

Abstract
Background
Leptin (LEP) has been consistently associated with angiogenesis and tumor growth. Leptin exerts its physiological action through its specific receptor (LEPR). We have investigated whether genetic variations in LEP and LEPR have implications for susceptibility to and prognosis in breast carcinoma.
Methods
We used the polymerase chain reaction and restriction enzyme digestion to characterize the variation of the LEP and LEPR genes in 308 unrelated Tunisian patients with breast carcinoma and 222 healthy control subjects. Associations of the clinicopathologic parameters and these genetic markers with the rates of the breast carcinoma-specific overall survival (OVS) and the disease free survival (DFS) were assessed using univariate and multivariate analyses.
Results
A significantly increased risk of breast carcinoma was associated with heterozygous LEP (-2548) GA (OR = 1.45; P = 0.04) and homozygous LEP (-2548) AA (OR = 3.17; P = 0.001) variants. A highly significant association was found between the heterozygous LEPR 223QR genotype (OR = 1.68; P = 0.007) or homozygous LEPR 223RR genotype (OR = 2.26; P = 0.001) and breast carcinoma.
Moreover, the presence of the LEP (-2548) A allele showed a significant association with decreased disease-free survival in breast carcinoma patients, and the presence of the LEPR 223R allele showed a significant association with decreased overall survival.
Conclusion
Our results indicated that the polymorphisms in LEP and LEPR genes are associated with increased breast cancer risk as well as disease progress, supporting our hypothesis for leptin involvement in cancer pathogenesis.
Background
Breast cancer is the most frequently diagnosed cancer among women, with a lifetime incidence of about 10–13% [1,2] but its etiology is still not fully understood. Besides, age at menarche and menopause, diet, reproductive history, estrogen administration and genetic factors have been suggested as risk factor for breast cancer [3-7]. Germline mutations in dominant, highly penetrant susceptibility genes, such as BRCA1 and BRCA2, have been identified to be responsible for less than 10% of breast carcinoma. In addition, low-penetrance genes, that also increase susceptibility to breast cancer, have been suggested. For instance, candidate low-penetrance breast cancer susceptibility genes include those involved in the complex mechanisms of carcinogenesis [8].
Higher body weight appears to play a role in the development of breast/mammary tumors. Obesity has been identified as a risk factor for breast cancer in postmenopausal women [9-11], and higher body weight is associated with increased incidence of both spontaneous and chemically induced mammary tumors in rodents [12-15].
Leptin, a 16 kDa polypeptide hormone produced predominantly by white adipose tissue [16], plays an important role in body weight homeostasis through effects on food intake and energy expenditure [17,18]. In addition to the regulation of body weight, leptin also influences hematopoiesis, reproduction, angiogenesis, and immune processes [19-22]. The leptin gene, the human homologue of the rat obese gene (OB), has been cloned and sequenced by Zhang et al. [16]. It is located on chromosome 7q31.3 [23,24] and expresses a 4.5 kb mRNA in adipose tissue [16,25].
Leptin exerts its physiological action through the leptin receptor (LEPR), a member of the class 1 cytokine receptor family. LEPR was initially identified in the brain, which explains the negative feedback mechanism of controlling food intake and body weight [26]. However, further studies have demonstrated that LEPR is also expressed in many other tissues and cells, including placenta, pancreas, hematopoietic cells, liver, lung and gastric mucosal cells [27-32].
The human LEPR gene is on chromosome 1. Six isoforms derived from LEPR transcription have been identified, and a long isoform, LEPR-b, is reported to be responsible for signal transduction [33].
In animal and human cell lines, leptin and leptin receptors have also been clearly associated with enhanced in vitro tumor proliferation and/or to in vitro and in vivo promotion of angiogenesis. These effects have been documented in embryonic cells [34], adipocytes [35], glia [36], endothelial cells [37], hematopoietic cells [38], and in benign and malignant epithelial breast cells [39-42], kidney, colon [43,44], liver [45], and pancreas [46].
In breast cancer tissue, it was shown that leptin and leptin receptor are both expressed and that they act to favour cancer proliferation and metastasis [47,48]. Controversial results have been reported regarding the detection of serum leptin levels in breast cancer patients [49,50]. However, the most recent reports indicate that higher leptin serum levels are associated with advanced stage breast cancer [51,52].
In humans, several polymorphisms have been identified in the LEP and LEPR genes: a G to A substitution at nt -2548 upstream of the ATG start site [53] in the LEP gene 5' promoter region, and an A to G substitution at nt 668 from the start codon 223 in exon 6 (Q223R) of the LEPR gene coding for the extracellular region common to all isoforms of LEPR [54].
The G to A substitution at -2548 of the LEP gene was associated with leptin production [55,56]. The glutamine to arginine substitution occurs on the LEPR gene within the first of two putative leptin-binding regions and may be associated with impaired signalling capacity of the leptin receptor [57].
Enhanced gene expression and increased circulating leptin levels have been reported in subjects carrying the LEPR 223R or LEP (-2548) A alleles [55-57]. The LEP (-2548) A allele has also been associated with a two-fold increase of leptin secretion adipocytes when compared to secretion by adipocytes bearing only the LEP (-2548) G allele [56].
We hypothesize that these leptin and leptin receptor polymorphisms associated with higher leptin serum levels and overexpression of leptin in adipocytes, favour breast cancer development and aggressiveness. In the present study, we analysed LEP (-2548) G/A and LEPR Q223R genotypes in a series of breast cancer cases and normal controls.
Methods
Patients and controls
The gene and allele frequencies of the LEP and LEPR genes were determined in a group of 222 control subjects and 308 patients with breast carcinoma. Controls and patients were selected from the same population living in the middle coast of Tunisia. Both the control and patients groups include unrelated subjects.
Data on patient, tumour and treatment characteristics at the study entry for each subject were collected from the department of Radiation Oncology and Medical Oncology of Sousse Hospital (Sousse, Tunisia) between 1996 and 2003. They were selected consecutively whenever practically feasible.
All patients included in this study had primary breast carcinoma, with unilateral breast tumours and with no family history for the disease. The diagnosis of cancer was confirmed by histopathology analyses. The patients (n = 308) had a mean age of 50 ± 24 years. The median of follow-up was 36 months (range, 1–120 months). At time of analysis, 66 patients experienced recurrence (local or distant). Among them, 35 died from breast carcinoma (54.5 %). Table 1 shows the treatment description of all patients. A detailed description of the clinicopathologic characteristic of this cohort has been reported elsewhere [58] and data on tumor size at diagnosis, nodal status and histologic grade are briefly included in Table 3.
