

Abstract
The pleiotropic effects of statins, inhibitors of 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase, have been recently extended to the modulation of angiogenesis. Here, to get more insight into the statins action, the authors have investigated the effect of atorvastatin on the expression of several angiogenic and inflammatory genes in human umbilical endothelial cells (HUVECs). Atorvastatin was proangiogenic at the dose of 10 nM, and antiangiogenic at the concentrations of 1 to 10 μM. Moreover, these higher concentrations inhibited also the proliferation of HUVECs induced by vascular endothelial growth factor (VEGF). Lower doses of atorvastatin did not influence endothelial cell proliferation. Importantly, atorvastatin at the micromolar concentrations diminished the production of interleukin (IL)-8, a proinflammatory and proangiogenic chemokine, and inhibited the synthesis of urokinase plasminogen activator (uPA), a potent proinflammatory mediator. However, it decreased also the expression of plasminogen activator inhibitor-1 (PAI-1) and thrombospondin-1 (TSP-1), the inhibitors of angiogenesis. Atorvastatin stimulated the expression of angiopoietin (Ang)-2 and moderately enhanced the expression of endothelial nitric oxide synthase (eNOS), whereas heme oxygenase-1 (HO-1) was not significantly affected. In conclusion, the present findings points to other angiogenesis-related effects of atorvastatin, which may be of relevance to the beneficial influence of statins in cardiovascular system.
Keywords: Atherosclerosis, Cancer, Heme Oxygenase-1, Interleukin 8, Vascular Endothelial Growth Factor
Statins are potent inhibitors of the 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase via blocking the substrate accessibility to the enzyme and thereby effectively subverting cholesterol metabolism (for reviews see Kaushal et al. 2003; Undas et al. 2004; Liao and Laufs 2004). Those efficient drugs have, however, the spectrum of activities much broader than could be explained only by decrease in cholesterol synthesis. They constitute the pleiotropic effects, which have been demonstrated to influence the production of inflammatory cytokines and other mediators, such as reactive oxygen species (for a recent review see Wassmann and Nickening 2003; Liao and Laufs 2004). Those pleiotropic, beneficial effects of statins in cardiovascular diseases have been recently extended to the modulation of angiogenesis. A biphasic influence has been observed, i.e., stimulation of angiogenesis at low, nanomolar concentrations, and inhibition at higher, micromolar concentrations (Weis et al. 2002).
Among others, the proangiogenic activities of statins are due to their effects on endothelial progenitor cells, which are protected from senescence and apoptosis by nanomolar concentrations of the drugs (Assmus et al. 2003; Llevadot et al. 2001). At the molecular level this protection is mostly ascribed to the stimulation of the inositol triphosphate (PI3)-Akt kinase pathway, resulting in the phosphorylation of endothelial nitric oxide synthase (eNOS), a critical mediator of angiogenic activity of endothelial cells (Kureishi et al. 2000). The phosphorylation of eNOS at Ser1177 by Akt is dependent on statin-mediated recruitment of Akt to eNOS complex by heat shock protein 90 (hsp90) chaperone protein. Statins promote tyrosine phosphorylation of hsp90 and direct interaction of hsp90 with Akt (Brouet et al. 2001). Antiapoptotic effects are due to inhibition of p21 and p27 cyclin-dependent-kinase inhibitors (Assmus et al. 2003). On the other hand antiangiogenic effect of higher, micromolar concentrations of statins is due to the induction of apoptosis in endothelial cells and inhibition of the synthesis of vascular endothelial growth factor (VEGF) (Frick et al. 2003; Weis et al. 2002). Inhibitory influence of statins on the production of VEGF has been observed both in vitro (Frick et al. 2003; Dulak et al. 2001) and in vivo (Alber et al. 2002, 2005).
Nevertheless, although widely investigated, the field is far from clarity. For example, antiapoptotic effect of simvastatin on differentiated endothelial cells (human umbilical vein endothelial cells; HUVECs) has been claimed by some studies to occur at 1 μM concentration (Kureishi et al. 2000). On the contrary, others reported the proapoptotic activity of simvastatin at the same low- micromolar concentration (Urbich et al. 2002; Assmus et al. 2003). Antiangiogenic effect has been also ascribed to occur due to the inhibition of VEGF synthesis at micromolar doses of statins (Weis et al. 2002; Frick et al. 2003). However, studies demonstrated also the stimulation of VEGF synthesis at high-micromolar concentrations of the drugs (Frick et al. 2003). Therefore, to get more insight into the angiogenic action of statins, we performed the analysis of the effect of atorvastatin, a representative of this class of drugs, on angiogenic gene expression in HUVECs.
MATERIALS AND METHODS
Reagents
M199 medium, l-glutamine, epithelial growth factor (EGF), hydrocortisone, and carboxymethylcellulose were purchased from Sigma. Fetal calf serum (FCS) was procured from Invitrogen. CytoTox-96 assay, Reverse Transcription System, PCR Core System were obtained from Promega. Human recombinant VEGF165 and basic fibroblast growth factor (bFGF), as well as enzyme-linked immunosorbent assay (ELISA) kits for human VEGF and interleukin (IL8)-proteins were purchased from R&D Systems. The cell proliferation ELISA was obtained from Roche Diagnostic. GEArray expression arrays were purchased from SuperArray Bioscience Corporation. Primers for reverse transcriptase–polymerase chain reaction (RT-PCR) of eNOS, heme oxygenase (HO)-1 and EF2 genes have been designed using the sequences deposited in GeneBank and synthesized at Institute of Biochemistry and Biophysics in Warsaw, Poland. For amplification of angiopoitin (Ang)-1, Ang-2, and VEGF-D, the commercially available primers from R&D Systems (Abingdon, UK) were used according to vendor's protocol.
Cell Culture and Incubation Experiments
HUVECs were freshly isolated from umbilical veins by collagenase digestion. Cells were cultured in M199 medium supplemented with FCS (10%), endothelial cell grow supplement (ECGS), heparin, l-glutamine (2 mM), hydrocortisone (1 μg/mL), and antibiotics. Experiments were performed on confluent cell cultures at second or third passages. Angiogenic activities of HUVECs were stimulated by supplementation of cells with VEGF165 or bFGF (10 to 30 ng/mL).
