

Abstract
Human prolactin (hPRL) promotes the proliferation and differentiation of mammary epithelial cells during mammary gland development and has been linked to breast tumor development. The receptor for hPRL (hPRL-R) is elevated in a majority of human breast tumors, suggesting the overexpression of hPRL-R makes cancer cells highly sensitive to the mitogenic and anti-apoptotic activity of hPRL. These findings provide the rationale for the development of hPRL-R targeted breast cancer therapeutics. Previously, an hPRL antagonist, G129R, was developed that competitively binds to hPRL-R resulting in growth inhibition and the induction of apoptosis in certain types of breast cancer cells. To further increase the potency of G129R, we fused G129R to a truncated form of Pseudomonas exotoxin A (PE40) that lacks the cell recognition domain of the toxin but retains the domains necessary for PE40 to translocate into the cytosol and inhibit protein synthesis. We postulated that the fusion of G129R with PE40-KDEL would 1) deliver the recombinant toxin to breast cancer cells where hPRL-R is overexpressed; 2) block hPRL signaling via its G129R moiety; and 3) inhibit protein synthesis via its PE40-KDEL moiety. We demonstrate that the fusion toxin can competitively displace hPRL from T-47D human breast cancer cells and inhibit STAT5 phosphorylation induced by hPRL. In addition, we show that G129R-PE40-KDEL is selectively cytotoxic to breast cancer cell lines expressing the PRL-R and that cell death is associated with the inhibition of protein synthesis rather than caspase mediated apoptosis.
Keywords: antagonist, breast cancer, exotoxin A, fusion toxin, G129R, prolactin, prolactin receptor
Introduction
Human prolactin (hPRL) promotes the proliferation and differentiation of mammary epithelial cells during mammary gland development and is necessary for the stimulation of lactogenesis. The receptor for hPRL (hPRL-R) is up regulated in mammary tissue during development and studies have shown that a majority of human breast tumors have higher hPRL-R levels than surrounding normal mammary tissue [1, 2, 3]. It has also been shown that hPRL mRNA and protein are present in both normal and cancerous human mammary tissue [4, 5] and that it originates primarily from the mammary epithelium [3]. Based on these observations, it has been hypothesized: 1) that elevated hPRL-R levels in malignant breast tissue make these cells highly sensitive to the mitogenic and anti-apoptotic activity of hPRL; 2) that hPRL released from the epithelial cells acts locally via the hPRL-R in an autocrine/paracrine loop; and 3) that alterations in hPRL signaling may play a role in malignant transformation and/or tumor development.
Since both hPRL and hPRL-R are expressed widely in breast cancers, it was proposed that targeted inhibition of signaling via the hPRL-R would have potential as a therapy for breast cancers [6]. In vitro, anti-hPRL antibodies and anti-sense RNA directed against the hPRL gene have been used to successfully inhibit T-47Dco cell proliferation [7, 8] as have antibodies against hPRL-R in multiple breast cancer cell lines [9]. An antibody raised against the PRL-R has also been shown to reduce the incidence of carcinogen induced breast cancers in vivo in mice [10]. These findings validate hPRL/hPRL-R signaling as a target for the development of breast cancer therapeutics.
An hPRL antagonist, G129R, was developed by substituting a single glycine residue at position 129 of hPRL with a bulky arginine residue [11, 12]. This amino acid substitution disrupts the structural integrity of the lower affinity binding site of hPRL so that it retains the ability to bind to the first receptor with high affinity but can no longer interact with a second receptor to initiate signal transduction [12, 13]. We demonstrated that G129R binds with hPRL-R with similar affinity as hPRL and is able to competitively antagonize the effects of hPRL on hormone responsive human breast cancer cell lines [12]. We further demonstrated that the antagonist competitively blocks hPRL-induced Janus-associated kinase/signal transducers and activators of transcription (JAK/STAT) signaling and by doing so results in the up-regulation of a tumor suppressor (TGF-β) and the down-regulation of a proto-oncogene (bcl-2); leading to the arrest of cell cycle progression and in some circumstances the induction of apoptosis [14, 15, 16, 17]. Since the blocking of hPRL signaling by G129R appears to be more cytostatic than cytotoxic to breast cancer cells in vitro and in vivo using mouse xenografts [18], we proposed to enhance the cytotoxicity of G129R by fusing it to a toxin that potently inhibits protein synthesis.
Pseudomonas exotoxin A (PE) is a bacterial toxin with three structurally and functionally different domains [19, 20]. Domain I is responsible for cell recognition, domain II for translocation, and domain III for inhibition of protein synthesis. PE binds to the membrane-associated heavy chain of α2-macroglobulin receptor and undergoes receptor-mediated endocytosis [21, 22, 23]. Within the endosome PE is acidified and processed [24, 25, 26, 27]; the low pH induces conformational changes in PE necessary for translocation [28] and for processing by furan [29, 30], which cleaves PE between arginine 279 and glycine 280 [31]. The mechanism by which the 37 kDa C-terminal fragment of PE translocates to the cytosol remains unclear; however, both the amino-terminal [32, 33] and carboxy-terminal regions of PE37 are necessary for translocation. The carboxy-terminal five amino acids, REDLK, resemble an endoplasmic reticulum (ER) retention signal [34] and are necessary for cytotoxicity [32]. After removal of the C-terminal lysine, this carboxyl-terminal fragment is thought to exploit the KDEL receptor retrieval system by binding to KDEL receptors present in the Golgi, which retrograde transport the toxin to the lumen of the ER [35, 36]. In the ER, PE may be reduced, unfolded and reverse translocated across the membrane into the cytosol where it can inactivate elongation factor 2 (EF-2), thereby inhibiting protein synthesis and ultimately causing cell death.
Here we describe the development of a fusion toxin intended to specifically target and kill hPRL-R-positive breast cancers using G129R and a recombinant form of Pseudomonas exotoxin A that lacks the cell recognition domain. Our data demonstrates that G129R-PE40-KDEL drastically increases the potency of G129R even though it has a lower affinity for the hPRL-R when fused with PE40-KDEL. We show that G129R-PE40-KDEL has the attributes of a dual function protein; it retains the ability to antagonize the hPRL-R by partially inhibiting PRL-induced STAT5 phosphorylation and it drastically reduces the viability of PRL-R positive human breast cancer cells by inhibiting protein synthesis.
Material and methods
Cell lines and reagents
The human breast cancer cell lines T-47D, MCF-7, BT-474, BT-483, MDA-MB-134, MDA-MB-453, MDA-MB-231 and the murine breast cancer cell line 4T1 were purchased from the American Type Culture Collection (Manassas, VA) and were maintained in medium containing 10 μg/ml gentamicin at 37°C with humidity and 5% CO2 unless otherwise mentioned. All media and supplements were purchased from Invitrogen (Carlsbad, CA) unless otherwise mentioned. T-47D and MCF-7 cells were maintained in RPMI Medium 1640 supplemented with 10% fetal bovine serum (FBS). BT-474 cells were maintained in RPMI Medium 1640 supplemented with 10% FBS, 1 mM sodium pyruvate, and 10 μg/ml insulin (Sigma, St. Louis, MO). BT-483 cells were maintained in RPMI Medium 1640 supplemented with 20% FBS, 2.5 mg/ml glucose (Sigma), 10 mM HEPES, 1 mM sodium pyruvate, and 10 μg/ml insulin. MDA-MB-134 cells were maintained in Leibovitz's L-15 Medium supplemented with 20% FBS without CO2. MDA-MB-453 and MDA-MB-231 cells were maintained in Leibovitz's L-15 Medium supplemented with 10% FBS without CO2. 4T1 cells were maintained in RPMI Medium 1640 supplemented with 10% FBS, 2.5 mg/ml glucose (Sigma), 10 mM HEPES, and 36.4 ml of 7.5% (w/v) sodium bicarbonate per liter of medium.