Table 1.
Treatment description of the 308 patients with Breast Carcinoma
| Surgery | No surgery | |
| Radiotherapy | 33 | 14 |
| Chemotherapy | 28 | 14 |
| Radiotherapy + Chemotherapy | 137 | 18 |
| Radiotherapy + endocrine therapy | 14 | 0 |
| Chemotherapy + endocrine therapy | 7 | 4 |
| Radiotherapy + Chemotherapy + endocrine therapy | 39 | 0 |
Table 3.
Genotype frequencies of LEP and LEPR polymorphisms in relation to pathological indices of breast cancer severity.
| Pathological feature | Number of patients (%) | P-value | |
| LEP (-2548) G/A | GG | GA + AA | |
| Tumor size | |||
| T1-T2 | 82 (42.05) | 113 (57.95) | 0.05 |
| T3-T4 | 21 (26.92) | 57 (73.08) | |
| Histological grade | |||
| 1–2 | 68 (39.08) | 106 (60.92) | 0.3 |
| 3 | 28 (32.56) | 58 (67.44) | |
| Lymph node metastases | |||
| Negative | 64 (42.38) | 87 (57.62) | 0.13 |
| Positive | 51 (34.00) | 99 (66.00) | |
| Hormone receptor status | |||
| Negative | 34 (38.20) | 55 (61.80) | 0.36 |
| Positive | 77 (44.00) | 98 (56.00) | |
| LEPR Q223R | QQ + RR | ||
| Tumor size | |||
| T1-T2 | 56 (29.17) | 136 (70.83) | 0.57 |
| T3-T4 | 27 (32.53) | 56 (67.47) | |
| Histological grade | |||
| 1–2 | 58 (32.95) | 118 (67.05) | 0.19 |
| 3 | 21 (25.00) | 63 (75.00) | |
| Lymph node metastases | |||
| Negative | 55 (36.67) | 95 (63.33) | 0.07 |
| Positive | 41 (27.15) | 110 (72.85) | |
| Hormone receptor status | |||
| Negative | 40 (41.53) | 49 (58.47) | 0.57 |
| Positive | 85 (34.33) | 90 (65.67) | |
OR, odds ratio; CI, confidence interval. The χ2 test was used to determine whether significant differences (P-value) were observed when the patient group was compared with the control group. f, frequencies.
Healthy women (n = 222) having a mean age of 48 ± 14 years, were blood donors with no evidence of any personal or family history of cancer (or other serious illness). Samples from healthy controls were collected consecutively between 1996 and 2003. Both cases and controls were informed and gave written consent to participate in the study and to allow their biological samples to be genetically analyzed. Approval for the study was given by the National Ethical Committee.
Patients treated by chemotherapy as a primary treatment
Among the 308 patients, 121 had chemotherapy as an anticancer primary treatment. The chemotherapy induction was based in all cases on the combination of cyclophosphamide (100%), 5-fluorouracil (100%) with adriamycin (34%) or epirubicin (52%) or methotrexate (14%). For neoadjuvant treatment, patients received four or six chemotherapy cycles before surgery. The clinical response to induction chemotherapy for all cases was defined according to the following criteria: complete response when regression of the tumour was total, partial response when reduction of tumour size was greater then 50% and poor response when the reduction of tumour size was less then 50% (tumour size was measured by the bidimensional product of the horizontal and vertical dimensions).
Genomic DNA extraction
Genomic DNA was extracted from peripheral blood leukocytes by a salting procedure [59]. Briefly, 10 ml of blood was mixed with triton lysis buffer (0.32M sucrose, 1% Triton X-100, 5 mM MgCl2, H2O, 10 mM Tris-HCl, PH 7.5). Leukocytes were spun down and washed with H2O. The pellet was incubated with proteinase K at 56°C and subsequently salted out at 4°C using a substrate NaCl solution. Precipated proteins were removed by centrifugation. The DNA in supernatant fluid was precipated with ethanol. The DNA pellet was dissolved in 400 μl H2O.
Polymorphism analysis of the -2548 G/A LEP gene
The -2548 G/A polymorphism of the LEP gene was analysed by restriction fragment length polymorphism-polymerase chain reaction (RFLP-PCR). This method was carried out by PCR amplification using forward primer 5'-TTTCTGTAATTTTCCCGTGAG-3' and reverse primer 5'-AAAGCAAAGACAGGCATAAAAA-3' in a 25 μl reaction mixture containing genomic DNA samples (100 ng), 200 μmol/L dNTPs, 1.5 mM MgCl2, 1 X Taq polymerase buffer, 50 pmol of each primer, and 0.5 unit of Taq DNA polymerase (Amersham, Paris, France). Reaction conditions used with the thermal cycler (Biometra, GÖttingem, Germany) were as follows: an initial incubation at 94°C for 5 minutes followed by 30 cycles of incubation at 94°C for 45 seconds, 52°C for 45 seconds and 72°C for 45 seconds with a final extension at 72°C for 7 minutes. The PCR product (242 bp) was verified by DNA sequencing.
The amplified products were digested with the addition of 2 U CfoI for 3 hours at 37°C. The digested samples were separated by electrophoresis through a 2% agarose gels and stained with ethidium bromide. The polymorphism was defined by presence (G) or absence (A) of the CfoI restriction site. To assess reliability of genotyping we performed double sampling RFLP-PCR in more than 10% of the samples and found no differences.
Polymorphism analysis of the LEPR gene
Based on the method described by Gotoda et al. [54], a PCR followed by digestion with endonuclease MspI was used to detect the A to G transition polymorphism at codon 223 of LEPR gene. Two sequence specific oligonucleotides primers were used for the polymerase chain reaction (PCR): the 3' primer 5'-AAACTCAACGACACTCTCCTT- 3' and 5' primer 5'-TGAACTGACATTAGAGGTGAC- 3'. PCR was performed by using 100 ng genomic DNA as template, 0.01 mM dNTPs, 1.5 mM MgCl2, 1 X Taq polymerase buffer, 0.75μM of each primer and 0.5 U of Taq DNA polymerase in a total reaction of 25 μl. Reaction conditions used with the thermal cycler were as follows: 94°C for 5 min followed by 35 cycles at 94°C for 30 seconds, 52°C for 45 seconds and 72°C for 45 seconds. A final extension step was carried out at 72°C for 5 min.