Atorvastatin was dissolved in DMSO (stock 10 mM) and added to the cells at indicated concentrations for the whole incubation period. DMSO was added at the same amount to control the effect of diluent. The final concentration of DMSO never exceeded 0.1% and did not affect cell viability (not shown). Mevalonic acid (dissolved in ethanol) was used at 100 μM concentration.
Proliferation Assay
Experiments were performed on HUVECs cultured in 96-well plates in media with 10% FCS but without ECGS. After a 48-h incubation period, BrdU (10 μM) was added for 2 h and proliferation was measured by BrdU incorporation assay according to the vendor's protocol. In brief, the cells were fixed and peroxidase-labeled anti-BrdU antibodies were added for 90 min. Then the wells were washed and a substrate solution was added and incubated at 15°C to 25°C until color development was sufficient for photometric detection (usually 5 to 30 min). Reaction was stopped by addition of 1 mM sulphuric acid and the absorbance was measured at 450 nm.
Capillary Sprouting
Experiments were performed as previously described (Jozkowicz et al. 2003, 2004) according to the procedure established by Korff and Augustin (1998) using medium containing 10% FCS, but without ECGS. In order to generate HUVEC spheroids, 750 cells were suspended in culture medium containing 0.25% (w/v) carboxymethylcellulose. During the first 24 h of culture, all the suspended cells contributed to the formation of a single spheroid, which was then embedded in a collagen gel. Under such conditions spheroids formed capillary-like sprouts, which were measured in the following 24 h of culture using a digitized imaging system connected to an inverted microscope.
Cell Viability Assay
Cell viability was assessed by colorimetric measurement of lactate dehydrogenase (LDH) release according to vendor's protocol (Promega, Madison, USA).
RT-PCR
Total RNA was isolated from the cells by acid guanidinum thiocyanate-phenol-chloroform extraction. Synthesis of cDNA was performed on 2 μg of total RNA with oligo-dT primers for 1 h at 37°C using MMLV reverse transcriptase, according to vendor's instruction. Then PCR with Taq polymerase was performed on cDNA for 22 to 35 cycles using the following protocol: 95°C 40 s, 58°C 40 s and 72°C 50 s. The primers recognizing VEGF (5′-CAC CGC CTT GGC TTG TCA CAT and 5′-CTG CTC TCT TGG GTG CAC TG), eNOS (5′-GTG ATG GCG AAG CGA GTG AA and 5′-CCG AGC CCG AAC ACA CAG AA), HO-1 (5′-CTT TCA GAA GGG TCA GGT GTC C-3′ and 5′-GTG GAG ACG CTT TAC GTA GTG C-3′), and EF2 (5′-GCG GTC AGC ACA ATG GCA TA and 5′-GAC ATC ACC AAG GGT GTG CAG) as a reporter gene have been used. For amplification of Ang-1, Ang-2, and VEGF-D, the commercially available primers from R&D Systems (Abingdon, UK) have been used. The positive control (plasmid DNA containing relevant sequences flanked by the primers used) provided by the vendor was amplified in each reaction. PCR products were analyzed by electrophoresis in 2% agarose gel. The product length for the VEGF121 was 431 bp, for VEGF165 563 bp, 421 bp for eNOS, 250 bp for HO-1, and 218 bp for EF2.
Detection of Gene Expression by Macroaray Hybridization
For analysis of the differential expression of the multiple angiogenesis-associated genes, we used gene expression arrays (SuperArray, Inc., Frederick, MD, USA). Each GEArray membrane consists of 23 cDNA fragments from genes associated with angiogenesis as well as positive Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and negative (pUC18 DNA) controls printed in two-spot configuration on specialized nylon membranes. cDNA was prepared from 2 μg RNA by reverse transcription with MMLV reverse transcriptase, labeled with biotin—16-dUTP then hybridized to membranes overnight with continuous agitation at 60°C. After washing, the chemiluminescent detection was done and the arrays were exposed to x-ray film. The intensity of each of the gene-specific spots within an individual array was normalized by expressing values as percentages of total gene-specific spot intensity. This allowed comparisons between array experiments. Each experiment was performed twice.
ELISA Assays
ELISA for IL-8 and eNOS were from R&D Systems and were performed according to vendor's protocol. ELISA for urokinase plasminogen activator (uPA) was purchased from Oncogene Science (Cambridge, MA, USA). ELISA for HO-1 from Stressgen Biotechnologies, Canada. VEGF, IL-8, and uPA concentrations were determined in conditioned media. HO-1 and eNOS protein were detected in cell lysates prepared according to the vendor's protocol.
Western Blotting
Cells were lysed in ice-cold lysis buffer (1% Triton, 10 mM phenylmethylsulfonyl fluoride [(PMSF)], 10 mM aprotinin, and 10 mM leupeptine). Samples were centrifuged for 20 min at 8000 × g at 4°C and clear supernatants were collected. Protein concentration was determined using bicinchoninic acid protein assay kit (BCA, Sigma). Fifteen microgram of each protein samples were subjected on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis followed by transfer to nitrocellulose membrane Hybond ECL (Amersham Pharmacia Biotech, Buckinghamshire, UK). Membranes were probed with polyclonal antibodies against HO-1 (Stressgen Biotechnologies) followed by biotin-conjugated secondary antibodies (Stressgen Biotechnologies) at the dilution 1:2500 in Tris-buffered saline (TBS) with 3% albumin. Alkaline phosphatase–conjugated streptavidin (Dak, Denmark) at the dilution 1:5000 in TBS was used and the visualization was performed using the 5-bromo-4-chloro-3-indolyl phosphate/p-nitroblue tetrazolic chloride (BCIP/NBT)Blue liquid substrate for membranes.
Statistical Analysis
All experiments were performed in duplicates or triplicates and were repeated at least two times. Data are presented as mean ± standard deviation (SD). Statistical evaluation was done with analysis of variance (ANOVA) followed by a posteriori Tukey test. Differences were accepted as statistically significant at p < .05.
RESULTS
Proangiogenic Effect of Atorvastatin at Nanomolar Concentration Is Not Dependent on Cell Proliferation
Recently we have observed that atorvastatin at nanomolar concentration was proangiogenic (Frick et al. 2003). Also in the present study, atorvastatin at the dose of 10 nM enhanced the capillary sprouting from HUVEC spheroids (Figure 1). This effect disappeared at higher concentrations (Figure 1).
FIG. 1.
