

Abstract
Vertebrate photoreceptor outer segment (OS) morphogenesis requires peripherin/rds (P/rds). We have characterized this protein’s C-terminus and present evidence that suggests it is intrinsically disordered. We propose that structural flexibility may underlie the multifunctionality proposed for this domain previously. The extremely short C-termini present in other tetraspanin family members suggest that intrinsic disorder may also play a role for those proteins.
Keywords: peripherin/rds, tetraspanin, intrinsically disordered, natively unstructured, photoreceptor, outer segment
INTRODUCTION
The molecular basis for the dynamic laminar structure of the vertebrate photoreceptor rod, is not well understood. Disruptions in the regular ordering and/or turnover of this light-sensitive membranous organelle can result in serious vision loss and are therefore of interest from both basic science and clinical standpoints. One player known to be centrally involved in OS architecture is the 39 kDa integral membrane protein P/rds [1-3]. Genetic defects affecting this protein cause a variety of progressive retinal degenerations in humans and mice, and illustrate its importance for the formation and long-term stability of photoreceptor OSs [4,5]. Although several hypotheses have been advanced for P/rds molecular function, a definitive role has not yet been assigned. As a member of the tetraspanin protein family, it stands within a group of molecules displaying diverse and critical functions in eukaryotic biology [6].
Although a relatively distant relative to the tetraspanins (∼41% homology with its nearest neighbor, tetraspanin NET-7), P/rds shares several common structural features. It spans the lipid bilayer four times, has cytoplasmically exposed amino and carboxy termini, and contains several distinctive cysteine motifs that are highly conserved within the family [7,8]. Like other tetraspanins, it also possesses a large hydrophilic loop (extracellular loop 2, EC2); this domain resides within the intradiskal space in rod photoreceptors, a location that corresponds to an extracellular exposure for cell-surface tetraspanins. Finally, P/rds is known to function as an oligomer - a developing theme amongst other family members [9].
Crystal structures for the EC2 domain of the tetraspanin CD81 depict a mushroom-like silhouette; mushrooms crystallize in a dimeric form, with the stalk regions appearing to provide the major interaction interfaces [9,10]. Disulfide bonds play a clear role in providing structural rigidity to the mushroom head and likely contribute to oligomer affinity by decreasing entropic factors. The EC2 region in P/rds has similarly been shown to contain numerous intramolecular disulfide bonds proposed to constrain its structure [11]. This domain also contributes specific determinants for oligomeric subunit assembly and is important for global protein folding [12]. In each case, findings describe a highly-ordered and relatively rigid disulfide-bonded domain important for protein folding and oligomerization. The significance of the P/rds EC2 region was predicted by early studies that found that pathogenic mutations were common in this domain [5].
In contrast, the structure and function of tetraspanin C-terminal domains are understood less well; although, a recent report suggests that even relatively small C-termini can play significant roles [13-15]. In contrast to most tetraspanins, P/rds possesses a sizable (63 amino acid) C-terminus and the available molecular genetic evidence suggests that this region is important for protein function [4]. This domain does not appear to contribute to global folding or subunit assembly, but instead appears to have functional import for P/rds. Battaglia et al. have presented a series of reports that demonstrate that in vitro fusogenic activity is associated with the C-terminus, and indicate that it is driven via an amphipathic α-helix [16-18]. More recent reports have identified an OS targeting and localization signal within the C-terminus, and demonstrate that it overlaps, yet is separable from the amphipathic C-terminal helical region (CHR) associated with fusogenic activity [19,20].
We use a variety of biochemical and biophysical approaches here to further characterize the structure of the P/rds C-terminus. We find a highly extended structure in aqueous solution that largely lacks tertiary structure and exhibits a conformational flexibility associated with intrinsically disordered domains [21-23]. Our findings suggest that although the P/rds C-terminus is largely disordered, it may acquire additional structure by interacting with membranes or other proteins, and we hypothesize that the observed conformational flexibility may be important for the regulation of overlapping activities contained within this region.