The PCR products (80 bp) were electrophoresed on a 3% agarose gel containing ethidium bromide to monitor amplification and possible contamination. The PCR product (80 bp) was verified by DNA sequencing. The 80 bp PCR products were digested with MspI and analysed on 4% (3:1 Nusieve: Normal) agarose gels because of the small DNA products size. The presence of MspI site was indicated by the cleavage of the amplified product into two fragments of 57 and 23 bp. The two allelic forms of LEPR, corresponding to the absence or the presence of the MspI site, are referred to as 223Q and 223R, respectively.
Statistical analyses
The allele frequencies of LEP and LEPR were tested for the Hardy-Weinberg equilibrium for both patient and control groups using the chi-square test. The same test was used to evaluate for significant association between disease (breast carcinoma versus controls) and LEP or LEPR genotypes or alleles.
Disease-free survival (DFS) was defined as the time from the date of diagnosis to the first local or distant recurrence or to last contact. Breast carcinoma-specific overall survival (OVS) was defined as the time from the date of diagnosis to death if the patient died from breast carcinoma or to last contact. Six-year survival rates were estimated, and survival curves were plotted according to Kaplan and Meier [60]. The differences between groups were calculated by the log-rank test [61].
Clinicopathological parameters were dichotomised as follows: nodal status (≥ 1 versus no positive lymph node), SBR (Scarff, Bloom and Richardson) tumour grade (1–2 versus 3), clinical tumour size (T1-T2 versus T3-T4).
Statistics were performed using SEM-STATISTIQUES software (centre Jean Perrin, Clermont-Ferrand, France).
Results
Polymorphisms in the LEP and LEPR genes as risk factor for breast carcinoma
The genotype distribution and allele frequencies for the LEP (-2548) G/A and LEPR Q223R polymorphisms in all patients with breast carcinoma and in the control group are presented in Table 2. The allele frequencies of LEP and LEPR genes were in Hardy-Weinberg equilibrium in both patient and control groups (P = 0.55, P = 0.7; P = 0.17, P = 0.376 respectively).
Table 2.
The LEP (-2548) G/A and LEPR Q223R Genotype distributions in Control Subjects and in Patients with Breast Carcinoma.
| Genotypes | Patients (n = 308) | Controls (n = 222) | OR | Confidence interval | P-value | ||
| n | f | n | f | ||||
| LEP (-2548) G/A | |||||||
| GG | 119 | (0.386) | 112 | (0.504) | 1 | ||
| GA | 152 | (0.494) | 99 | (0.446) | 1.45 | [0.99–2.11] | 0.04 |
| AA | 37 | (0.120) | 11 | (0.050) | 3.17 | [1.47–6.96] | 0.001 |
| Alleles | |||||||
| G-allele | 390 | (0.633) | 323 | (0.727) | |||
| A-allele | 226 | (0.367) | 121 | (0.273) | 1.55 | [1.18–2.04] | 0.001 |
| LEPR Q223R | |||||||
| 98 | (0.318) | 102 | (0.460) | 1 | |||
| QR | 145 | (0.471) | 90 | (0.405) | 1.68 | [1.12–2.50] | 0.007 |
| RR | 65 | (0.211) | 30 | (0.135) | 2.26 | [1.31–3.90] | 0.001 |
| Alleles | |||||||
| Q-allele | 341 | (0.554) | 294 | (0.662) | |||
| R-allele | 275 | (0.446) | 150 | (0.338) | 158 | [1.22–2.05] | 0.0003 |
OR, odds ratio; CI, confidence interval. The χ2 test was used to determine whether significant differences (P-value) were observed when the patient group was compared with the control group. f, frequencies.
A significantly higher risk for breast cancer was observed for carriers of LEP (-2548) AA genotype (Odds Ratio (OR) = 3.17; P = 0.001) and carriers of LEP (-2548) GA genotype (OR = 1.45; P = 0.04). The LEP (-2548) A allele frequency was significantly higher in the patient group compared to the control group (OR = 1.55; P = 0.001).
The allelic frequency of the LEPR 223R was 0.446 in patients with breast carcinoma and 0.338 in control subjects (OR = 1.57; P = 0.0003). The frequency of heterozygous LEPR 223QR was 0.471 in the patient group and 0.405 in the control population resulting in a significantly increased OR in the patient group bearing this genotype (OR = 1.68; P = 0.007). The frequency of the homozygous LEPR 223 RR was 0.211 in the patient group and only 0.135 in controls (OR = 2.26; P = 0.001). These data, taken together, suggest that an increased risk of developing breast carcinoma is associated with the inheritance of the LEP (-2548) A and LEPR 223R alleles in a dose-dependant manner.
When we stratified the patients according to the menopause status (190 pre-menopausal and 118 post-menopausal) no significant changes in the LEP and LEPR genotype distributions were seen in breast carcinoma subgroups (data not shown).
Prognostic significance of polymorphism in LEP and LEPR genes
Table 3 shows the distributions of LEP and LEPR polymorphisms according to the clinic-pathologic indices of breast carcinoma severity.
We stratified patients according to tumor size in two subgroups. The first group includes tumor size less than 5 cm (T1-T2) and the second group includes patients with locally advanced breast cancer (T3-T4). The frequency of the LEP (-2548) A allele was significantly higher in patients with larger tumor size (P = 0.05). No association was found with the other clinic-pathologic parameters.
When we tested the relationship between the presence of the LEP (-2548) A allele in all the 308 patients and the survival (OVS and DFS), significant differences were observed between the DFS Kaplan-Meier survival curves for the different polymorphisms.
As shown in figure 1, the breast carcinoma specific DFS was significantly shorter in the patient population carrying the LEP (-2548) A allele. The estimated 3- and 6- year breast carcinoma specific DFS rates in the group of patients carrying the LEP (-2548) A allele were, respectively, 85.29 % and 60.29 % versus the DFS rates of 95.59 % and 79.41 % for those carrying the LEP (-2548) G allele (log rank test, P < 0.03). No statistical difference in overall survival between both groups of patients was observed.