Pseudomonas exotoxin A was purchased from List Biological Laboratories (Campbell, CA). PE40, which retains the enzymatic activity of the wild type toxin but has low cytotoxicity due to the lack of the cell recognition domain Ia [37], was prepared and purified as described previously from the periplasmic fraction [38]. PRL and G129R were prepared in house as previously described [18]. Cbz-Val-Ala-Asp-(Ome)-fluoromethyl ketone (zVAD-fmk) was purchased from Enzyme Systems Products (Livermore, CA) and dissolved in DMSO (Fisher Scientific, Pittsburgh, PA) to a final concentration of 50 mM, aliquoted, and stored at -20°C. Working solutions were made in culture medium immediately before use.
Construction of PRL-PE40-KDEL and G129R-PE40-KDEL expression vectors
Previously, PRL and G129R were cloned by inserting their cDNAs between the Nde I and Xho I sites of the expression vector pET22b obtained from Novagen (Madison, WI). Plasmid JH8, which encodes a recombinant form of Pseudomonas exotoxin A (PE40), was obtained from the American Type Culture Collection (Manassas, VA). All oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA), all restriction and modifying enzymes were purchased from New England BioLabs (Beverly, MA), and all PCR reactions were performed using the Advantage-HF™ Kit (BD Biosciences, Palo Alto, CA). Internal BamH I and Xho I sites were removed from the DNA encoding PE40 using the QuickChange® XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The carboxy-terminal codons of PRL, G129R, and PE40 were modified and restriction sites were added by PCR using oligonucleotides; the stop codons of PRL and G129R were replaced with codons encoding GGGGS and the codons of PE40 encoding REDLK were replaced with codons encoding KDEL since it has been shown to significantly increase the cytotoxicity of PE and its derivatives [37]. The PCR products were ligated with pCR2.1 T/A cloning vector and transformed into E. coli TOP10 cells obtained from Invitrogen. Positive clones were identified by plating the cells on Luria-Bertani (LB) agar plates containing 100 μg/ml ampicillin and 80 μg/ml 5-bromo-4-chloro-3-indolyl-bD-galactoside. The plasmids were isolated using the QIAprep® Spin Miniprep Kit (Qiagen, Valencia, CA) and the Nde I-PRL-G4S-BamH I, Nde I-G129R-G4S-BamH I, and BamH I-PE40-KDEL-Xho I DNA fragments were isolated by restriction digestion, separated by electrophoresis, purified from the agarose gel using the QIAquick® Gel Extraction Kit (Qiagen), and ligated with Nde I and Xho I cut pET22b to create pET22b-PRL-PE40-KDEL and pET22b-G129R-PE40-KDEL. Various oligonucleotides linkers were inserted at the BamH I site located between G129R and PE40-KDEL to increase the distance between the two moieties.
Production and purification of fusion toxins
Chemically competent E. coli BL21 (DE3) Star cells (Invitrogen) were transformed with pET22b-PRL-PE40-KDEL or pET22b–G129R-PE40-KDEL and propagated overnight at 37°C with agitation in LB broth containing 100 μg/ml ampicillin. The next morning 160 ml of each of the starter cultures was used to inoculate 4 liters of LB broth and the cultures were grown to an O.D.600 of 0.9 before induction of protein production with 1 mM isopropyl β-thiogalactoside (Alexis Biochemicals, San Diego, CA). After five hours of induction the cells were harvested by centrifugation at 5000 x g for 5 min and resuspended in Solution 1 (0.2M NaPO4; 10 mM EDTA, pH 8.0; 0.5% Triton X-100; pH adjusted to 8.0) to which 0.1 mg/ml lysozyme was added. The cells were incubated at room temperature for one hour to weaken the cell membranes before the cells were sheared on ice with five 1-min ultrasonic pulses (30% duty at setting 7) using a 550 Sonic Dismembrator (Fisher Scientific). The insoluble fraction containing the inclusion bodies was recovered by centrifugation at 12,000 x g for 30 min at 4°C and resuspended in Solution 2 (0.2M NaPO4; 10 mM EDTA, pH 8; 0.5% Triton X-100; 1 M urea; pH adjusted to 7.0). The inclusion bodies were collected by centrifugation at 12,000 x g for 30 min at 4°C and were solubilized and denatured in Solution 3 (0.2M NaPO4; 8 M urea; 1% v/v β-mercaptoethanol; pH adjusted to 8.0). The proteins were refolded by dialysis against baths of 20 mM Tris·HCl, pH 8.0 containing decreasing amounts of urea (4M urea, 2 M urea, 0 M urea, 0 M urea). The refolded fusion toxins were filtered through 0.45 μm filters and purified by anion-exchange chromatography using a 5 ml HiTrap Q Sepharose XL column (Amersham Biosciences, Piscataway, NJ) on an ÄKTA fast protein liquid chromatography system (Amersham Biosciences). The purified fusion toxins were separated by SDS-PAGE along with BSA standards (Pierce, Rockford, IL) and stained with SYPRO® Orange (Molecular Probes, Eugene, OR) to determine the concentration and purity of the fusion toxins. The identity of the fusion toxins were confirmed by immunoblotting.
Immunoblot analysis
PE, G129R, and G129R-PE40-KDEL were separated on a 12% polyacrylamide gel by SDS-PAGE. The proteins were transferred by Western blot to a Hybond™ enhanced chemiluminescence (ECL) nitrocellulose membrane (Amersham Biosciences) at 16 W for 1 h. The nitrocellulose membrane was incubated in a blocking solution containing TBS with 0.05% Tween 20 (TBS-T) and 5% milk for 1 h at room temperature. The membrane was incubated overnight at 4°C with agitation in blocking solution containing a 1:2000 dilution of anti-exotoxin A (List Biological Laboratories). The blots were washed three times with TBS-T, and incubated for 2 h at room temperature with agitation in blocking solution containing a 1:2000 dilution of swine anti-goat horseradish peroxidase (HRP) conjugate (Roche, Indianapolis, IN). Blots were washed three times with TBS-T and incubated for 1 min with ECL™ Western Blotting Detection Reagents (Amersham Biosciences). Immunoblots were exposed to Kodak Biomax MR film (Fisher Scientific, Pittsburgh, PA) and the film was developed with a Konica SRX-101A processor (Konica Minolta Medical Imagining, Wayne, NJ). The membrane was stripped and re-probed with a 1:4000 dilution of rabbit anti-hPRL antiserum (National Hormone and Pituitary Program, NIH, Bethesda, MD) and 1:2000 dilution of goat anti-rabbit conjugated horseradish peroxidase (Bio-Rad, Hercules, CA).
Radio-receptor binding assay
One million cells per well of T-47D human breast cancer cells were seeded in six-well tissue culture plates and grown to confluency. Cells were starved in serum-free RPMI Medium1640 for 1 h, and incubated for 2 h at room temperature in serum-free medium containing 125I-labeled hPRL (specific activity, 40 μCI/μg; NEN Perkin-Elmer, Boston, MA) with or without various concentrations of G129R or G129R-PE40-KDEL. Cells were washed three times with serum-free RPMI Medium 1640 and were lysed in 0.5 ml of 0.1 N NaOH/1% SDS. Bound radioactivity was determined by scintillation counting and the percentage of specific displacement was calculated and compared among the samples.
