Marty S, Zafra F, da Penha Berzaghi M, Thoenen H, Lindholm D. GABAergic stimulation switches from enhancing to repressing BDNF expression in rat hippocampal neurons during maturation in vitro. Development. 1995;121:2327–2335. doi: 10.1242/dev.121.8.2327. [DOI] [PubMed] [Google Scholar]
- Blondel O, Collin C, McCarran WJ, Zhu S, Zamostiano R, Gozes I, et al. A glia-derived signal regulating neuronal differentiation. Journal of Neuroscience. 2000;20:8012–8020. doi: 10.1523/JNEUROSCI.20-21-08012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton MM, Lo DC, Sherwood NT. Long-term regulation of excitatory and inhibitory synaptic transmission in hippocampal cultures by brain-derived neurotrophic factor. Progress in Brain Research. 2000;128:203–218. doi: 10.1016/s0079-6123(00)28018-7. [DOI] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. European Journal of Neuroscience. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Allendoerfer KL, Radeke MJ, Welcher AA, Feinstein SC, Shatz CJ. Changing patterns of expression and subcellular localization of TrkB in the developing visual system. Journal of Neuroscience. 1996;16:7965–7980. doi: 10.1523/JNEUROSCI.16-24-07965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. Journal of Physiology. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona MA, Martinez A, Soler A, Blasi J, Soriano E, Aguado F. Ca(2+)-evoked synaptic transmission and neurotransmitter receptor levels are impaired in the forebrain of trkb (−/−) mice. Molecular and Cellular Neuroscience. 2003;22:210–226. doi: 10.1016/s1044-7431(03)00038-1. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nature Neuroscience. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Carter AR, Chen C, Schwartz PM, Segal RA. Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. Journal of Neuroscience. 2002;22:1316–1327. doi: 10.1523/JNEUROSCI.22-04-01316.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proceedings of the National Academy of Sciences of the USA. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nature Reviews Neuroscience. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S. The developing synapse: construction and modulation of synaptic structures and circuits. Science. 2002;298:770–776. doi: 10.1126/science.1075510. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- Collin C, Vicario-Abejon C, Rubio ME, Wenthold RJ, McKay RD, Segal M. Neurotrophins act at presynaptic terminals to activate synapses among cultured hippocampal neurons. European Journal of Neuroscience. 2001;13:1273–1282. doi: 10.1046/j.0953-816x.2001.01500.x. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Dell’Albani P, Mudo G, Timmusk T, Belluardo N. Expression of neurotrophins and their receptors in primary astroglial cultures: induction by cyclic AMP-elevating agents. Journal of Neurochemistry. 1994;63:509–516. doi: 10.1046/j.1471-4159.1994.63020509.x. [DOI] [PubMed] [Google Scholar]
- Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science. 2002;298:1245–1248. doi: 10.1126/science.1076595. [DOI] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, et al. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B. Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. Journal of Cell Biology. 2000;150:1423–1434. doi: 10.1083/jcb.150.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Crumling MA, Parsons TD, Balice-Gordon RJ. Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. Journal of Neuroscience. 2004;24:2380–2393. doi: 10.1523/JNEUROSCI.4112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. Journal of Neuroscience. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. Journal of Neuroscience. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S, Furukawa Y, Satoyoshi E, Hayashi K. Synthesis/secretion of nerve growth factor is associated with cell growth in cultured mouse astroglial cells. Biochemistry and Biophysics Research Communications. 1987;142:395–402. doi: 10.1016/0006-291x(87)90287-7. [DOI] [PubMed] [Google Scholar]
- Garner CC, Zhai RG, Gundelfinger ED, Ziv NE. Molecular mechanisms of CNS synaptogenesis. Trends in Neurosciences. 2002;25:243–251. doi: 10.1016/s0166-2236(02)02152-5. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, et al. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- Hama H, Hara C, Yamaguchi K, Miyawaki A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron. 2004;41:405–415. doi: 10.1016/s0896-6273(04)00007-8. [DOI] [PubMed] [Google Scholar]
- Haydon PG. Glia: listening and talking to the synapse. Nature Reviews Neuroscience. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Houlgatte R, Mallat M, Brachet P, Prochiantz A. Secretion of nerve growth factor in cultures of glial cells and neurons derived from different regions of the mouse brain. Journal of Neuroscience Research. 1989;24:143–152. doi: 10.1002/jnr.490240204. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annual Review of Neuroscience. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, et al. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. Journal of Neuroscience. 2004;24:522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nature Neuroscience. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. Journal of Neuroscience. 2002;22:1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proceedings of the National Academy of Sciences of the USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Kim AH, Khursigara G, Chao MV. The uniqueness of being a neurotrophin receptor. Current Opinion in Neurobiology. 2001;11:281–286. doi: 10.1016/s0959-4388(00)00209-9. [DOI] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proceedings of the National Academy of Sciences of the USA. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Differential effects of NGF and BDNF on voltage-gated calcium currents in embryonic basal forebrain neurons. Journal of Neuroscience. 1995;15:3084–3091. doi: 10.1523/JNEUROSCI.15-04-03084.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Selective role for trkB neurotrophin receptors in rapid modulation of hippocampal synaptic transmission. Brain Research Molecular Brain Research. 1996;38:300–303. doi: 10.1016/0169-328x(96)00025-3. [DOI] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. Journal of Neuroscience. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H. Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain—implications for neuronal plasticity. Journal of Neurobiology. 1994;25:1362–1372. doi: 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- Liu QY, Schaffner AE, Chang YH, Vaszil K, Barker JL. Astrocytes regulate amino acid receptor current densities in embryonic rat hippocampal neurons. Journal of Neurobiology. 1997;33:848–864. [PubMed] [Google Scholar]