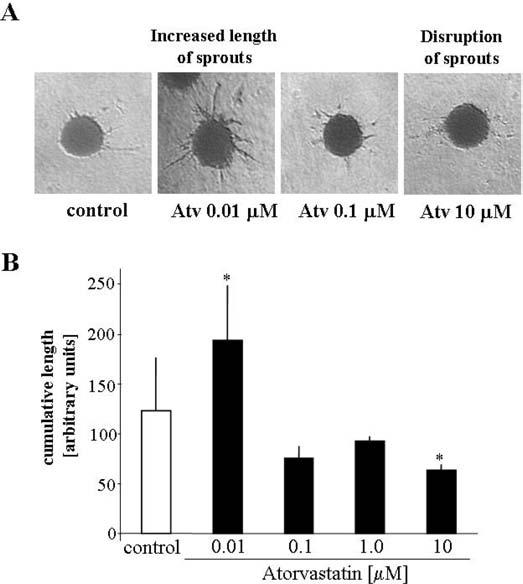
Spheroid assay. Effect of atorvastatin (0.01 to 10 μM) on outgrowth of capillaries. (A) Representative pictures of capillary sprouting, 24 h after embedding HUVEC spheroids in collagen gel. (B) Comparison of cumulative length of capillaries. Each bar represents mean ± SD of three independent experiments. p < .05 versus control.
Interestingly, this proangiogenic effect was not dependent on the endothelial cell proliferation, as no further increase in VEGF-induced BrdU incorporation was observed in the presence of nanomolar concentrations of atorvastatin (Figure 2A). However, 1 to 10 μM concentrations of atorvastatin decreased significantly HUVEC proliferation (Figure 2A). Similar influence has been exerted on bFGF-induced proliferation (Figure 2B). No significant toxicity of atorvastatin on HUVECs was observed at tested concentrations (not shown).
FIG. 2.
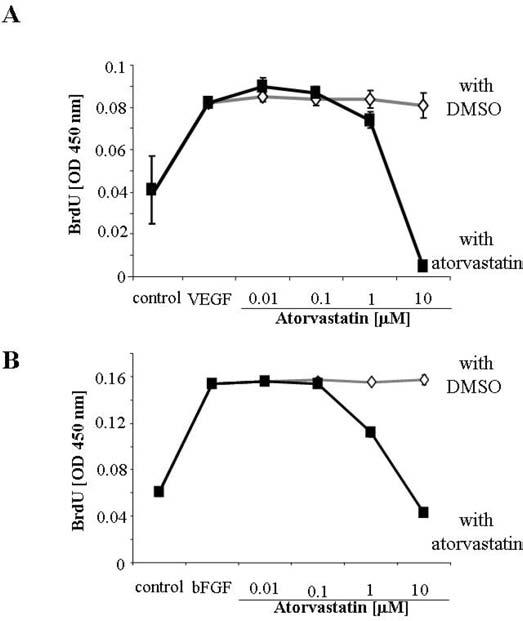
Inhibition of VEGF-induced and bFGF-induced HUVEC proliferation by atorvastatin. Effect of atorvastatin (0.001 to 10 μM) on the VEGF-induced (A) and bFGF-induced (B) proliferation of HUVECs. Control cells were treated with solvent (DMSO) added at the same final amount as was used for atorvastatin. The highest solvent concentration was 0.1% (when 10 μM atorvastatin was used). Proliferation was measured colorimetrically after 48 h incubation, by BrdU incorporation assay. Each bar represents mean ± SD of three independent experiments.
Atorvastatin Decreases IL-8 Production in HUVECs
IL-8 is another potent proangiogenic mediator, which can enhance endothelial cell proliferation and survival (Li et al. 2003). Interestingly, atorvastatin at proangiogenic concentrations (0.01 to 0.1 μM) did not affect IL-8 synthesis in HUVECs. On the contrary, higher, micromolar concentrations of atorvastatin decreased synthesis of this cytokine (Figure 3).
FIG. 3.
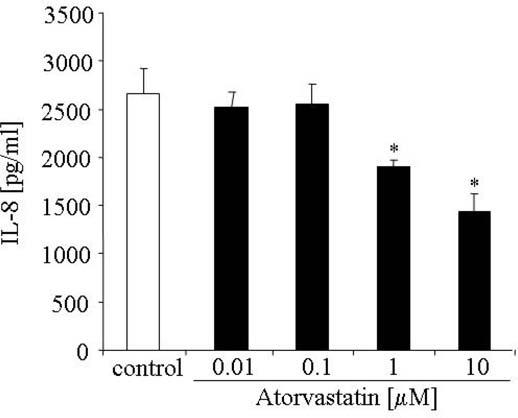
Effect of atorvastatin on IL-8 production in HUVECs. Concentrations of IL-8 protein in media harvested after a 24-h incubation period were measured by ELISA. Representative results of five independent experiments, * p < .05 versus control.
Atorvastatin Decreases uPA Production in HUVECs
Angiogenic effect of VEGF requires the activity of uPA (Heymans et al. 1999). Therefore, impairment of uPA synthesis may also result in attenuation of angiogenesis. Interestingly, in the present study, synthesis of uPA was diminished already at nanomolar concentrations of atorvastatin (Figure 4A). Treatment with mevalonic acid reversed the inhibitory effect of atorvastatin (Figure 4B).
FIG. 4.
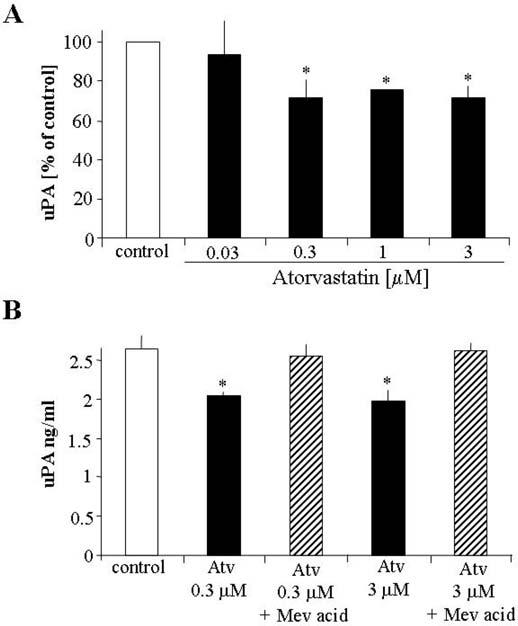
Effect of atorvastatin on uPA production in HUVECs. Highnanomolar and low-micromolar concentrations of atorvastatin attenuate uPA release by HUVECs determined by ELISA after 24 h stimulation (A). The inhibitory effect was reversed by mevalonic acid (B). Representative result of three independent experiments, * p < .05 versus control.