STAT5 phosphorylation assay
T-47D cells were grown to 90% confluency in six-well plates in RPMI Medium 1640 supplemented with 10% (v/v) charcoal-stripped fetal bovine serum (CSS; HyClone, Logan, UT) and 10 μg/ml gentamycin. The cells were depleted for 1 h in RPMI Medium 1640 supplemented with 0.5% CSS and then treated for 20 min with various concentrations and combinations of PRL, G129R, PRL-PE40-KDEL, and G129R-PE40-KDEL. The cells were washed with ice cold phosphate buffered saline (PBS) and lysed in 200 μl of Lysis Buffer (20 mM Tris·HCl, pH 7.4; 1% Igepal CA630; 6 mM deoxycholic acid; 150 mM NaCl; 1 mM EDTA, pH 8.0) containing protease inhibitors (1 μg/ml aprotinin; 1 μg/ml leupeptin; 170 μg/ml PMSF; 180 μg/ml sodium orthovanadate). The lysates were agitated for 10 min on an orbital shaker, transferred to 1.5 ml tubes, passed six times through 21-gauge needles, and incubated on ice for 20 minutes. The lysates were clarified by centrifugation for 10 min at 12,000 x g at 4°C and 30 μl of the supernatants were used for immunoblot analysis as described previously using 4-15% polyacrylamide gels. Blots were performed with 1.5 μg/ml mouse anti-phosphate STAT5A/B antibody (Upstate, Lake Placid, NY) and a 1:2000 dilution of goat anti-mouse-HRP conjugate (Bio-Rad). The membranes were re-probed with 3 μg/ml rabbit anti-STAT5 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and a 1:2000 dilution of goat ant-rabbit-HRP conjugate (Bio-Rad).
Cell viability assay
The various cell lines to be assayed were seeded in 96-well culture plates at a density of 5,000 cells per well in 200 μl of appropriate culture medium and grown as recommended. After incubation for 24 hours, the medium was discarded and replaced with 200 μl of medium containing various concentrations of PE, G129R, or G129R-PE40-KDEL and the cells were incubated for an additional 48 hours. The culture medium was discarded and replaced with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium/phenazine methosulfate (MTS/PMS) solution diluted in PBS (CellTiter 96 AQueous non-radioactive cell proliferation kit; Promega, Madison, WI). The viability of the cells was determined by colorimetric measurement of the reduction of MTS by the living cells using a Benchmark microplate reader (Bio-Rad) measuring absorbance at 495 nM. All assays were performed in quadruplicate and the cell viability was calculated as a percentage of control treatments.
Protein synthesis inhibition assay
Approximately 1 x 105 cells were seeded in each well of a 24-well plate in a final volume of 0.5 ml of appropriate growth medium and the cells were incubated overnight. The medium was replaced with 0.5 ml of appropriate treatments and incubated for 44 h after which each well was supplemented with 1 μCi of 3H-leucine (Amersham Biosciences) in 100 μl of medium and incubated an additional 4 h. The monolayers were washed with 1 ml PBS and the protein was precipitated with 0.5 ml of 26% trichloroacetic acid (TCA) overnight at -20 °C. The monolayers were solubilized with 0.5 ml of 0.2 nM NaOH and loaded onto GF/C glass fiber filters (Whatman, Clifton, NJ) pre-rinsed with 1 ml of 10% TCA. The filters were washed twice with 1 ml of 10% TCA and once with 3 ml of 95% ethanol and dried by vacuum. The dried filters were placed in scintillation vials and aqueous scintillation cocktail was added before counting with a liquid scintillation counter. The amount of 3H-leucine incorporated into precipitable protein was determined and expressed as a percent of control.
Poly-(ADP-ribose)-polymerase (PARP) cleavage assay
Approximately 3 x 105 MCF-7 were resuspended in 5 ml of RPMI Medium 1640 supplemented with 10% CSS and seeded into T-25 flasks and allowed to grow 24 hours. For experiments that included zVAD-fmk, 50 μM of the peptide was added 1 h prior to treatment with 1 μg/ml of PE, PRL, G129R, or G129R-PE40-KDEL. After 24 h treatment, the medium and cells were collected and transferred to 5 ml Falcon tubes and centrifuged at 5,000 x g. The pellets were washed once with ice-cold PBS and resuspended in 100 μl of Lysis Buffer with inhibitors and lysed for 30 min at 4°C. The lysates were clarified by centrifugation for 10 min at 12,000 x g at 4°C and the protein content of the lysates were assayed using the Coomassie Plus Protein Assay Reagent (Pierce). Thirty micrograms of the lysates were resolved by SDS-PAGE and were immunoblotted as previously described using a blocking solution containing 10% milk. A 1:2000 dilution of rabbit anti-PARP (Roche) and 1:2000 dilution of goat anti-rabbit-HRP conjugate (Bio-Rad) were used.
Results
Construction, expression, and purification of PRL-PE40-KDEL and G129R-PE40-KDEL
To target the PE40 toxin to the hPRL-R, we fused cDNA encoding hPRL or G129R to the N-terminus of DNA encoding PE40 and replaced the DNA sequence encoding the native ER retention sequence (REDLK) of the toxin with a sequence encoding KDEL to increase its cytotoxicity (Figure 1). Linkers of increasing length were inserted between hPRL or G129R and PE40-KDEL since the fusion toxins had little cytotoxicity with the Gly-Gly-Gly-Gly-Ser (G4S) linker. For all studies we used the fusion toxins containing the Gly-Gly-Gly-Gly-Ser-Thr-Leu-Arg-Asp-Leu-Phe-Asp-Arg-Ala-Val-Val-Leu-Ser-His-Tyr-Ile-His-Asn-Leu-Ser-Ser-Glu-Met-Phe-Ser-Glu-Gly-Ser linker since they showed greater potency. PRL-PE40-KDEL and G129R-PE40-KDEL were produced in E. coli and found to accumulate in the insoluble fraction as inclusion bodies. The inclusion bodies were isolated and denatured and the fusion toxins were refolded and purified by anion-exchange chromatography to yield preparations of greater than 90% purity. Purified G129R-PE40-KDEL was analyzed by SDS-PAGE and was found to have a size corresponding to the predicted molecular mass of 66 kDa (Figure 2a). Immunoblots with antibodies that detect the G129R and PE40-KDEL moieties confirmed the identity of purified protein to be G129R-PE40-KDEL (Figure 2b-c).
Figure 1.