Figure 1.
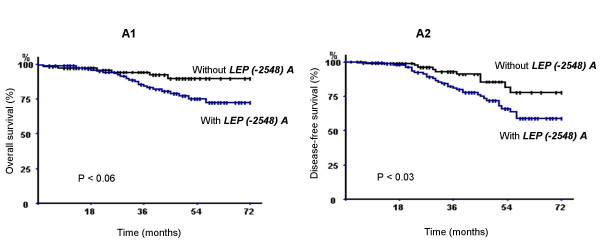
Breast carcinoma specific overall survival (A1) and specific disease-free survival (A2) of 308 breast carcinoma patients according to the presence or absence of LEP (-2548) A allele (p denotes the log-rank test value).
The breast carcinoma specific OVS was significantly shorter in the group of patients carrying LEPR 223R allele (Fig. 2). The estimated 3- and 6- year breast carcinoma OVS rate in the group of patients carrying LEPR 223R allele were, respectively, 86.76 % and 75 % versus 98.5 and 97.06% for those carrying the 223Q allele (log rank test, P < 0.01). In contrast, no association was found between LEPR 223R and the DFS rate in this population of patients with breast carcinoma.
Figure 2.
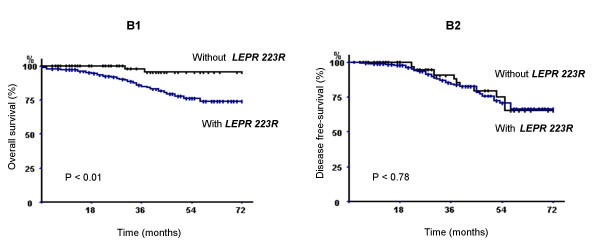
Breast carcinoma specific overall survival (B1) and specific disease-free survival (B2) of 308 breast carcinoma patients according to the presence or absence of LEPR 223R allele (p denotes the log-rank test value).
Multivariate analyses were undertaken to evaluate the importance of LEP and LEPR markers in the risk of the recurrence and death compared with the clinicopathologic parameters. Introducing the genetic and the clinicopathologic parameters bearing prognostic significance has tested the Cox model. No genetic and clinicopathologic parameters were selected for OVS and DFS.
Discussion
Leptin is a cytokine produced mainly by adipose tissue. Leptin has recently been reported to stimulate the proliferation of various cell types and is considered to be a new growth factor. Hyperleptinemia is a common feature of obese women who have a risk of breast cancer higher than those with normal weight and epidemiologic studies have suggested a positive correlation between obesity and breast cancer risk [62].
In addition, recent studies indicate that leptin is a mitogenic [63], as well as pro-angiogenic factor in various cell models. New data documented that human breast cancer cell lines and breast tumors may express leptin and leptin receptors [40,42,47,48]. These characteristics of leptin prompted us to evaluate whether genetic variations of the LEP gene promoter and/or LEPR exon 6 affect susceptibility to and prognosis of breast cancer.
The present case/control study showed a substantially increased risk of breast carcinoma associated in a dose-dependent manner, with the inheritance of the LEP (-2548) A allele. Individuals homozygous for LEP (-2548) A have more than 3-fold risk to develop breast carcinoma (OR = 3.17; P = 0.001) compared with individuals homozygous for LEP (-2548) G allele. The LEP (-2548) GA heterozygotes patients have an intermediate risk for this cancer (OR = 1.45; P = 0.04).
Comparison of genotype frequencies of the LEPR Q223R alleles in patients with breast carcinoma and control subjects indicated an increase of LEPR 223QR and RR genotypes. Consequently, the frequency of the LEPR 223R allele was found to be significantly higher in patients group compared with controls.
In this study, we investigate the prognostic significance evaluation of LEP and LEPR polymorphism by examining the potential association of LEP -2548 and LEPR exon 6 variations and the clinical response to chemotherapy induction. This evaluation indicated that LEP and LEPR polymorphisms do not have a predictive value for clinical responses to chemotherapy.
The prognostic evaluation of the LEP and LEPR genetic markers in breast carcinoma indicated that LEP (-2548) A allele is associated with a larger tumor size at diagnosis and a shorter disease-free survival, and that the LEPR 223R allele is associated with shorter overall survival, and therefore with a poor prognosis in breast carcinoma.
Both functional studies and genetic analyses have highlighted the role of LEP and LEPR in cancer pathogenesis and disease progression [64-67].
Previous reports demonstrated that LEP (-2548) A allele was significantly associated with higher leptin level [55,56]. However, the relationship between leptin level and the LEPR Q223R polymorphism are conflicting. Quinton et al. have indicated that LEPR 223R allele was associated with lower circulating leptin level [68]. In contrast, Yiannakouris et al. have found that carriers of the LEPR 223R allele had significantly higher leptin level than non carriers [57].
Several studies have shown the relationship between LEP and LEPR gene polymorphisms and human cancer [66,67,69]. The LEP (-2548) A allele, which result in high leptin secretion, was associated with increased risk of prostate cancer [66]. Recently, a study conducted on 45 patients has reported that LEPR Q223R polymorphism is not associated with an increased risk of breast cancer [69]. This result is in contrast with our findings.
The relationship between leptin serum level and breast cancer is still controversial. Mantzoros et al. were observed that leptin did not appear to increase the risk of pre-menopausal breast cancer in situ [70]. In addition, Petridou et al. have found no relationship between leptin serum levels and breast cancer in post-menopausal women [49]. A recent large study has demonstrated that subjects with elevated serum leptin levels displayed increased risk of developing breast cancer than those with the normal levels [52]. Furthermore, in the same study, the serum level of leptin was not associated with menopausal status in patients with mammary disease. This result along with our findings suggests that leptin increased the risk of breast cancer independently from menopausal status.
Additionnaly, in breast cancer it has been shown that a high leptin serum level is associated with reduced OVS [71]. Recently, it has been reported that the levels of expression of leptin and leptin receptor correlate with distant metastasis [47]. Patients with LEPR negative and low leptin-expressing tumors have shown an extremely good outcome, and the survival rate tended to be lower for patients with LEPR positive or high leptin-expressing tumors [47].