EXPERIMENTAL
Limited proteolysis. Proteinase K (Roche, Inc), a non-specific endoproteinase was used to probe the conformation of the P/rds C-terminus in bovine rod OS membranes. Membranes were prepared from bovine retinas according to the method of McDowell, et al. [24], and were stored as pellets at -80°C until use. Since initial studies demonstrated that light bleaching had no significant effects on rod OS P/rds Proteinase K sensitivity, rod OS were routinely bleached for 15min under bright white illumination prior to further manipulation. Rod OS membranes were subjected to denaturing treatments (see Results) at 0.4 mg/ml total protein for 30min at RT, then were pelleted at 90k × g for 30 minutes at 4°C, and washed once with a large excess of 50mM Tris-HCl, 150mM NaCl, pH 7.5. The membranes were resuspended at 0.4 mg/ml total protein in 50mM TRIS-HCl, 150mM NaCl, pH 7.5 for digestion. Proteinase K (Roche, Inc.) was added to a final concentration of 2 μg/ml and digests were allowed to proceed at 37°C. Alternatively, trypsin digests utilized membranes resuspended at 0.13 mg/ml total rod OS protein in 100mM Tris-HCl, pH 8.5. Sequencing grade bovine pancreatic trypsin (Roche, Inc.) was added to a final concentration of 0.3 μg/ml and digests were allowed to proceed at 37°C. In each case, aliquots were removed from digests at the indicated time points and were quenched by heating to 98°C in reducing Laemmli sample buffer for 5min. Samples were analyzed by Western blotting using MAbC6 as described previously [12].
Expression and purification of recombinant protein. A glutathione S-transferase (GST) fusion protein encoding the predicted 63 C-terminal amino acids of bovine P/rds joined to a GST header via a PreScission Protease site was derived from the previously described pGSTCTER [18]. The pGSTCTER plasmid was digested with BamHI, and the 260 bp insert was ligated into BamHI-cut and purified pGEX-6P (Amersham Pharmacia Biotech, Inc.) to generate pGEX6CTER. Insert and junction sequences were verified by double-stranded DNA sequencing, using a Big Dye terminator cycle sequencing kit (Applied Biosystems, Inc.). Expression of the predicted 33 kDa P/rds fusion protein was confirmed by induction of BL21 bacterial cultures with 1 mM isopropylthio-β-galactosidase (IPTG) for 2 hrs at 37°C.
Milligram quantities of fusion protein were prepared by innoculating 30 mls of pGEX6CTER transformed and saturated BL21 E. coli culture into 900 mls of Luria broth (LB) containing 100 μg/ml ampicillin. Cultures were grown with agitation at 37°C for 1.5 hrs, and then were induced with 0.1mM IPTG for an additional 3 hrs. Bacteria were collected by centrifugation and stored at -70°C until use. The frozen cell pellets were thawed on ice and resuspended by vortexing in 35 mls of ice-cold phosphate-buffered saline (PBS), pH 7.4, containing 1 μg/ml Leupeptin, 1 μg/ml Pepstatin A, 1 mM phenylmethylsulfonyl fluoride, and 1mM dithiothreitol (DTT). Bacteria were lysed using a French pressure cell at 1000 psi. Triton X-100 was added to final concentration of 1% and the lysate was stirred for 20 min on ice, and then was centrifuged at 15k × g for 20 min at 4°C to remove particulates. GST fusion protein was purified from clarified supernatants using Glutathione Sepharose 4B, essentially as described by the manufacturer (Amersham Biosciences, Inc.). GST6-CTER fusion protein was dialyzed at 4 °C against Tris-buffered saline (TBS), pH 7.0, containing 1 mM ethylenediaminetetraacetic acid (EDTA), 1mM DTT and was stored in aliquots at -70°C until use.
Isolated C-terminal domain was prepared by cleavage of GST6-CTER and removal of GST. PreScission Protease (Amersham Biosciences, Inc.) was added to fusion proteins thawed on ice (1 unit/100μg protein) and the digest was incubated 1.5 hrs in an ice water bath with occasional gentle agitation. The reaction was quenched for 10 min on ice by an addition of 1mg/ml Pefabloc SC. The cleaved protein was applied to a 6ml column of Glutathione Sepharose 4B and the eluent was collected along with a two bed volume wash of TBS. Eluents were combined and concentrated in a stirred ultrafiltration cell (Millipore Corp.) and dialyzed at 4°C against TBS containing 1mM tris(carboxyethyl) phosphine (TCEP). Protein sample, “CTER” was frozen and stored at -70°C until use.
Purified CTER was subjected to quantitative amino acid analysis to confirm the fidelity of expression and cleavage, and an extinction coefficient of 14,104 M-1cm-1 at 280 nm was determined using the average value for duplicate hydrolyzates. CTER concentration was routinely determined thereafter using this extinction coefficients in combination with A280 measurement.
SDS-PAGE and Western blot analysis. 16.5% polyacrylamide tricine and 10% polyacrylamide glycine gels (BioRad, Inc) were utilized in conjunction with the manufacturer-supplied loading buffers and GelCode Blue stain (Pierce, Inc) to visualize the ∼7 kDa recombinant CTER domain. Samples containing bacterially expressed proteins were heated to 98°C for 5 min prior to gel loading. Samples containing rod OS membranes were heated to 37°C for 5 min prior to gel loading. Samples SDS-PAGE run in standard glycine buffers and Western blot analysis were performed essentially as described previously [12].