Atorvastatin Decreases the Expression of Thrombospondin (TSP)-1 and Plasminogen Activator Inhibitor (PAI)-1 and Enhances the Expression of VEGF-D and Ang-2
Macroarray hybridization has been used to find more angiogenic genes whose expression is influenced by atorvastatin. It has been shown, that atorvastatin at micromolar concentrations down-regulates TSP-1 and PAI-1, whereas increases the expression of VEGF-D and Ang-2 (Figure 5A and B). The enhancement in expression of Ang-2 has been confirmed by RT-PCR (Figure 5C). We were not able, however, to validate the upregulation of VEGF-D. The level of expression of VEGF-D in HUVECs is, probably, very low as changes in the expression of VEGF-D mRNA could be detected only after 38 rounds of PCR amplification (Figure 5C). Moreover, ELISA for VEGF-D did not demonstrate any VEGF-D protein in conditioned media harvested from HUVEC cultures (not shown).
FIG. 5.
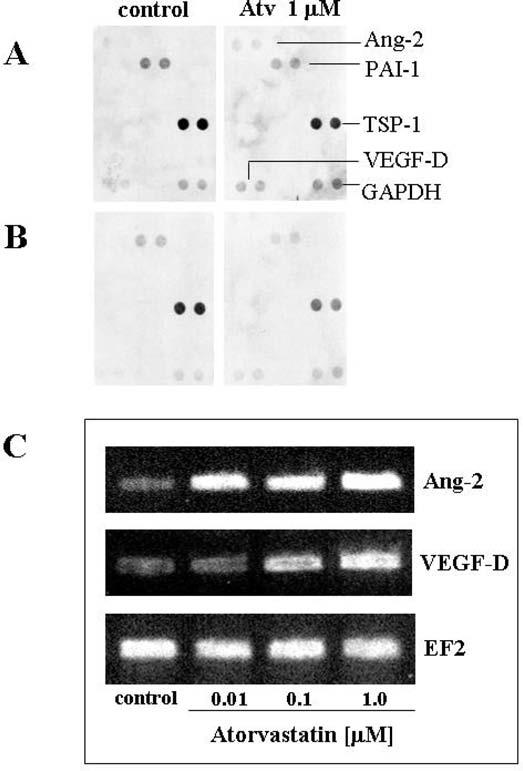
Effect of atorvastatin on PAI-1, TSP-1, Ang-2, and VEGF-D in HUVECs. (A and B) Macroarray analysis demonstrated enhanced expression of Ang-2 and VEGF-D, and decreased PAI-1 and TSP-1, at 6 h after stimulation with atorvastatin. (results of two independent hybridizations are shown). (C) RTPCR confirmed the induction of VEGF-D; however, this effect was visible only after 38 cycles of amplification. Ang-2 expression was detected after 30 cycles.
Effect of Atorvastatin on eNOS and HO-1 Expression
eNOS and HO-1 are involved in angiogenesis and protection of endothelial cells from apoptosis and oxidative injury (for review and references see Dulak and Jozkowicz 2003; Dulak et al. 2004). Statins are known to up-regulate eNOS (Laufs et al. 1997, 1998). Here we determined the effect of atorvastatin on eNOS and HO-1 generation.
Under basal conditions, mRNAs, for eNOS and HO-1 were detected by RT-PCR, although the eNOS expression was higher than HO-1 and could be easily detected after fewer (i.e., 22) cycles than HO-1 amplicons, which were visible after 30 cycles of PCR (Figure 6A). Atorvastatin moderately increased eNOS and HO-1 mRNA expression (Figure 6A). ELISA confirmed the enhanced synthesis of eNOS protein in HUVEC (Figure 6B). However, the protein expression of HO-1 did not change significantly, as shown by ELISA (Figure 6C) or western blotting (Figure 6C, insert) for HO-1.
FIG. 6.
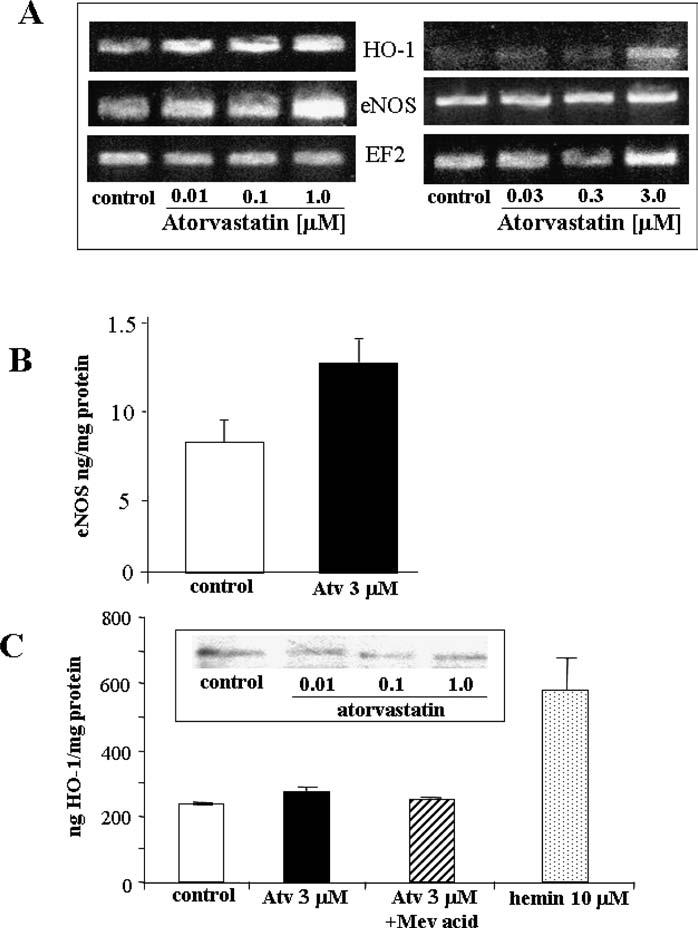
Atorvastatin modulates the expression of eNOS and HO-1. (A) RT-PCR results showing the increased expression of eNOS (22 cycles performed) and HO-1 mRNA (30 cycles performed) in HUVECs 24 h after treatment with increasing concentrations of atorvastatin (results of two representative experiments are shown). (B) ELISA for eNOS protein confirmed the induction of eNOS synthesis after atorvastatin treatment. In contrast, HO-1 protein concentration did not change, as shown (B) ELISA (C) and Western blotting (C, insert).