Some authors have demonstrated that leptin increase cell proliferation which is an essential element for tumor metastasis, through cell progression in MCF-7 human breast cancer cells with up-regulation of PKC-α, PPARγ and PPARα [41]. Moreover, Laud et al. have suggested that leptin stimulated the proliferation of the mitogen-activated protein kinase (MAP kinase) pathway [40]. Leptin may also modulate apoptosis, since it has been reported in leukaemia cell lines that leptin acts as an inhibitor of apoptosis [72]. In addition, leptin has been shown to promote invasiveness of renal and colonic epithelial cells via the phosphatidylinositol 3'-kinase, rho-dependent, and rac-dependent cascades [43]. Others have also reported that leptin act synergistically with VEGF and fibroblast growth factor 2 (FGF-2) to promote angiogenesis [73,74]. Moreover, leptin has been shown to increase the expression of many other genes involved in angiogenesis, for example, MMP-2 and MMP-9 gene products [75].
Other lines of evidence derive from studies on markers of tumoral risk that are elevated in breast cancer, that can upregulate leptin production. Factors such as TNF-alpha [76,77], IL-1alpha [78], IL-1 beta [79], estrogens [79], all raise leptin levels, promoting growth and differentiation of tumors.
The results of the current study, which show the association between the LEP (-2548) G/A and LEPR Q223R polymorphisms and breast carcinoma susceptibility and survival, suggest that the genetic basis of the potential tumor promoter role attributed to LEP and its receptor may result from certain LEP and LEPR polymorphisms. Therefore, these polymorphisms may play a casual role in tumor development as well as in governing poor prognosis of breast carcinoma.
Conclusion
In conclusion, this study suggests that genetic variation in the tandem LEP and its receptor LEPR may be attractive susceptibility markers for breast carcinoma. These genetic markers represent also, prognostic variables for predicting recurrence and death from breast carcinoma.
The role of the leptin and its receptor as genetic markers for breast cancer can be completed with other SNPs and a haplotype analysis.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
SK conceived of the study, conducted data analysis, and drafted the manuscript. AD-S contributed to the design and management of data. NB and S-BA provided samples and clinical information. LC conceived, designed and participated in the data analysis and interpretation of the study. AN-H contributed to reviewing the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
This work was supported by le Ministère de la Recherche Scientifique et de Technologie et du Développement des Compétences, by le Ministère de l'Enseignement Supérieur, by le Ministère de la Santé Publique de la République Tunisienne.
Contributor Information
Kaouther Snoussi, Email: naftikaouther@yahoo.fr.
A Donny Strosberg, Email: strosber@scripps.edu.
Noureddine Bouaouina, Email: noueddine.bouaouina@rns.tn.
Slim Ben Ahmed, Email: slim.benahmed@rns.tn.
A Noureddine Helal, Email: Ahmed.Helal@fphm.rnu.tn.
Lotfi Chouchane, Email: lotfi.chouchane@planet.tn.
References
- Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- Ries LAG, Kosary CI, Hankey BF, eds . SEER Cancer Statistics Review, 1973–1996. Bethesda, MD: National Cancer Institute; 1999. [Google Scholar]
- Collaborative Group on Hormonal Factors in Breast Cancer Breast Cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52, 705 women with breast cancer and 108, 411 women without breast cancer. Lancet. 1997;350:1047–1059. doi: 10.1016/S0140-6736(97)08233-0. [DOI] [PubMed] [Google Scholar]
- Collaborative Group on Hormonal Factors in Breast Cancer Breast cancer and Hormonal contraceptives: collaborative reanalysis of individual data on 53, 297 women with breast cancer and 100, 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996;347:1713–1727. doi: 10.1016/S0140-6736(96)90806-5. [DOI] [PubMed] [Google Scholar]
- Hulka BS, Stark AT. Breast cancer: cause and prevention. Lancet. 1995;346:883–887. doi: 10.1016/S0140-6736(95)92713-1. [DOI] [PubMed] [Google Scholar]
- Kelsey JL. Breast cancer epidemiology: summary and future directions. Epidemio Rev. 1993;15:256–263. doi: 10.1093/oxfordjournals.epirev.a036112. [DOI] [PubMed] [Google Scholar]
- World Cancer research Fund-American Institute of Cancer Research Food, nutrition and the prevention of cancer: A global perspective. Mebesha USA: Banta Book Group. 1997;S.252 [Google Scholar]
- Antoniou AC, Pharoah PD, McMullan G, Day NE, Ponder BA, Easton D. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet Epidemiol. 2001;89:51–62. doi: 10.1002/gepi.1014. [DOI] [PubMed] [Google Scholar]
- Clearly MP, Maihle NJ. The role of body mass index in the relative risk of developing premenopausal versus postmenopausal breast cancer. Proc Soc Exp Biol Med. 1997;216:28–43. doi: 10.3181/00379727-216-44153b. [DOI] [PubMed] [Google Scholar]
- Willett WC. Fat, energy and breast cancer. J Nutr. 1997;127:921S–3S. doi: 10.1093/jn/127.5.921S. [DOI] [PubMed] [Google Scholar]
- Galanis DJ, Kolonel LN, Lee J, Le Marchand L. Anthropometric predictors of breast cancer incidence and survival in a multi-ethnic cohort of female residents of Hawaii, United States. Cancer Causes Control. 1998;9:217–224. doi: 10.1023/A:1008842613331. [DOI] [PubMed] [Google Scholar]