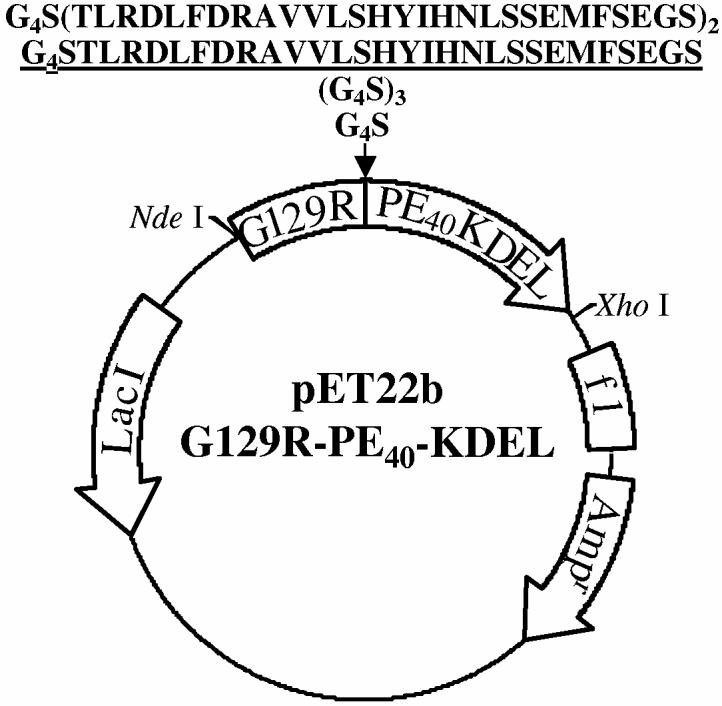
: Schematic representation of plasmid used to produce G129R-PE40-KDEL in E. coli. The cDNA encoding G129R was fused to the DNA encoding an enzymatically active form of PE (PE40-KDEL) with a BamH I site separating the two. In an effort to increase the activity of the fusion toxin, linkers of increasing length were inserted at the BamH I site between G129R and PE40-KDEL to decrease steric hindrance caused by their close proximity. The diagram above shows the amino acids encoded by the linkers inserted between G129R and PE40-KDEL. The fusion toxin used for all studies is shown underlined. The selection was based on its superior potency at inhibiting protein synthesis.
Figure 2.

: SDS-PAGE (A) and immunoblot analysis (B and C) of PE (Lane 1), G129R (Lane 2), and G129R-PE40-KDEL (Lane 3). The purity of G129R-PE40-KDEL was confirmed after SDS-PAGE by staining with SYPRO® Orange (A). The identity of the protein was confirmed by immunoblotting with antibodies that detect the G129R (B) and PE40-KDEL moieties (C) as indicated by arrows.
G129R-PE40-KDEL binds to T-47D human breast cancer cells expressing PRL-R
The ability of G129R and G129R-PE40-KDEL to specifically bind to hPRL-Rs expressed on the surface of T-47D human breast cancer cells was demonstrated using a radio-receptor binding assay. Figure 3 shows that increasing doses of G129R or G129R-PE40-KDEL were able to competitively displace 125I-hPRL from hPRL-Rs. This confirmed that G129R-PE40-KDEL retains the ability to bind to hPRL-Rs; however, the lower percent specific displacement suggests that it has a much lower affinity than G129R for hPRL-Rs.
Figure 3.
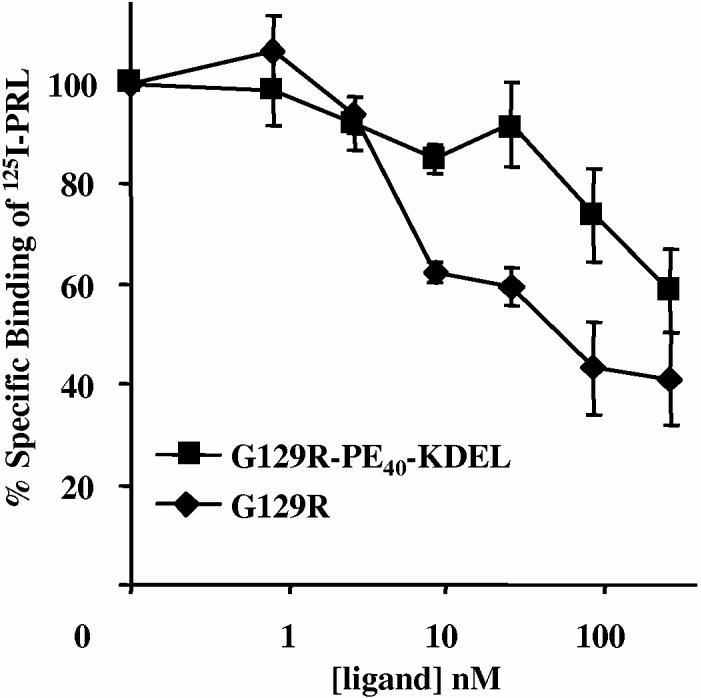
: Prolactin receptor binding assay using T-47D human breast cancer cells. The ability of G129R and G129R-PE40-KDEL to bind to the hPRL-Rs was determined by measuring their ability to displace the binding of 125I-labeled hPRL. Each data point represents the mean + SD of at least three independent experiments.
PRL-PE40-KDEL activates STAT5 and G129R-PE40-KDEL inhibits its phosphorylation in T-47D human breast cancer cells
Human PRL induces hPRL-R dimerization and leads to the phosphorylation of STAT5 via receptor associated JAK2. The PRL-PE40-KDEL fusion toxin retained the ability of the hPRL moiety to bind to hPRL-Rs on T-47D breast cancer cells and activate STAT5 phosphorylation (Figure 4), albeit with only a fraction (1/10 to 1/20th) of the activity of wild type hPRL. The ability of this fusion toxin to activate STAT5 suggested that G129R-PE40-KDEL should be able to bind to and antagonize the hPRL-R. Like G129R, G129R-PE40-KDEL did not activate STAT5 (Figure 5a) and was able to compete with hPRL for occupancy of the hPRL-R as shown by its ability to partially block STAT5 phosphorylation (Figure 5b). G129R-PE40-KDEL was unable to completely block the action of hPRL at a ratio of 1:100 and had antagonistic activity less than 1/10th that of G129R, which corresponds with the reduced activity observed for PRL-PE40-KDEL. These findings suggested the fusion toxin was still capable of binding to hPRL-R but with a much reduced affinity.
Figure 4.

: Stimulation of STAT5 phosphorylation by PRL and PRL-PE40-KDEL using T-47D human breast cancer cells. The ability of PRL and PRL-PE40-KDEL to bind to hPRL-Rs and activate STAT5 was determined by treatment of the cells with 4 nM of PRL or increasing concentrations of PRL-PE40-KDEL. STAT5 was detected by immunoblotting using an anti-phosphate STAT5A/B antibody as indicated by an arrow.
Figure 5.

: Competitive inhibition of STAT5 phosphorylation by G129R and G129R-PE40-KDEL in T-47D human breast cancer cells. The inability of G129R and G129R-PE40-KDEL to activate STAT5 (A) and their ability to competitively block hPRL-induced STAT5 activation (B) was determined by immunoblotting with antibodies against either STAT5A/B or phospho-STAT5A/B as indicated.
The PE40-KDEL moiety of G129R-PE40-KDEL retains the ability to cleave PARP in MCF-7 cells
Having confirmed that the G129R moiety retains its ability to bind and block receptor activation, we wanted to determine if the PE40-KDEL moiety retained its activity. Previously, it has been shown that treatment of MCF-7 cells with PE results in the activation of caspases that cleave PARP into 89 kDa and 24 kDa fragments [39]. Figure 6a shows that PE cleaved this DNA repair enzyme and that this cleavage was inhibited by treating the cells with the broad spectrum caspase inhibitor, zVAD-fmk. Neither PRL nor G129R caused cleavage of PARP, confirming that the ability of G129R-PE40-KDEL to cleave PARP is due the PE40-KDEL moiety of the fusion toxin (Figure 6a).
Figure 6.