DISCUSSION
In the present study we have confirmed that atorvastatin, a representative of HMG-CoA reductase inhibitors, exerts dual effect on angiogenic activity of endothelial cells, being stimulatory at low, nanomolar concentrations and inhibitory at higher, i.e., micromolar doses. Additionally, we have shown that atorvastatin affects the synthesis of several proangiogenic mediators, such as uPA, IL-8, eNOS, VEGF-D, and Ang-2. Interestingly, it inhibits the production of uPA and IL-8, while enhances the expression of Ang-2 and moderately stimulates eNOS and VEGF-D.
Current hypotheses on the mechanisms of development of cancer, atherosclerosis, or Alzheimer disease point to the crucial role of the formation of new blood vessels in those conditions. Although the consequence of atherosclerosis is very often insufficient blood supply and the resulting heart and peripheral muscle ischemia, the development of the atherosclerotic plaque is undoubtedly dependent on the formation of blood vessels. Analyses have convincingly demonstrated the increased number of blood vessels in plaques or myocardium of hypercholesterolemic animals (Rodriguez-Porcel et al. 2000). Importantly, treatment with statins preserved the structure of the vascular wall, the effect ascribed to decreased expression of VEGF, hypoxia-inducible factor-1 (HIF-1), and decreased number of vasa vasorum capillaries (Wilson et al. 2002). It is hypothesized that the process of development of the plaque shows important analogies to the tumor growth and it can be conceivably considered that antiangiogenic therapy may be beneficial in both situations (Moulton et al. 1999, 2003).
Interestingly, recent in vitro (Frick et al. 2003) and in vivo (Weis et al. 2002) experiments demonstrated the proangiogenic activity of statins, which occurs particularly at low, picomolar or nanomolar concentrations (Weis et al. 2002; Urbich et al. 2002). Such effects have been also investigated in the certain clinical settings, where ex vivo experiments and in vivo studies demonstrated the enhancement of endothelial progenitor cell survival and differentiation in patients undergoing statin therapy (Dimmeler et al. 2001). Thus, it has been suggested that in vivo statins may be mostly proangiogenic, as such effects have been exerted at low concentrations of the drugs, which are equivalent to the levels of statins detected in plasma of patients (Weis et al. 2002; Lennernas 2003). Nevertheless, recent report demonstrated that cerivastatin induced proangiogenic effect also at high concentration (6 mg/kg body weight/day), stimulating blood vessel formation in ischemic tissues (Sata et al. 2004). Thus, the effect of statins on angiogenesis and molecular mechanisms are far from understanding and require further studies.
The proangiogenic effects of statins have been ascribed to the phosphorylation of Akt kinase, the effect observed already at 10 nM concentration of atorvastatin (Urbich et al. 2002). That activation of Akt is suggested to be responsible for enhanced endothelial cell proliferation and survival. It may also prevent the senescence and apoptosis of endothelial progenitors (Assmus et al. 2003). Higher, micromolar doses of statins may exert weak effect or no influence on Akt kinase phosphorylation (Urbich et al. 2002), although Kureishi et al. noted that 1 μM concentration of simvastatin enhanced Akt phosphorylation in HUVECs, the effect claimed to be responsible for inhibition of apoptosis (Kureishi et al. 2000).
Proangiogenic effects of statins are abolished in eNOS knockout mice (Sata et al. 2001). Interestingly, the antiangiogenic effect of atorvastatin occurs at the concentrations which enhance the expression of eNOS (this study and Assmus et al. 2003), the crucial gene involved in the angiogenic activity of endothelial cells. Moreover, NO generation is enhanced in endothelial cells stimulated with VEGF and endothelial cell migration relies on NO synthesis (Jozkowicz et al. 2004).
NO protects endothelial cells from apoptosis induced by several stimuli, such as tumor necrosis factor alpha (TNFα)or serum withdrawal (for a review see Dimmeler and Zeiher 1999). Similar effect is exerted by VEGF (for a review see: Zachary and Gliki 2001). However, induction of eNOS expression by micromolar concentration of statin appears to be not sufficient to enhance the angiogenesis.
HO-1 is a stress-inducible enzyme that degrades heme to carbon monoxide, iron, and biliverdin (for review see Sikorski et al. 2004). Besides removal of pro-oxidant heme, the products of HO-1 activity have been recently demonstrated to be involved in numerous protective processes. In vascular system HO-1 expression is proangiogenic (Deramaudt et al. 1998; Dulak et al. 2002, 2004). CO, biliverdin, and its derivative, bilirubin, as well as ferritin induced by iron are regarded as protective, and their influence may result, among others, in prevention of endothelial cells from apoptosis (for reviews see Dulak and Jozkowicz 2003; Dulak et al. 2004). Hence, it was reasonable to determine the potential effect of statins on HO-1 expression. However, in our hands atorvastatin at wide range of concentrations tested did not affect significantly HO-1 synthesis. Interestingly, HO-1 mRNA expression has been enhanced by micromolar concentrations of atorvastatin, whereas the protein production did not change. To that extent our results are in partial agreement with a recent study that demonstrated the induction of HO-1 mRNA and protein expression by simvastatin in vascular smooth muscle cells but not endothelial cells nor macrophages (Lee et al. 2004). Thus, the effect of statins may be cell-type dependent, but further studies are necessary for better understanding of those interactions.
Moreover, antiangiogenic effects of atorvastatin at micromolar concentrations can derive from other pathways that are affected by this compound. In our hands atorvastatin decreased uPA synthesis and IL-8 production. Indeed, uPA activity is required for the VEGF-induced angiogenesis and in animals devoid of uPA gene angiogenesis was significantly impaired in comparison to the wild-type counterparts (Heymans et al. 1999).