- Klurfeld DM, Lioyd LM, Welch CB, Davis MJ, Tulp OL, Kritchevsky D. Reduction of enhanced mammary carcinogenesis in LA/N-cp (corpulent) rat by energy restriction. Proc Soc Exp Biol Med. 1992;196:381–384. doi: 10.3181/00379727-196-43202. [DOI] [PubMed] [Google Scholar]
- Waxler SH. Obesity and cancer susceptibility in mice. Am J Clin Nutr. 1960;8:760–766. [Google Scholar]
- Heston WE, Vlahakis G. Genetic obesity and neoplasia. J Natl Cancer Inst. 1962;29:197–209. [PubMed] [Google Scholar]
- Wolff GL, Kodell RL, Cameron AM, Medina D. Accelerated appearance of chemically induced mammary carcinomas in obese yellow (Avy/A) (BALB/c x VY) F1 hybrid mice. J Toxicol Environ Health. 1982;10:131–142. doi: 10.1080/15287398209530237. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guiez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- Hallaas J, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman J. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Benett BD, Solar GP, Yuan JQ, Mathias J, Thomas JR, Matthews W. A role for leptin and its cognate receptor in hematopoiesis. Cur Biol. 1996;6:1170–1180. doi: 10.1016/S0960-9822(02)70684-2. [DOI] [PubMed] [Google Scholar]
- Hoggard N, Hunter L, Trayhurn P, Williams LM, Mercer JG. Leptin and reproduction. Proc Nutr Soc. 1998;57:421–427. doi: 10.1079/PNS19980061. [DOI] [PubMed] [Google Scholar]
- Bouloumie A, Drexler HC, Lafontan M, Busse R. Leptin the product of Ob gene, promotes angiogenesis. Cir Res. 1998;269:546–549. doi: 10.1161/01.res.83.10.1059. [DOI] [PubMed] [Google Scholar]
- Loffreda S, yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM. Leptin regulates proinflammatory immune responses. FASEB J. 1998;12:57–65. [PubMed] [Google Scholar]
- Green ED, Maffei M, Braden VV, Proenca R, DeSilva U, Zhang Y, Chua SC, Jr, Leibel RL, Weissenbach J, Friedman JM. The human obese (OB) gene: RNA expression pattern and mapping on the physical, cytogenetic, and genetic maps of chromosome 7. Genome Res. 1995;5:5–12. doi: 10.1101/gr.5.1.5. [DOI] [PubMed] [Google Scholar]
- Isse N, Ogawa Y, Tamura N, Masuzaki H, Mori K, Okazaki T, Satoh N, Shigemoto M, Yoshimasa Y, Nishi S, Hosoda K, Inazawa J, Nakao K. Structural organization and chromosomal assignment of the human obese gene. J Biol Chem. 1995;270:27728–27733. doi: 10.1074/jbc.270.46.27728. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Ogawa Y, Isse N, Satoh N, Okazaki T, Shigemoto M, Mori K, Tamura N, Hosoda K, Yoshimasa Y, Jingami H, Kawada T, Nakao K. Human obese gene expression: Adipocyte-specific expression and regional differences in the adipose tissue. Diabetes. 1995;44:855–858. doi: 10.2337/diab.44.7.855. [DOI] [PubMed] [Google Scholar]
- Malik KF, Young WS., 3rd Localisation of binding sites in the central nervous system for leptin (OB protein) in normal, obese (ob/ob), and diabetic (db/db) C57BL/6J mice. Endocrinology. 1996;137:1497–1500. doi: 10.1210/en.137.4.1497. [DOI] [PubMed] [Google Scholar]
- Hoggard N, hunter L, Duncan JS, Williams LM, Trayhurn P, Murcer JG. Leptin and leptin recptor mRNA and protein expression in the murine foetus and placenta. Proc Natl Acad Sci USA. 1997;94:11073–11078. doi: 10.1073/pnas.94.20.11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V, Liu YL, Awthorne MA, Morton NM, Davenport M. Expression of the functional leptin receptor mRNA in pancreatic islet and direct inhibitory action of leptin on insulin secretion. Diabetes. 1997;46:313–316. doi: 10.2337/diab.46.2.313. [DOI] [PubMed] [Google Scholar]
- Cioffi JA, Shafer AW, Zupancic TJ, Smith-Gbur J, Mikhail A, Platika D, Snodgrass HR. Novel B219/OB receptor isoforms: possible role of leptin hematopoiesis and reproduction. Nat Med. 1996;2:585–589. doi: 10.1038/nm0596-585. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Hanif S, Arch JR, Tadayyon M. Leptin receptor long-form signalling in a human liver cell line. Cytokine. 2001;14:225–229. doi: 10.1006/cyto.2001.0871. [DOI] [PubMed] [Google Scholar]
- Tsuchiya T, Shimiu H, Hori T, Mori M. Expression of leptin receptor in lung: leptin as a growth factor. Eur J Pharmacol. 1999;365:273–279. doi: 10.1016/S0014-2999(98)00884-X. [DOI] [PubMed] [Google Scholar]
- Mix H, Widjaja A, Jandl O, Cornberg M, Kaul A, Goke M, Beil W, Kuske M, Brabant G, Manns MP, Wagner S. Expression of leptin receptor isoforms in the human stomach. Gut. 2000;36:457–461. doi: 10.1136/gut.47.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T, Nakao K. No adipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3:1029–1033. doi: 10.1038/nm0997-1029. [DOI] [PubMed] [Google Scholar]
- Oliveira AM, Nashimento AG, LIoyd RV. Leptin and leptin receptor mRNA are widely expressed in tumors of adipocytic differenciation. Mod Pathol. 2001;14:549–555. doi: 10.1038/modpathol.3880349. [DOI] [PubMed] [Google Scholar]
- Morash B, Johnstone J, Leopold C, Li A, Murphy P, Ur E, Wilkinson M. The regulation of leptin gene expressioin in the C6 glioblastoma cell line. Mol Cell Endocrinol. 2000;165:97–105. doi: 10.1016/S0303-7207(00)00259-8. [DOI] [PubMed] [Google Scholar]
- Quehenberger P, Exner M, Sunder-Plassmann R, Ruzicka K, Bieglmayer C, Endler G, Muellner C, Speiser W, Wagner O. Leptin induces endothelin-1 in endothelial cells in vitro. Circ Res. 1998;83:1059–1066. doi: 10.1161/01.res.0000014226.74709.90. [DOI] [PubMed] [Google Scholar]
- Umemoto Y, Tsuji K, Yang F-C, Ebihara Y, Kaneko A, Furukawa S, Nakahata T. Leptin stimulates the proliferation of murine myelocytic and primitive hematopoietic progenitor cells. Blood. 1997;90:3438–3443. [PubMed] [Google Scholar]