Velocity and equilibrium sedimentations. Analytical ultracentrifugation was performed at the National Analytical Centrifugation Facility (University of Connecticut), using a Beckman XL-I centrifuge. For velocity measurements, 500 μl aliquots of CTER diluted to 1.0, 0.3 and 0.1 mg/ml in 50 mM Tris-HCL, 150 mM NaCl, 1 mM TCEP, pH 7.5 were prepared and each solution was loaded into the centrifuge rotor and equilibrated under vacuum at 20°C. After reaching temperature equilibrium, the rotor was accelerated to 50k rpm and scans were immediately started and recorded at one-minute intervals for a total of seven hours. These data were analyzed using the program Sedfit (version 8.5) using the model of a continuous distribution, c(s), of sedimentation coefficients [25].
For equilibrium measurements, CTER was diluted in 50 mM Tris-HCL, 150 mM NaCl, 1 mM TCEP, pH 7.5 to prepare 150 μl samples at 1.0, 0.33 mg/ml and 0.11 mg/ml. The samples were loaded into a 6-channel external loading cell and buffer was loaded in the three reference channels. Centrifugation was performed at 28k rpm at 20°C and scans were taken at 15-minute intervals. After running overnight the data were checked to ascertain that equilibrium had been reached and a set of final scans were collected. The rotor was accelerated to 40k rpm and centrifugation was continued overnight. Again, the data were checked to determine that the material had reached equilibrium and then a final set of scans at 40k rpm was recorded. The data were analyzed using the program WinNonlin version 1.06 (Dr. David Yphantis, University of Connecticut).
Size exclusion chromatography (SEC). Purified CTER (0.5 to 1.5 mg) was chromatographed on a 1cm × 46cm column of Sephacryl S-100HR. The column was developed at RT with 50mM TRIS-HCl, 150mM NaCl, 1 mM TCEP, pH 7.5 at 0.2 ml/min. CTER elution was monitored online by absorbance measurement at 280nm and was confirmed by SDS-PAGE analysis and Coomassie blue staining of trichloroacetic acid precipitated fractions. Calibration of the SEC column utilized the buffer system and running conditions described above. A standard curve was derived from a 1st order regression analysis of protein standards included in gel filtration kits obtained from Amersham Biosciences, Inc and Sigma Chemical Co. Apparent MW was determined according to the analysis of Laurent and Killander, using equation (3) to calculate Kav [26].
Intrinsic tryptophan fluorescence. CTER was diluted to ∼0.01 OD280 units (5 μg/ml) in 50 mM TRIS-HCL, 150 mM NaCl, 1 mM TCEP, pH 7.5 at 25°C. After 10min of equilibration, fluorescence determinations were made on either Perkin Elmer LS-5B or Shimadzu RF-5000 recording spectrofluorimeters. Excitation spectra were initially collected to confirm a single absorption peak corresponding to the tryptophan maximum at 295nm. Excitation was thereafter performed at 295nm (slit width of 5nm), and emission scans were routinely collected from 295 to 430nm (slit width of 10 nm). Instrument software was utilized to subtract background spectra collected immediately prior to the acquisition of experimental scans.
Circular dichroism spectroscopy. Far UV CD was performed on a Jasco J-715 instrument, using 0.3 mg/ml CTER in a 0.1 cm cell situated in a Peltier temperature controller. Typically, ten spectra collected at 10 nm/min were averaged. Signals were converted to mean residue ellipticity, after solvent subtraction, using the protein concentration, path length, and mean residue weight. Thermal melts were performed at 220 nm using a scan rate of 20 °C/h. Effects of dextran (40,000) and trifluoroethanol (TFE) were measured using 0.08 mg/ml CTER in a 0.05cm cell. Secondary structure estimates using spectral deconvolution by the method of Bohm et al. [27] were performed using CDNN (version 2.1).
RESULTS
Predicted disorder. We have used secondary sequence modeling of P/rds previously to assist targeted mutagenesis studies and were surprised by the extent to which random coil elements dominate predictions for the protein’s C-terminus. Fig. 1 shows the results of the application of two commonly used secondary structure prediction methods to the P/rds C-terminus. Modeling by both the GOR4 [28] and JNET [29] methods predicts more than half of the C-terminal sequence to be unstructured. In contrast, one strongly predicted helical element, centered at Leu320, is also suggested, and a previous FTIR evaluation of a 15 amino acid synthetic peptide encoding Val312-Leu326 supports the prediction of helical content in this region [17]. An additional short region of helix is also predicted by each method; however, the locations assigned do not concur. Beyond the agreement of the helical region centered at Leu320, the most obvious commonality is the extent of random coil structure. Given this suggestion of limited secondary structure, we applied a Predictor of Natural Disordered Regions (PONDR) VL-XT analysis [30], to further evaluate this region. This approach uses a series of neural network predictors that utilize amino acid sequence data to predict disorder for a given sequence; it defines disordered regions as entire proteins or regions of proteins which lack a fixed tertiary structure, essentially being partially or fully unfolded in situ [22]. The prediction generated by the PONDR VL-XT program for the C-terminus is qualitatively similar to both the GOR4 and JNET results. As well, the bulk of predicted disorder is clearly confined to the C-terminus; whereas roughly 55% of the C-terminus is predicted to be disordered, only ∼14% disorder is found for P/rds overall. Moreover, when PONDR VL-XT is applied to P/rds regions other than the C-terminus, it predicts only 3.6% disorder. Overall, both secondary structure and intrinsic disorder prediction methods suggest that, with the exception of a single helix, the P/rds C-terminus is anticipated to lack fixed structure.