: PARP cleavage and the effect of zVAD-fmk on the viability of MCF-7 breast cancer cells. Cells were treated for 24 h with 1 μg/ml of PE, PRL, G129R, or G129R-PE40-KDEL in the absence or presence of a broad spectrum caspase inhibitor, zVAD-fmk (A, B). The cell lysate was separated by SDS-PAGE, and immunblotted with anti-PARP antibody. The caspase mediated cleavage of PARP is indicated by the presence of a 24 kDa PARP fragment as shown by the arrow (A). The role of activated caspases in cell killing caused by PE or G129R-PE40-KDEL was examined by measuring the reduction of MTS by living cells in the absence or presence of zVAD-fmk (B). Data presented is representative of the mean + SD from assays performed in quadruplicate on two separate occasions.
A cell viability assay was used to examine the cytotoxic effects of PE and G129R-PE40-KDEL on MCF-7 cells in the presence or absence of zVAD-fmk. Both wild type PE and G129R-PE40-KDEL were found have cytotoxic effects, confirming that at least a portion of the fusion toxin was taken up and processed correctly by MCF-7 cells (Figure 6b). Interestingly, pretreatment with zVAD-fmk had little effect on the viability of the cells, suggesting that caspase mediated apoptosis plays a minor role in cell death associated with protein synthesis inhibition (Figure 6b).
G129R-PE40-KDEL inhibits protein synthesis and decreases the viability of multiple human breast cancer cell lines expressing hPRL-R
Human breast cancer cell lines expressing various levels of hPRL-R were treated for 48 h with increasing doses of G129R-PE40KDEL and the inhibition of protein synthesis (Figure 7a) and its effect on viability of the cells (Figure 7b) was determined by measuring the incorporation of 3H-leucine into precipitable protein and by colorimetrically measuring the reduction of MTS by living cells, respectively. The extrapolated IC50 concentrations for the inhibition of cell proliferation and protein synthesis are summarized in Table 1. Cells expressing hPRL-Rs were more sensitive to G129R-PE40-KDEL than cell lines such as MDA-MB231 and MDA-MB453 which lack or have few hPRL-Rs (Figure 7a-b).
Figure 7.
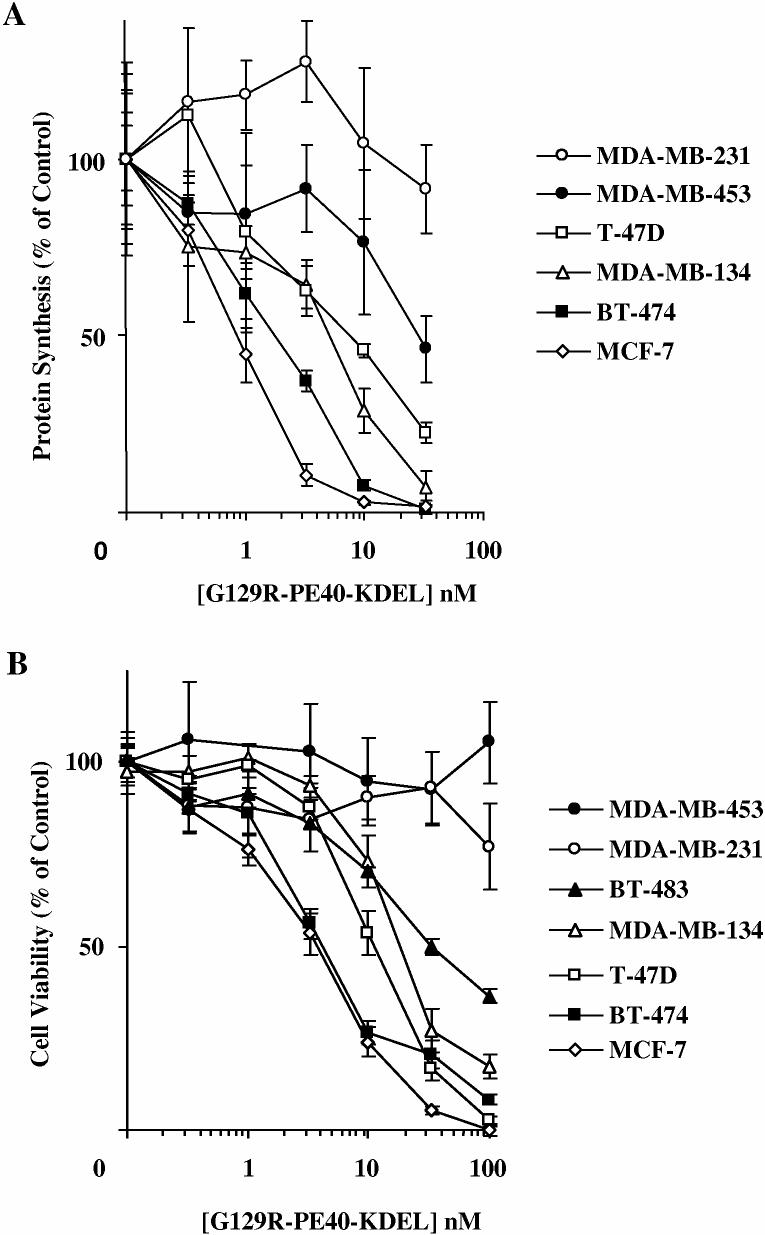
: The effect of G129R-PE40KDEL on protein synthesis and the viability of various human breast cancer cell lines expressing different levels of hPRL-R. Cells were treated for 48 h with increasing doses of G129R-PE40KDEL and the inhibition of protein synthesis was determined by measuring the incorporation of 3H-leucine into precipitable protein (A). The cell viability was determined colorimetrically by measuring the reduction of MTS by living cells. Data points presented are representative of the mean + SD from three assays performed in quadruplicate.
Table 1.
Extrapolated concentrations of G129R-PE40-KDEL necessary for 50% inhibition of protein synthesis (A) and proliferation (B) of human breast cancer cell lines
| A | B | |
|---|---|---|
| Cell Line | IC50 (nM) | IC50 (nM) |
| MCF-7 | 0.9 | 4.75 |
| BT-474 | 2.1 | 4.8 |
| BT-134 | 6 | 22.7 |
| T-47D | 8.2 | 12.5 |
| MDA-MB-453 | 29.6 | >100 |
| MDA-MB-231 | >33 | >100 |
The inhibitory effect of G129R-PE40-KDEL is mediated via the G129R moiety and is due to the inhibition of protein synthesis by the PE40-KDEL moiety
Previously, we have shown that G129R can inhibit the proliferation of breast cancer cells. At the concentrations used, G129R appeared to be cytostatic and did not decrease the viability of T-47D, MCF-7, or BT-474 breast cancer cells which have elevated levels of the hPRL-R (Figure 8). Even with the greater than ten fold reduced receptor binding affinity and reduced antagonistic activity as indicated by STAT5 phosphorylation, G129R-PE40-KDEL was extremely cytotoxic in comparison to G129R. Fusion of G129R with PE40-KDEL drastically decreased the viability of the cells at concentrations well below those observed for G129R, suggesting that the increased cytotoxicity was due to the PE40-KDEL moiety of the fusion toxin.
Figure 8.

: Comparison of the response of breast cancer cells to G129R (◇), G129R-PE40-KDEL (□), or wild type PE (△). The viability of three cell lines was examined after 48 h treatment by measuring the reduction of MTS by living cells. Data points presented are representative of the mean + SD from assays performed in quadruplicate on three separate occasions.