IL-8 is a CXC chemokine generated in a significant amounts by endothelial cells (Jozkowicz et al. 2001). It is a proinflammatory and proangiogenic factor, whose effects are mostly exerted by the chemotactic activity toward polymorphonuclear cells. IL-8 production is increased in atherosclerosis and statins have been reported to decrease IL-8 synthesis both in vitro (Rezaie-Majd et al. 2003) and in vivo (Waehre et al. 2003). Recent data indicate also that IL-8 exerts direct proangiogenic activity on endothelial cells, by stimulation of their proliferation and inhibition of the starvation-induced apoptosis (Li et al. 2003). Therefore, inhibitory effect of atorvastatin on IL-8 production may contribute to the antiangiogenic activities of statins at micromolar concentrations.
Besides influencing angiogenesis, the decrease in the production of IL-8 can exert anti-inflammatory activity. This effect may add to the attenuation of inflammation caused by decrease in PAI-1 synthesis (Wiesbauer et al. 2002). Similar effect on PAI-1 has been observed in our study.
Interestingly, we have observed for the first time that TSP-1 expression in endothelial cells is decreased in cells treated with atorvastatin, and this effect has been already observed at 100 nM concentration. TSP-1 is known as inhibitor of angiogenesis and the progression of tumors is dependent on down-regulation of TSP-1 and TSP-2 (Lawler and Detmar 2004; de Fraipont et al. 2001). Therefore, inhibition of TSP-1 expression could result in enhancement of angiogenesis. Hypoxia was also shown to inhibit TSP-1 generation (Laderoute et al. 2000). Inhibition of TSP-1 expression is thus regarded as proangiogenic whereas TSP-1 overexpression as antiangiogenic (Weinstat-Saslow et al. 1994). Therefore, it may be surprising that down-regulation of TSP-1 expression by atorvastatin is paralleled by the inhibition of angiogenic activity of endothelial cells. However, this again points to the complexity of statin-dependent regulation of angiogenic gene expression and angiogenic activity of endothelial cells.
It should be noticed, however, that a stimulatory effect of hypoxia on TSP-1 expression in cultured endothelial cells has been also reported (Phelan et al. 1998). Similarly, the role of TSP-1 in tumor growth is still enigmatic. It has been for example shown that the expression of TSP-1 and TSP-2 was significantly increased in invasive breast carcinoma as compared to benign or normal tissue (Bertin et al. 1997; Wang-Rodriguez et al. 2003). Therefore, inhibition of TSP-1 synthesis may be also considered as beneficial, at least in certain types of tumors. This has been demonstrated in advanced epithelial ovarian carcinoma or breast cancer, although that effect of TSP-1 down-regulation may not be necessarily related to the angiogenesis (Clezardin et al. 1993). Additionally, low-microgram concentration of TSP-1 have been reported to be proangiogenic, whereas higher, i.e., more than 25 μg/mL per ml are claimed to be antiangiogenic (Motegi et al. 2002). TSP-1 has been also shown to increase uPA and PAI-1 and promote metastasis of breast cancer cells (Arnoletti et al. 1995). Thus, further studies should elucidate what is the role of TSP-1 in the growth and angiogenesis of specific types of tumors.
Finally, macroarray analysis, which demonstrated the changes in PAI-1 and TSP-1 expression, revealed also an effect of atorvastatin on Ang-2 and VEGF-D in HUVECs. However, RT-PCR demonstrated only modest enhancement of Ang-2 and VEGF-D expression. Additionally, we were unable to detect any VEGF-D protein production by HUVECs using the commercially available ELISA. Those discrepancies may reflect the technical drawbacks of the kit and arrays used and require further validation.
Extrapolation of the results of experiments in vitro to the clinical settings has to be done cautiously. In the in vitro studies the high concentrations of statins have been very often used, although the micromolar doses can induce endothelial cell apoptosis (Muck et al. 2004; Kaneta et al. 2003; Newton et al. 2003; Frick et al. 2003; Urbich et al. 2002). Importantly, plasma concentrations of statins in patients treated with HMG-CoA reductase inhibitors are in the picomolar and nanomolar ranges (Desager and Horsmans 1996), although some other studies reported higher concentrations (Wong et al. 2002). Moreover, it may be hypothesized that the local concentrations of statins in certain tissues are in the range of those used in the in vitro experiments. It is also interesting that the antiangiogenic activities of statins are exerted at those doses that induce apoptosis of tumor cells, which might constitute the background for novel approaches in anticancer therapy. Further studies are, however, required to elucidate that point of the actions of statins.
Footnotes
Supported by Pfizer, Poland, PBZ-KBN-107/P04/2004 and by the Polish-Austrian Collaborative Grant (17/2002). Dr Jozkowicz is an International Senior Research Fellow of Wellcome Trust.