- Liu QY, Schaffner AE, Li YX, Dunlap V, Barker JL. Upregulation of GABAA current by astrocytes in cultured embryonic rat hippocampal neurons. Journal of Neuroscience. 1996;16:2912–2923. doi: 10.1523/JNEUROSCI.16-09-02912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD. Neurotrophic factors increase neuregulin expression in embryonic ventral spinal cord neurons. Journal of Neuroscience. 1997;17:1416–1424. doi: 10.1523/JNEUROSCI.17-04-01416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Hmadcha A, Fischbach GD, Land SJ, Zakarian VL. Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. Journal of Neuroscience. 2002;22:2206–2214. doi: 10.1523/JNEUROSCI.22-06-02206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, et al. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. Journal of Neuroscience. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty S, Wehrle R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. Journal of Neuroscience. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Jr, et al. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nature Neuroscience. 2003;6:153–160. doi: 10.1038/nn994. [DOI] [PubMed] [Google Scholar]
- Nagler K, Mauch DH, Pfrieger FW. Glia-derived signals induce synapse formation in neurones of the rat central nervous system. Journal of Physiology. 2001;533:665–679. doi: 10.1111/j.1469-7793.2001.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H. Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience. 1999;88:1009–1014. doi: 10.1016/s0306-4522(98)00496-5. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Iwakura Y, Kawamura M, Araki K, Kozaki S, Takei N, et al. Brain-derived neurotrophic factor regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors by enhancing the N-ethylmaleimide-sensitive factor/GluR2 interaction in developing neocortical neurons. Journal of Biological Chemistry. 2002;277:40901–40910. doi: 10.1074/jbc.M202158200. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Current Opinion in Neurobiology. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nature Reviews Neuroscience. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, et al. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. Journal of Neuroscience. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Xu B, Reichardt LF. TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nature Neuroscience. 2002;5:225–233. doi: 10.1038/nn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Blum R, Pichler B, Lepier A, Kafitz KW, Konnerth A. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature. 2003;426:74–78. doi: 10.1038/nature01983. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Alderson RF, Pasnikowski E, McClain J, Ip NY, Lindsay RM. Expression of Ciliary Neurotrophic Factor and the Neurotrophins – Nerve Growth Factor, Brain-Derived Neurotrophic Factor and Neurotrophin 3 – in Cultured Rat Hippocampal Astrocytes. European Journal of Neuroscience. 1992;4:459–471. doi: 10.1111/j.1460-9568.1992.tb00896.x. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. Journal of Neuroscience. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R. TrkB receptor ligands promote activity-dependent inhibitory synaptogenesis. Journal of Neuroscience. 2000;20:5367–5373. doi: 10.1523/JNEUROSCI.20-14-05367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop R, Poo MM. Potentiation of transmitter release by ciliary neurotrophic factor requires somatic signaling. Science. 1995;267:695–699. doi: 10.1126/science.7839148. [DOI] [PubMed] [Google Scholar]
- Stoop R, Poo MM. Synaptic modulation by neurotrophic factors. Progress in Brain Research. 1996;109:359–364. doi: 10.1016/s0079-6123(08)62118-4. [DOI] [PubMed] [Google Scholar]
- Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. Journal of Neurochemistry. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B. Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. Journal of Biological Chemistry. 2001;276:37585–37593. doi: 10.1074/jbc.M101683200. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Righi M, Cattaneo A. Activity-dependent dendritic targeting of BDNF and TrkB mRNAs in hippocampal neurons. Journal of Neuroscience. 1997;17:9492–9505. doi: 10.1523/JNEUROSCI.17-24-09492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Current Opinion in Neurobiology. 2000;10:358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nature Reviews Neuroscience. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. Journal of Neuroscience. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Harris BT, Wu A, Chan JR, Barres BA. Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Molecular and Cellular Neuroscience. 2004;25:241–251. doi: 10.1016/j.mcn.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. Journal of Neuroscience. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejon C, Owens D, McKay R, Segal M. Role of neurotrophins in central synapse formation and stabilization. Nature Reviews Neuroscience. 2002;3:965–974. doi: 10.1038/nrn988. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature. 2002;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, et al. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. Journal of Neuroscience. 2002;22:7580–7585. doi: 10.1523/JNEUROSCI.22-17-07580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Castren E, Thoenen H, Lindholm D. Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proceedings of the National Academy of Sciences of the USA. 1991;88:10037–10041. doi: 10.1073/pnas.88.22.10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, et al. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]