To confirm that the killing observed with G129R-PE40-KDEL was not due to non-specific binding and to confirm that it was due to the inhibition of protein synthesis by the PE40-KDEL moiety, we measured the incorporation of 3H-leucine into precipitable protein and the cell viability in response to G129R-PE40-KDEL in the absence or presence of competitors (Figures 9a-b). Treatment of T-47D cells with increasing doses of G129R-PE40-KDEL decreased protein synthesis (Figure 9a); this inhibition was reduced by co-treatment of the cells with excessive amounts of G129R, suggesting that the cytotoxic effects of the toxin rely heavily on cell binding. Similarly, cells co-treated with anti-PE had no reduction in protein synthesis, suggesting that the PE moiety was responsible for the inhibition of protein synthesis. Treatment of T-47D cells with increasing doses of G129R-PE40-KDEL also decreased the viability of the cells (Figure 9b). Co-treatment of the cells with G129R-PE40-KDEL and G129R or PRL revealed that the viability of the cells was increased by G129R and PRL. This confirmed that the inhibition of protein synthesis was mediated through the hPRL-R and that the decreased viability was due to the PE40-KDEL moiety. We also examined the non-specific effects of G129R-PE40-KDEL in mouse 4T1 cells which are extremely sensitive to wild type PE and the non-specific effects of PE40; G129R-PE40-KDEL was found to have lower cytotoxicity than PE40 by itself, suggesting that the fusion toxin has lower non-specific cytotoxicity than PE40 alone (Figure 10).
Figure 9.

: Competition assays. The ability of G129R-PE40-KDEL to inhibit protein synthesis and decrease the viability of T-47D cells in the absence or presence of competitors, G129R, PRL or anti-PE antibody, was determined by measuring the incorporation of 3H-leucine into precipitable protein (A) and by measuring the reduction of MTS by living cells (B). Data points presented in B are representative of the mean + SD from assays performed in quadruplicate on two separate occasions.
Figure 10.

: Cytotoxicity assay using 4T1 mouse breast cancer cells. The consequence of fusing G129R to PE40-KDEL was examined to determine its effect on non-specific cytotoxicity. Cells were treated with PE, PE40, G129R-PE40-KDEL, or G129R and their viability was examined after 48 h by measuring the reduction of MTS. Data points presented are representative of the mean + SD from assays performed in quadruplicate on two separate occasions.
Discussion
Most growth factors and hormones are taken up by receptor-mediated endocytosis, a broadly named process that can involve a variety of different internalization pathways. For this reason, the ligand chosen for making a fusion toxin is not only important for determining its host-range, but also the ability of the toxin to be properly internalized and processed. We chose to make a fusion toxin that targets the hPRL-R since it is often overexpressed by breast cancer cells. The binding of hPRL and its antagonist, G129R, to the hPRL-R is well established; however, little is known of the intracellular fate of the PRL/PRL-R and/or G129R/PRL-R complexes. Previously, we constructed G129R-interleukin 2 and G129R-endostatin fusion proteins and found that their G129R moieties retained the ability to bind to the hPRL-R and competitively block hPRL-induced signal transduction [40, 41]. In these studies we also showed that the interleukin 2 and endostatin portions of the fusion proteins retained their ability to bind to and activate receptors on T-cells and endothelial cells, respectively. The intracellular fate of these fusion proteins was inconsequential since there functions were believed to be served on the surface of cells. Unlike G129R-interleukin 2 and G129R-endostatin, the fusion of G129R to PE40-KDEL would require hPRL-R mediated internalization and processing of the toxin so that it could enter the cytoplasm and inhibit protein synthesis. Since numerous fusion toxins have been constructed by gene fusion or conjugation of PE40 with cytokines, growth factors, hormones, or antibodies, we did not anticipate that the fusion of PRL or G129R with PE40-KDEL would behave any differently or require nanomolar concentrations rather than picomolar concentrations to kill PRL-R positive breast cancer cell lines (Table 1). There are multiple explanations that may explain the relatively higher IC50 values for protein synthesis inhibition (Table 1) using PRL or G129R as the targeting agent.
Human PRL and G129R may have unanticipated intracellular fates other than degradation in endosomes/lysosomes, the organelle that PE exploits to eventually gain access to the cytoplasm. A recent study showed that human growth hormone (hGH), a hormone closely related to hPRL, binds to its receptor and is rapidly internalized and can enter into the nuclease; whereas the hGH antagonist, G120R, appeared to be internalized but failed to enter the nuclease [42]. This suggests that hGH is not necessarily degraded in endosomes/lysosomes and that it may have an intracellular function. Evidence suggests that PRL can interact with cyclophilin B to undergo nuclear retrotranslocation and can actually modulate transcriptional events [43, 44]. This suggests that the translocation domain and the ER retention sequence of PE40-KDEL may interfere with the normal means by which hPRL is internalized or vice versa. In accordance with this, we observed large differences between the cytotoxicity of G129R-PE40-KDEL and wild type PE in many of the breast cancer cell lines examined (Figure 8), suggesting the effectiveness of the toxin moiety was influenced by the different binding moiety and not due to an inherent defect in the machinery of the cells to properly process the PE40 moiety to inhibit protein synthesis.
The sensitivity of target cells to the cell killing activity of G129R-PE40KDEL may reflect the reduced binding affinity of the fusion toxin. G129R-PE40-KDEL had a much lower receptor binding affinity than G129R (Figure 3) and the previous fusion proteins we constructed (data not shown). Both PRL-PE40-KDEL and G129R-PE40-KDEL had nearly twenty fold reductions in their ability to activate or competitively inhibit STAT5 phosphorylation, respectively (Figure 4 and 5b). This suggests that the PE40-KDEL moiety may sterically hinder the ability of the PRL and G129R moieties to bind to the hPRL-R. Another possibility is that the number of hPRL-Rs expressed on the surface of breast cancer cells may be low in comparison to other receptors that have been targeted with other fusion toxins. We have only measured the PRL-R mRNA expression levels for the breast cancer cell lines using real time PCR [45] and have yet to measure the actual PRL-R levels, which may be lower or exist in intracellular pools that are not expressed on the surface of the cells. Variations in the PRL-R have also been reported that may impact the effectiveness of PRL and G129R fusion toxins. One of the predominant isoforms of hPRL-R in normal and malignant breast tissue has an extracellular truncation that reduces its binding affinity [46]. An isoform with an intracellular truncation has also been identified in rodents which has a slower internalization rate than the long isoform [47]. These variations in binding affinity and internalization rate may affect the effectiveness of the fusion toxins too. The expression levels of the various isoforms of hPRL-R in the breast cancer cell lines we examined have not been explored.
For most of our assays we utilized T-47D breast cancer cells since they are PRL responsive and express relatively high levels of hPRL-R [48]. We showed that G129R-PE40-KDEL was toxic to T-47D cells and that the cytotoxic effects were reduced drastically by treatment with excess amounts of G129R or antibody against PE (Figure 9), suggesting that the cytotoxic effects of the fusion toxin relied heavily on cell binding and was mediated via the toxin moiety. The ability of G129R-PE40-KDEL to specifically displace 125I-labeled hPRL from T-47D cells confirmed that it was binding specifically to hPRL-Rs (Figure 3). Even though G129R-PE40-KDEL had a reduced binding affinity for the hPRL-R, it drastically increased the cytotoxicity of G129R in T-47D cells (Figure 8). Although higher concentrations of G129R-PE40-KDEL were necessary for cytotoxicity than expected (Table 1), these concentrations were orders of magnitudes lower than the concentrations of G129R needed to observe a decrease in the viability of T-47D cells.