REFERENCES
- Alber HF, Dulak J, Frick M, Dichtl W, Schwarzacher SP, Pachinger O, Weidinger F. Atorvastatin decreases vascular endothelial growth factor in patients with coronary artery disease. Journal of American College of Cardiology. 2002;39:1951–1955. doi: 10.1016/s0735-1097(02)01884-3. [DOI] [PubMed] [Google Scholar]
- Alber H, Frick M, Dulak J, Dörler J, Zwick R, Dichtl W, Pachinger O, Weidinger F. Vascular endothelial growth factor (VEGF) plasma levels in coronary artery disease. Heart. 2005;91:365–366. doi: 10.1136/hrt.2003.021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoletti JP, Albo D, Granick MS, Solomon MP, Castiglioni A, Rothman VL, Tuszynski GP. Thrombospondin and transforming growth factor-beta 1 increase expression of urokinase-type plasminogen activator and plasminogen activator inhibitor-1 in human MDA-MB-231 breast cancer cells. Cancer. 1995;76:998–1005. doi: 10.1002/1097-0142(19950915)76:6<998::aid-cncr2820760613>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Assmus B, Urbich C, Aicher A, Hofmann WK, Haendeler J, Rossig L, Spyridopoulos I, Zeiher AM, Dimmeler S. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circulation Research. 2003;92:1049–1055. doi: 10.1161/01.RES.0000070067.64040.7C. [DOI] [PubMed] [Google Scholar]
- Bertin N, Clezardin P, Kubiak R, Frappart L. Thrombospondin-1 and -2 messenger RNA expression in normal, benign, and neoplastic human breast tissues: Correlation with prognostic factors, tumor angiogenesis, and fibroblastic desmoplasia. Cancer Research. 1997;57:396–399. [PubMed] [Google Scholar]
- Brouet A, Sonveaux P, Dessy C, Moniotte S, Balligand JL, Feron O. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circulation Research. 2001;89:866–873. doi: 10.1161/hh2201.100319. [DOI] [PubMed] [Google Scholar]
- Clezardin P, Frappart L, Clerget M, Pechoux C, Delmas PD. Expression of thrombospondin (TSP1) and its receptors (CD36 and CD51) in normal, hyperplastic, and neoplastic human breast. Cancer Research. 1993;53:1421–1430. [PubMed] [Google Scholar]
- de Fraipont F, Nicholson AC, Feige JJ, Van Meir EG. Thrombospondins and tumor angiogenesis. Trends in Molecular Medicine. 2001;7:401–407. doi: 10.1016/s1471-4914(01)02102-5. [DOI] [PubMed] [Google Scholar]
- Deramaudt BM, Braunstein S, Remy P, Abraham NG. Gene transfer of human heme oxygenase into coronary endothelial cells potentially promotes angiogenesis. Journal of Cellular Biochemistry. 1998;68:121–127. doi: 10.1002/(sici)1097-4644(19980101)68:1<121::aid-jcb12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Desager JP, Horsmans Y. Clinical pharmacokinetics of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. Clinical Pharmacokinetics. 1996;31:348–371. doi: 10.2165/00003088-199631050-00003. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, Rutten H, Fichtlscherer S, Martin H, Zeiher AM. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. Journal of Clinical Investigation. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Zeiher AM. Nitric oxide-an endothelial cell survival factor. Cell Death and Differentiation. 1999;6:964–968. doi: 10.1038/sj.cdd.4400581. [DOI] [PubMed] [Google Scholar]
- Dulak J, Jozkowicz A. Regulation of vascular endothelial growth factor synthesis by nitric oxide: Facts and controversies. Antioxidants and Redox Signaling. 2003;5:123–132. doi: 10.1089/152308603321223612. [DOI] [PubMed] [Google Scholar]
- Dulak J, Jozkowicz A, Foresti R, Kasza A, Frick M, Huk I, Green CJ, Pachinger O, Weidinger F, Motterlini R. Heme oxygenase activity modulates vascular endothelial growth factor synthesis in vascular smooth muscle cells. Antioxidants and Redox Signaling. 2002;4:229–240. doi: 10.1089/152308602753666280. [DOI] [PubMed] [Google Scholar]
- Dulak J, Jozkowicz A, Frick M, Alber HF, Dichtl W, Schwarzacher SP, Pachinger O, Weidinger F. Vascular endothelial growth factor: Angiogenesis, atherogenesis or both? Journal of American College of Cardiology. 2001;38:2137–2138. doi: 10.1016/s0735-1097(01)01624-2. [DOI] [PubMed] [Google Scholar]
- Dulak J, Loboda A, Zagorska A, Jozkowicz A. Complex role of heme oxygenase-1 in angiogenesis. Antioxidants and Redox Signaling. 2004;6:858–866. doi: 10.1089/ars.2004.6.858. [DOI] [PubMed] [Google Scholar]
- Frick M, Dulak J, Cisowski J, Jozkowicz A, Zwick R, Alber H, Dichtl W, Schwarzacher SP, Pachinger O, Weidinger F. Statins differentially regulate vascular endothelial growth factor synthesis in endothelial and vascular smooth muscle cells. Atherosclerosis. 2003;170:229–236. doi: 10.1016/s0021-9150(03)00299-5. [DOI] [PubMed] [Google Scholar]
- Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nature Medicine. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- Jozkowicz A, Dulak J, Prager M, Nanobashvili J, Nigisch A, Winter B, Weigel G, Huk I. Prostaglandin-J2 induces synthesis of interleukin-8 by endothelial cells in a PPAR-gamma-independent manner. Prostaglandins and Other Lipid Mediators. 2001;66:165–177. doi: 10.1016/s0090-6980(01)00155-1. [DOI] [PubMed] [Google Scholar]
- Jozkowicz A, Dulak J, Nigisch A, Funovics P, Weigel G, Polterauer P, Huk I, Malinski T. Involvement of nitric oxide in angiogenic activities of vascular endothelial growth factor isoforms. Growth Factors. 2004;22:19–28. doi: 10.1080/0897719041682863. [DOI] [PubMed] [Google Scholar]
- Józkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, Dulak J. Heme oxygenase and angiogenic activity of endothelial cells: Stimulation by carbon monoxide, inhibition by tin protoporphyrin-X. Antioxidants and Redox Signaling. 2003;5:155–162. doi: 10.1089/152308603764816514. [DOI] [PubMed] [Google Scholar]
- Kaneta S, Satoh K, Kano S, Kanda M, Ichihara K. All hydrophobic HMG-CoA reductase inhibitors induce apoptotic death in rat pulmonary vein endothelial cells. Atherosclerosis. 2003;170:237–243. doi: 10.1016/s0021-9150(03)00301-0. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Kohli M, Mehta P, Mehta JL. Potential anticancer effect of statins: Fact or fiction. Endothelium. 2003;10:49–58. doi: 10.1080/10623320303358. [DOI] [PubMed] [Google Scholar]
- Korff T, Augustin HG. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. Journal of Cell Biology. 1998;143:1341–1352. doi: 10.1083/jcb.143.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature Medicine. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderoute KR, Alarcon RM, Brody MD, Calaoagan JM, Chen EY, Knapp AM, Yun Z, Denko NC, Giaccia AJ. Opposing effects of hypoxia on expression of the angiogenic inhibitor thrombospondin 1 and the angiogenic inducer vascular endothelial growth factor. Clinical Cancer Research. 2000;6:2941–2950. [PubMed] [Google Scholar]
- Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. Journal of Biological Chemistry. 1997;272:31725–31729. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- Lawler J, Detmar M. Tumor progression: The effects of thrombospondin-1 and -2. International Journal of Biochemistry and Cell Biology. 2004;36:1038–1045. doi: 10.1016/j.biocel.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Lee TS, Chang CC, Zhu Y, Shyy JY. Simvastatin induces heme oxygenase-1: A novel mechanism of vessel protection. Circulation. 2004;110:1296–1302. doi: 10.1161/01.CIR.0000140694.67251.9C. [DOI] [PubMed] [Google Scholar]
- Lennernas H. Clinical pharmacokinetics of atorvastatin. Clinical Pharmacokinetics. 2003;42:1141–1160. doi: 10.2165/00003088-200342130-00005. [DOI] [PubMed] [Google Scholar]
- Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. Journal of Immunology. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- Liao JK, Laufs U. Pleiotropic effects of statins. Annual Reviews in Pharmacology and Toxicology. 2004 Aug 17;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, Walsh K, Isner JM, Asahara T. HMG-CoA reductase inhibitor mobilizes bone marrow–derived endothelial progenitor cells. Journal of Clinical Investigation. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi K, Harada K, Pazouki S, Baillie R, Schor AM. Evidence of a bi-phasic effect of thrombospondin-1 on angiogenesis. Histochemical Journal. 2002;34:411–421. doi: 10.1023/a:1023687505139. [DOI] [PubMed] [Google Scholar]
- Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726–1732. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, Lo KM, Gillies S, Javaherian K, Folkman J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proceedings of the National Acadaemy of Sciences of the United States of America. 2003;100:4736–4741. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muck AO, Seeger H, Wallwiener D. Class-specific pro-apoptotic effect of statins on human vascular endothelial cells. Zeischrift für Kardiologie. 2004;93:398–402. doi: 10.1007/s00392-004-0081-5. [DOI] [PubMed] [Google Scholar]
- Newton CJ, Xie YX, Burgoyne CH, Adams I, Atkin SL, Abidia A, McCollum PT. Fluvastatin induces apoptosis of vascular endothelial cells: Blockade by glucocorticoids. Cardiovascular Surgery. 2003;11:52–60. doi: 10.1016/s0967-2109(02)00117-5. [DOI] [PubMed] [Google Scholar]
- Phelan MW, Forman LW, Perrine SP, Faller DV. Hypoxia increases thrombospondin-1 transcript and protein in cultured endothelial cells. Journal of Laboratory and Clinical Medicine. 1998;132:519–529. doi: 10.1016/s0022-2143(98)90131-7. [DOI] [PubMed] [Google Scholar]
- Rezaie-Majd A, Prager GW, Bucek RA, Schernthaner GH, Maca T, Kress HG, Valent P, Binder BR, Minar E, Baghestanian M. Simvastatin reduces the expression of adhesion molecules in circulating monocytes from hypercholesterolemic patients. Arteriosclerosis, Thrombosis and Vascular Biology. 2003;23:397–403. doi: 10.1161/01.ATV.0000059384.34874.F0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Porcel M, Lerman A, Ritman EL, Wilson SH, Best PJ, Lerman LO. Altered myocardial microvascular 3D architecture in experimental hypercholesterolemia. Circulation. 2000;102:2028–2030. doi: 10.1161/01.cir.102.17.2028. [DOI] [PubMed] [Google Scholar]
- Sata M, Nishimatsu H, Osuga J, Tanaka K, Ishizaka N, Ishibashi S, Hirata Y, Nagai R. Statins augment collateral growth in response to ischemia but they do not promote cancer and atherosclerosis. Hypertension. 2004;43:1214–1220. doi: 10.1161/01.HYP.0000126186.29571.41. [DOI] [PubMed] [Google Scholar]
- Sata M, Nishimatsu H, Suzuki E, Sugiura S, Yoshizumi M, Ouchi Y, Hirata Y, Nagai R. Endothelial nitric oxide synthase is essential for the HMG-CoA reductase inhibitor cerivastatin to promote collateral growth in response to ischemia. FASEB Journal. 2001;15:2530–2532. doi: 10.1096/fj.01-0415fje. [DOI] [PubMed] [Google Scholar]
- Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: Molecular regulation of the heme oxygenase-1 gene in renal injury. American Journal of Physiology Renal Physiology. 2004;286:F425–F441. doi: 10.1152/ajprenal.00297.2003. [DOI] [PubMed] [Google Scholar]
- Undas A, Celinska-Lowenhoff M, Kaczor M, Musial J. New non-lipid effects of statins and their clinical relevance in cardiovascular disease. Thrombosis Haemostasis. 2004;91:1065–1077. doi: 10.1160/TH04-02-0064. [DOI] [PubMed] [Google Scholar]
- Urbich C, Dernbach E, Zeiher AM, Dimmeler S. Double-edged role of statins in angiogenesis signaling. Circulation Research. 2002;90:737–744. doi: 10.1161/01.res.0000014081.30867.f8. [DOI] [PubMed] [Google Scholar]
- Waehre T, Damas JK, Gullestad L, Holm AM, Pedersen TR, Arnesen KE, Torsvik H, Froland SS, Semb AG, Aukrust P. Hydroxymethylglutaryl coenzyme a reductase inhibitors down-regulate chemokines and chemokine receptors in patients with coronary artery disease. Journal of American College of Cardiology. 2003;41:1460–1467. doi: 10.1016/s0735-1097(03)00263-8. [DOI] [PubMed] [Google Scholar]
- Wang-Rodriguez J, Urquidi V, Rivard A, Goodison S. Elevated osteopontin and thrombospondin expression identifies malignant human breast carcinoma but is not indicative of metastatic status. Breast Cancer Research. 2003;5:R136–R143. doi: 10.1186/bcr620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmann S, Nickening G. Interrelationship of free radicals and endothelial dysfunction—modulation by statins. Endothelium. 2003;10:23–33. doi: 10.1080/10623320303357. [DOI] [PubMed] [Google Scholar]
- Weinstat-Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, Steeg PS. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Research. 1994;54:6504–6511. [PubMed] [Google Scholar]
- Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–745. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- Wiesbauer F, Kaun C, Zorn G, Maurer G, Huber K, Wojta J. HMG CoA reductase inhibitors affect the fibrinolytic system of human vascular cells in vitro: A comparative study using different statins. British Journal of Pharmacology. 2002;135:284–292. doi: 10.1038/sj.bjp.0704454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SH, Herrmann J, Lerman LO, Holmes DR, Jr, Napoli C, Ritman EL, Lerman A. Simvastatin preserves the structure of coronary adventitial vasa vasorum in experimental hypercholesterolemia independent of lipid lowering. Circulation. 2002;105:415–418. doi: 10.1161/hc0402.104119. [DOI] [PubMed] [Google Scholar]
- Wong WW, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16:508–519. doi: 10.1038/sj.leu.2402476. [DOI] [PubMed] [Google Scholar]
- Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovascular Research. 2001;49:568–581. doi: 10.1016/s0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]