- Dieudonne M-N, Machinal-quelin F, Serazin-Leroy V, Leneveu M-C, Pecquery R, Giudicelli Y. Leptin mediates a proliferative response in human MCF7 breast cancer cells. Biochem Biophys Res Commun. 2002;293:622–628. doi: 10.1016/S0006-291X(02)00205-X. [DOI] [PubMed] [Google Scholar]
- Laud K, Gourdou I, Pessemess L, Peyrat JP, Djiane J. Identification of leptin receptors in human breast cancer: Functional activity in the T47-D breast cancer cell line. Mol Cell Endocrinol. 2002;188:219–226. doi: 10.1016/S0303-7207(01)00678-5. [DOI] [PubMed] [Google Scholar]
- Okumura M, Yamamoto M, Sakuma H, Kojima T, Maruyama T, Jamali M, Cooper D, Yasuda K. Leptin and high glucose stimulate proliferation in MCF-7 human breast cancer cells: Reciprocal involvement of PKC-alpha and PPAR expression. Biochim Biophys Acta. 2002;1592:107–116. doi: 10.1016/s0167-4889(02)00276-8. [DOI] [PubMed] [Google Scholar]
- Hu X, Juneja SC, Maihle NJ, Cleary MP. Leptin a growth factor in normal and malignant breast cells and for normal mammary gland development. J Natl Cancer Inst. 2002;94:1704–1711. doi: 10.1093/jnci/94.22.1704. [DOI] [PubMed] [Google Scholar]
- Attoub S, Noe V, Pirola L, Bruyneel E, Chastre E, Mareel M, Wymann MP, Gespach C. Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3-kinase-, rho- and rac-dependent signalling pathways. FASEB J. 2000;14:2329–2338. doi: 10.1096/fj.00-0162. [DOI] [PubMed] [Google Scholar]
- Hardwick JCH, Van Den Brink GR, Offerhaus GJ, Van Deventer SIH, Peppelenbosh MP. Leptin is a growth factor for colonic epithelial cells. Gastroenterology. 2001;121:79–90. doi: 10.1053/gast.2001.25490. [DOI] [PubMed] [Google Scholar]
- Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y, Sato N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatol. 2001;34:288–297. doi: 10.1053/jhep.2001.26518. [DOI] [PubMed] [Google Scholar]
- Islam MS, Morton NM, Hansson A, Emilsson V. Rat insulinoma-derived pancreatic β-cells express a functional leptin receptor that mediates a proliferative response. Biochem Biophys Res Commun. 1997;238:851–855. doi: 10.1006/bbrc.1997.7399. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10:4325–4331. doi: 10.1158/1078-0432.CCR-03-0749. [DOI] [PubMed] [Google Scholar]
- Caldefie-Chézet F, Damez M, de Latour M, Konska G, Mishellani F, Fusillier C, Guerry M, Penault-Llorca F, Guillot J, Vasson M-P. Leptin: A proliferative factor for breast cancer? Study on human ductal carcinoma. Biochem Biophys Res Commu. 2005;334:737–741. doi: 10.1016/j.bbrc.2005.06.077. [DOI] [PubMed] [Google Scholar]
- Petridou E, Papadiamantis Y, Markopoulos C, Spanos E, Dessypris N, Trichopoulos D. Leptin and insulin growth factor I in relation to breast cancer (Greece) Cancer Causes Control. 2000;11:383–388. doi: 10.1023/A:1008903727238. [DOI] [PubMed] [Google Scholar]
- Coskun U, Gunel N, Toruner FB, Sancak B, Onuk E, Bayram O, Yilmaz E, Elbeg S, Ozkan S. Serum leptin, prolactin and vascular endothelial growth factor (VEGF) levels in patients with breast cancer. Neoplasma. 2003;50:41–46. [PubMed] [Google Scholar]
- Tessitore L, Visio B, Pesola D, Cecchini f, Mussa A, Argiles JM, Benedetto C. Adipocytes expression and circulating levels of leptin increase in both gynaecological and breast cancer patients. Int J Cancer. 2004;24:1529–1535. [PubMed] [Google Scholar]
- Han C, Zhang HT, Du L, Liu X, Jing J, Zhao X, Tian B. Serum levels of leptin, insulin, and lipids in relation to breast cancer in china. Endocrine. 2005;26:19–24. doi: 10.1385/ENDO:26:1:019. [DOI] [PubMed] [Google Scholar]
- Mammès O, Betoulle D, Aubert R, Giraud V, Tuzet S, Petiet A, Colas-Linhart N, Fumeron F. Novel polymorphisms in the 5' region of the LEP gene: Association with leptin levels and response to low-calorie diet in human obesity. Diabetes. 1998;47:487–489. doi: 10.2337/diabetes.47.3.487. [DOI] [PubMed] [Google Scholar]
- Gotoda T, Manning BS, Goldstone AP, Imrie H, Evans AL, Strosberg AD, McKeigue PM, Scott J, Aitman TJ. Leptin receptor gene variation and obesity; lack of association in a white British male population. Hum Mol Genet. 1997;6:869–876. doi: 10.1093/hmg/6.6.869. [DOI] [PubMed] [Google Scholar]
- Le Stunff C, Le Bihan C, Schrk NJ, Bougneres P. A common Promoter Variant of the Leptin Gene Is Associated With changes in the Relationship Between Serum Leptin and Fat Mass in Obese Girls. Diabetes. 2000;49:2196–2200. doi: 10.2337/diabetes.49.12.2196. [DOI] [PubMed] [Google Scholar]
- Hoffstedt J, Eriksson P, Mottagui-Tabar S, Arner P. A polymorphism in the leptin promoter region (-2548 G/A) influences gene expression and adipose tissue secretion of leptin. Horm Metab Res. 2002;34:355–359. doi: 10.1055/s-2002-33466. [DOI] [PubMed] [Google Scholar]
- Yiannakouris N, Yannakoulia M, Melistas L, Chan JL, Klimis-Zacas D, Mantrozos CS. The Q223R polymorphism of the leptin receptor gene is significantly associated with obesity and predicts a small percentage of body weight and body composition variability. J Clin Endocrinol Metab. 2001;86:4434–4439. doi: 10.1210/jc.86.9.4434. [DOI] [PubMed] [Google Scholar]
- Ben Ahmed S, Aloulou S, Bibi M, Landolsi A, Nouira M, Ben Fatma L, Kallel L, Gharbi O, khairi H, Kraiem C. Breast cancer detection in Tunisian women: an analysis of a hospital trial involving 729 patients. Santé Publique. 2002;14:231–241. [PubMed] [Google Scholar]