Similarly, G129R-PE40-KDEL drastically increased the potency of G129R in the other hPRL-R positive breast cancer cell lines we assayed and had little effect on cells lacking or having few hPRL-Rs (Figure 7). The treatment of the breast cancer cell lines with PE40 resulted in little or no killing of the cells (data not shown) and the fusion of PRL or G129R with PE40-KDEL restored the sensitivity of these breast cancer cells to the cytotoxic effects of the PE moiety. Even though nanomolar concentrations were necessary for achieving the IC50s for protein synthesis (Table 1), these higher concentrations had minimal non-specific effects on cells lacking or having few PRL-Rs, suggesting the fusion toxin was still acting specifically. In mouse 4T1 cells, which are extremely sensitive to the specific and non-specific effects of PE and PE40, respectively, we observed that fusion of G129R to PE40-KDEL actually reduced the non-specific side effects associated with PE40 (Figure 10), suggesting that G129R-PE40-KDEL decreased the non-specific killing associated with the PE40 moiety. These findings indicate that G129R-PE40-KDEL may be useful as a targeted therapy for breast cancers with elevated hPRL-Rs.
Acknowledgements
We would like to thank Karl Franek and Seth Tomblyn for critical reading of this manuscript. This work was supported in part by the Endowment Fund of the Greenville Hospital System and grants from the NIH/NCI (CA105479) and The Susan G. Komen Foundation (BCTR0402985).
References
- 1.Reynolds C, Montone KT, Powell CM, Tomaszewski JE, Clevenger CV. Expression of prolactin and its receptor in human breast carcinoma. Endocrinology. 1997;138:5555–5560. doi: 10.1210/endo.138.12.5605. [DOI] [PubMed] [Google Scholar]
- 2.Touraine P, Martini JF, Zafrani B, Durand JC, Labaille F, Malet C, Nicolas A, Trivin C, Postel-Vinay MC, Kuttenn F, Kelly PA. Increased expression of prolactin receptor gene assessed by quantitative polymerase chain reaction in human breast tumors versus normal breast tissues. J Clin Endocrinol Metab. 1998;83:667–674. doi: 10.1210/jcem.83.2.4564. [DOI] [PubMed] [Google Scholar]
- 3.Clevenger CV, Chang WP, Ngo W, Pasha TL, Montone KT, Tomaszewski JE. Expression of prolactin and prolactin receptor in human breast carcinoma. Evidence for an autocrine/paracrine loop. Am J Pathol. 1995;146:695–705. [PMC free article] [PubMed] [Google Scholar]
- 4.Fields K, Kulig E, Lloyd RV. Detection of prolactin messenger RNA in mammary and other normal and neoplastic tissues by polymerase chain reaction. Lab Invest. 1993;68:354–360. [PubMed] [Google Scholar]
- 5.Purnell DM, Hillman EA, Heatfield BM, Trump BF. Immunoreactive prolactin in epithelial cells of normal and cancerous human breast and prostate detected by the unlabeled antibody peroxidase-antiperoxidase method. Cancer Res. 1982;42:2317–2324. [PubMed] [Google Scholar]
- 6.Reynolds C, Montone KT, Powell CM, Tomaszewski JE, Clevenger CV. Expression of prolactin and its receptor in human breast carcinoma. Endocrinology. 1997;138:5555–5560. doi: 10.1210/endo.138.12.5605. [DOI] [PubMed] [Google Scholar]
- 7.Ginsburg E, Vonderhaar BK. Prolactin synthesis and secretion by human breast cancer cells. Cancer Res. 1995;55:2591–2595. [PubMed] [Google Scholar]
- 8.Ginsburg E, Vonderhaar BK. Prolactin: an autocrine growth factor in the mammary gland. In: Wilde CJ, Peaker M, Taylor E, editors. Biological Signalling and the Mammary Gland. Hannah Research Institute; Ayr, UK: 1997. pp. 47–58. [Google Scholar]
- 9.Fuh G, Wells JA. Prolactin receptor antagonists that inhibit the growth of breast cancer cell lines. J Biol Chem. 1995;270:13133–13137. doi: 10.1074/jbc.270.22.13133. [DOI] [PubMed] [Google Scholar]
- 10.Sissom JF, Eigenbrodt ML, Porter JC. Anti-growth action on mouse mammary and prostate glands of a monoclonal antibody to prolactin receptor. Am J Pathol. 1988;133:589–595. [PMC free article] [PubMed] [Google Scholar]
- 11.Goffin V, Kinet S, Ferrag F, Binart N, Martial JA, Kelly PA. Antagonistic properties of human prolactin analogs that show paradoxical agonistic activity in the Nb2 bioassay. J Biol Chem. 1996;271:16573–16579. doi: 10.1074/jbc.271.28.16573. [DOI] [PubMed] [Google Scholar]
- 12.Chen WY, Ramamoorthy P, Chen N, Sticca R, Wagner TE. A Human prolactin antagonist, hPRL-G129R, inhibits breast cancer cell proliferation through induction of apoptosis. Clinical Cancer Research. 1999;5:3583–3593. [PubMed] [Google Scholar]
- 13.Llovera M, Pichard C, Bernichtein S, Jeay S, Touraine P, Kelly PA, Goffin V. Human prolactin (hPRL) antagonists inhibit hPRL-activated signaling pathways involved in breast cancer cell proliferation. Oncogene. 2000;19:4695–705. doi: 10.1038/sj.onc.1203846. [DOI] [PubMed] [Google Scholar]
- 14.Cataldo L, Chen NY, Yuan Q, Li W, Ramamoorthy P, Wagner TE, Sticca RP, Chen WY. Inhibition of oncogene STAT3 phosphorylation by a prolactin antagonist hPRL-G129R, in T47D human breast cancer cells. International Journal of Oncology. 2000;17:1179–1185. doi: 10.3892/ijo.17.6.1179. [DOI] [PubMed] [Google Scholar]
- 15.Ramamoorthy P, Sticca RP, Wagner TE, Chen WY. In vitro studies of a prolactin antagonist, hPRL-G129R, in human breast cancer cells. Int J Oncology. 2001;18:25–32. doi: 10.3892/ijo.18.1.25. [DOI] [PubMed] [Google Scholar]
- 16.Beck MT, Peirce SK, Chen WY. Regulation of bcl-2 gene expression in human breast cancer cells by prolactin and its antagonist, hPRL-G129R. Oncogene. 2002;21:5047–5055. doi: 10.1038/sj.onc.1205637. [DOI] [PubMed] [Google Scholar]
- 17.Peirce SK, Chen WY. Human prolactin and its antagonist, hPRL-G129R, regulate bax and bcl-2 gene expression in human breast cancer cells and transgenic mice. Oncogene. 2004;23:1248–1255. doi: 10.1038/sj.onc.1207245. [DOI] [PubMed] [Google Scholar]
- 18.Chen NY, Li W, Cataldo L, Peirce S, Chen WY. In vivo anti-tumor activities of a human prolactin antagonist, hPRL-G129R. Int J Oncology. 2002;20:813–818. doi: 10.3892/ijo.20.4.813. [DOI] [PubMed] [Google Scholar]
- 19.Allured VS, Collier RJ, Carroll SF, McKay DB. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci USA. 1986;83:1320–1324. doi: 10.1073/pnas.83.5.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang J, FitzGerald DJ, Adhya S, Pastan I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell. 1987;48:129–136. doi: 10.1016/0092-8674(87)90363-1. [DOI] [PubMed] [Google Scholar]