- Oletup H, Zetterquiest H. HLA-DR typing by PCR amplification with sequence specific primers (PCR-SSP) in two hours: an alternative to serological DR typing in clinical practice including donor recipient matching in cadaveric transplantation. Tissue Antigens. 1992;39:225–235. doi: 10.1111/j.1399-0039.1992.tb01940.x. [DOI] [PubMed] [Google Scholar]
- Kaplan EL, Meir P0. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, Mantel N, McPherson K, Peto J, Smith PG. Design and analysis of randomised clinical trials requiring prolonged observation of each patient. Br J Cancer. 1977;35:1–39. doi: 10.1038/bjc.1977.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard-Barbash R, Schatzkin A, Carter CL, Kannel WB, Kreger BE, D'Agostino RB, Splansky GL, Anderson KM, Helsel WE. Body fat distribution and breast cancer in the Framingham study. J Natl Cancer Inst. 1990;82:286–290. doi: 10.1093/jnci/82.4.286. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Okimura Y, Mizuno I, Iida K, Takahashi T, Kaji H, Abe H, Chihara K. Leptin induces mitogen-activated proteinkinase-dependent proliferation of C3H10T1/2 cells. J Biol Chem. 1997;272:12897–12900. doi: 10.1074/jbc.272.20.12897. [DOI] [PubMed] [Google Scholar]
- Na Y, Dan W, Hua Z, Xia Y, Xiaojing S, Huijian W, Ge W, Xinjuan W, Younfeng S. Molecular Mechanisms Involved in the Growth Stimulation of Breast Cancer Cells by Leptin. Cancer Res. 2004;64:5870–5875. doi: 10.1158/0008-5472.CAN-04-0655. [DOI] [PubMed] [Google Scholar]
- Catalano S, Marsico S, Giordano C, Mauro L, Rizza P, Panno ML, Ando S. Leptin Enhances, via AP-1, Expression of Aromatase in the MCF-7 Cell Line. J Biol Chem. 2003;278:28668–28678. doi: 10.1074/jbc.M301695200. [DOI] [PubMed] [Google Scholar]
- Ribeiro R, Vasconcelos A, Costa S, Pinto D, Morais A, Oliveira J, Lobo F, Lopes C, Medeiros R. Overexpressing Leptin Genetic Polymorphism (-2548 G/A) Is Associated With Susceptibility to Prostate cancer and Risk of Advanced Disease. Prostate. 2004;59:268–274. doi: 10.1002/pros.20004. [DOI] [PubMed] [Google Scholar]
- Kote-Jerai Z, Singh R, Durocher F, Easton D, Edwards SM, Ardern-Jones A, Dearnaley DP, Houlston R, Kirby R, Eeles R. Association between leptin receptor gene polymorphisms and early-onset prostate cancer. BJU Int. 2003;92:109–112. doi: 10.1046/j.1464-410X.2003.04272.x. [DOI] [PubMed] [Google Scholar]
- Quinton ND, Alisson JL, Ross RJM, Eastell R, Blakemore AIF. A single nucleotide polymorphism (SNP) in the leptin receptor is associated with BMI, fat mass and leptin levels in postmenopausal Caucasian woman. Human Genet. 2001;108:233–236. doi: 10.1007/s004390100468. [DOI] [PubMed] [Google Scholar]
- Woo HY, Park H, Ki CS, Park YL, Bae WG. Rlationships among serum leptin, leptin receptor gene polymorphisms, and breast cancer in Korea. Cancer Lett. 2005;9:1–6. doi: 10.1016/j.canlet.2005.05.041. [DOI] [PubMed] [Google Scholar]
- Mantzoros CS, Bolhke K, Moschos S, Cramer CS. Leptin in relation to carcinoma in situ of the breast: a study of pre-menopausal cases and controls. Int J Cancer. 1999;80:523–526. doi: 10.1002/(SICI)1097-0215(19990209)80:4<523::AID-IJC7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Goodwin PJ, Ennis M, Fantus IG, Pritchard KI, Trudeau ME, Koo J, Hood N. Is leptin a mediator of adverse prognostic effects of obesity in breast cancer? J Clin Oncol. 2005;23:6037–6042. doi: 10.1200/JCO.2005.02.048. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Mikhail A, Estrov Z, Zhao S, Harris D, Sanchez-Williams G, Kornblau SM, Dong J, Kliche KO, Jiang S, Snodgrass HR, Estey EH, Andreef M. Expression and function of leptin receptor isoforms in myeloid leukaemia and myelodysplastic syndromes: proliferative and anti-apoptotic activities. Blood. 1999;93:1668–1676. [PubMed] [Google Scholar]
- Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, Madge LA, Schner JS, Scheabb MB, Polverini PJ, Flores-Riveros JR. Biological action of leptin as an angiogenic factor. Science. 1998;281:1583–1586. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- Ribatti D, Nico B, Belloni AS, Vacca A, Roncali L, Nussdorfer GG. Angiogenic activity of leptin in the chick embryo chorioallantoic membrane is in part mediated by endogenous fibroblast growth factor-2. Int J Mol Med. 2001;8:265–268. doi: 10.3892/ijmm.8.3.265. [DOI] [PubMed] [Google Scholar]
- Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, Jang Y, Cho SY, Kim HS. Potential role of leptin in angiogenesis: Leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. 2001;33:95–99. doi: 10.1038/emm.2001.17. [DOI] [PubMed] [Google Scholar]
- Kirchgessner TG, Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Tumor necrosis factor-α contributes to obesity-related hyperleptinemia by regulating leptin release from adipocytes. J Clin Invest. 1997;100:2777–2782. doi: 10.1172/JCI119824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbach MS, Boehme MWJ, Wahl P, Stremmel W, Ziegler R, Nawroth PP. Tumor necrosis factor increases serum leptin levels in humans. J Clin Endocrinol Metab. 1997;82:4080–4082. doi: 10.1210/jc.82.12.4080. [DOI] [PubMed] [Google Scholar]
- Janik JE, Curti BD, Considine RV, Rager HC, Powers GC, Alvord WG, Smith JW, II, Gause BI, Kopp WC. Interleukin 1α increases serum leptin concentrations in humans. J Clin Endocrinol Metab. 1997;82:3084–3086. doi: 10.1210/jc.82.9.3084. [DOI] [PubMed] [Google Scholar]
- Reichlin S, Chen G, Nicolson M. Blood to brain transfer of leptin in normal and interleukin-1β treated male rats. Endocrinol. 2000;141:1951–1954. doi: 10.1210/en.141.6.1951. [DOI] [PubMed] [Google Scholar]