- 21.Morris RE, Manhart MD, Saelinger CB. Receptor-mediated entry of Pseudomonas toxin: methylamine blocks clustering step. Infect Immun. 1993;40:806–811. doi: 10.1128/iai.40.2.806-811.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kounnas MZ, Morris RE, Thompson MR, FitzGerald DJ, Strickland DK, Saelinger CB. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J Biol Chem. 1992;267:12420–12423. [PubMed] [Google Scholar]
- 23.Zdanovsky AG, Zdanovskaia MV, Strickland D, FitzGerald DJ. Ligand-toxin hybrids directed to the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein exhibit lower toxicity than native Pseudomonas exotoxin. J Biol Chem. 1996;271:6122–6128. doi: 10.1074/jbc.271.11.6122. [DOI] [PubMed] [Google Scholar]
- 24.Jinno Y, Chaudhary VK, Kondo T, Adhya S, FitzGerald DJ, Pastan I. Mutational analysis of domain I of Pseudomonas exotoxin. Mutations in domain I of Pseudomonas exotoxin which reduce cell binding and animal toxicity. J Biol Chem. 1998;263:13203–13207. [PubMed] [Google Scholar]
- 25.Ogata M, Chaudhary VK, Pastan I, FitzGerald DJ. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J Biol Chem. 1990;265:20678–20685. [PubMed] [Google Scholar]
- 26.Siegall CB, Ogata M, Pastan I, FitzGerald DJ. Analysis of sequences in domain II of Pseudomonas exotoxin A which mediate translocation. Biochemistry. 1991;30:7154–7159. doi: 10.1021/bi00243a016. [DOI] [PubMed] [Google Scholar]
- 27.Fryling C, Ogata M, FitzGerald D. Characterization of a cellular protease that cleaves Pseudomonas exotoxin. Infect Immun. 1992;60:497–502. doi: 10.1128/iai.60.2.497-502.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Idziorek T, FitzGerald D, Pastan I. Low pH-induced changes in Pseudomonas exotoxin and its domains: increased binding of Triton X-114. Infect Immun. 1990;58:1415–1420. doi: 10.1128/iai.58.5.1415-1420.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiron MF, Fryling CM, FitzGerald DJ. Cleavage of Pseudomonas exotoxin and diphtheria toxin by a furin-like enzyme prepared from beef liver. J Biol Chem. 1994;269:18167–18176. [PubMed] [Google Scholar]
- 30.Gu M, Gordon VM, FitzGerald DJP, Leppla SH. Furin regulates both the activation of Pseudomonas exotoxin A and the quantity of the toxin receptor expressed on target cells. Infect Immun. 1996;64:524–527. doi: 10.1128/iai.64.2.524-527.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogata M, Fryling CM, Pastan I, FitzGerald DJ. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J Biol Chem. 1992;267:25396–25401. [PubMed] [Google Scholar]
- 32.Theuer CP, Buchner J, FitzGerald D, Pastan I. The N-terminal region of the 37-kDa translocated fragment of Pseudomonas exotoxin A aborts translocation by promoting its own export after microsomal membrane insertion. Proc Natl Acad Sci USA. 1993;90:7774–7778. doi: 10.1073/pnas.90.16.7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zdanovsky AG, Chiron M, Pastan I, FitzGerald DJ. Mechanism of action of Pseudomonas exotoxin. Identification of a rate-limiting step. J Biol Chem. 1993;268:21791–21799. [PubMed] [Google Scholar]
- 34.Chaudhary VK, Jinno Y, FitzGerald D, Pastan I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc Natl Acad Sci USA. 1990;87:308–312. doi: 10.1073/pnas.87.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hessler JL, Kreitman RJ. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry. 1997;36:14577–14582. doi: 10.1021/bi971447w. [DOI] [PubMed] [Google Scholar]
- 36.Jackson ME, Simpson JC, Girod A, Pepperkok R, Roberts LM, Lord JM. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J Cell Sci. 1999;112:467–475. doi: 10.1242/jcs.112.4.467. [DOI] [PubMed] [Google Scholar]
- 37.Seetharam S, Chaundhary VK, FitzGerald D, Pastan I. Increased cytotoxic activity of Pseudomonas exotoxin and two chimeric toxins ending in KDEL. J Biol Chem. 1991;266:17376–17381. [PubMed] [Google Scholar]
- 38.Kondo T, FitzGerald D, Chaudhary VK, Adhya S, Pastan I. Activity of immunotoxins constructed with modified Pseudomonas exotoxin A lacking the cell recognition domain. J Biol Chem. 1988;263:9470–9475. [PubMed] [Google Scholar]
- 39.Keppler-Hafkemeyer A, Brinkmann U, Pastan I. Role of caspases in immunotoxin-induced apoptosis of cancer cells. Biochemistry. 1998;37:16934–16942. doi: 10.1021/bi980995m. [DOI] [PubMed] [Google Scholar]
- 40.Zhang G, Li W, Holle L, Chen N, Chen WY. A novel design of targeted endocrine and cytokine therapy for breast cancer. Clin Cancer Res. 2002;8:1196–1205. [PubMed] [Google Scholar]
- 41.Beck MT, Chen NY, Franek KJ, Chen WY. Prolactin antagonist-endostatin fusion protein as a targeted dual-functional therapeutic agent for breast cancer. Cancer Res. 2003;63:3598–3604. [PubMed] [Google Scholar]
- 42.Mertani HC, Raccurt M, Abbate A, Kindblom J, Tornell J, Billestrup N, Usson Y, Morel G, Lobie PE. Nuclear translocation and retention of growth hormone. Endocrinology. 2003;144:3182–3195. doi: 10.1210/en.2002-221121. [DOI] [PubMed] [Google Scholar]
- 43.Rycyzyn MA, Reilly SC, O'Malley K, Clevenger CV. Role of cyclophilin B in prolactin signal transduction and nuclear retrotranslocation. Mol Endocrinol. 2000;14:1175–1186. doi: 10.1210/mend.14.8.0508. [DOI] [PubMed] [Google Scholar]
- 44.Rycyzyn MA, Clevenger CV. The intranuclear prolactin/cyclophilin B complex as a transcriptional inducer. Proc Natl Acad Sci U S A. 2002;99:6790–6795. doi: 10.1073/pnas.092160699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peirce SK, Chen WY. Quantification of prolactin receptor mRNA in multiple human tissues and cancer cell lines by real time RT-PCR. J Endocrinol. 2001;171:R1–4. doi: 10.1677/joe.0.171r001. [DOI] [PubMed] [Google Scholar]
- 46.Kline JB, Rycyzyn MA, Clevenger CV. Characterization of a novel and functional human prolactin receptor isoform (deltaS1PRLr) containing only one extracellular fibronectin-like domain. Mol Endocrinol. 2002;16:2310–22. doi: 10.1210/me.2001-0033. [DOI] [PubMed] [Google Scholar]
- 47.Lu JC, Scott P, Strous GJ, Schuler LA. Multiple internalization motifs differentially used by prolactin receptor isoforms mediate similar endocytic pathways. Mol Endocrinol. 2002;16:2515–2527. doi: 10.1210/me.2002-0077. [DOI] [PubMed] [Google Scholar]
- 48.Shiu RP, Murphy LC, Tsuyuki D, Myal Y, Lee-Wing M, Iwasiow B. Biological actions of prolactin in human breast cancer. Recent Prog Horm Res. 1987;43:277–303. doi: 10.1016/b978-0-12-571143-2.50013-7. [DOI] [PubMed] [Google Scholar]