Figure 1.
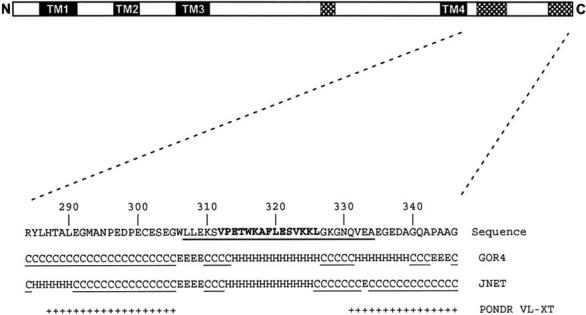
Modeling of CTER structural elements. Above: linear representation of full-length P/rds. Sequences predicted to form transmembrane helices (shaded) and disordered regions (hatched) are indicated. Below: the 63 C-terminal amino acids of bovine P/rds are shown. Region reported by Battaglia et al. as α-helical and fusogenic is illustrated in bold font [17]. Region reported by Tam et al. as important for OS targeting is underlined [19]. Results from GOR4 and JNET predictions are given as: random coil (C), helical (H), or extended strand (E) and regions given as disordered by these applications are underlined. Sequences predicted to be disordered by the PONDR VL-XT application are shown (+).
C-terminal structure in P/rds from rod OS membranes. We employed limited proteolysis to examine whether the disorder predicted is characteristic of the P/rds C-terminus in the context of its native membrane environment. Fig. 2A shows a Western blot analysis of bovine rod OS membranes probed with MAbC6, a monoclonal antibody that binds an epitope at the C-terminus of P/rds. The rod OS membranes were burst hypotonically to expose the P/rds cytoplasmic C-termini, then were pretreated with one of two denaturing conditions. The membranes were washed free of denaturants and then were digested with Proteinase K for the times indicated. Membranes pretreated with 5 mM Tris, pH 7.5 are shown for comparison (control). We found that approximately 70% of the C-terminal epitope was destroyed over 30 min in the control reaction. Highly structured, globular domains typically display an increased accessibility to proteolytic digestion upon denaturant induced unfolding. In contrast, the relatively harsh pretreatments used here, including 4M urea and 100 mM sodium carbonate, pH 11, had no significant effect on the time course of proteolysis. Similar results were obtained when trypsin, an enzyme with greater sequence specificity was employed (not shown). Thus, strong denaturants do not increase accessibility of the C-terminus to proteases, suggesting that an extended conformation is present in the native protein. We also tested the effect of pretreatment with 10 mM DTT, a strong, membrane-permeable reductant that is reported to disrupt P/rds interactions with other photoreceptor proteins [31], and found it also had no appreciable effect on the protease sensitivity of the P/rds C-terminus (Fig. 2B). These findings demonstrate that the C-terminus is relatively accessible to limited proteolysis and are consistent with the extensive regions of disorder predicted by structure modeling.
Figure 2.
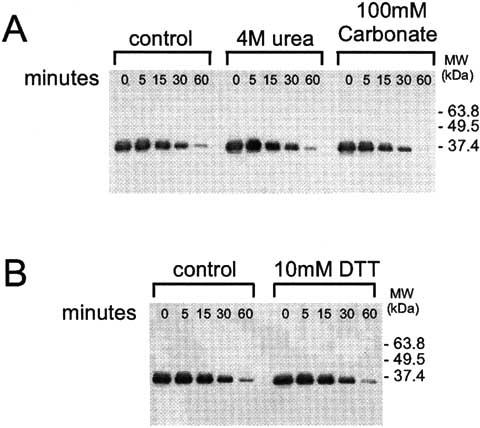
Protease sensitivity of the P/rds C-terminus in photoreceptor rod OS membranes. (A) Hypotonically lysed rod OS membranes were pretreated with 4M urea, 100 mM NaCarbonate, pH 11, or left untreated prior to limited digestion with Proteinase K at 37°C for the times indicated. Reactions were quenched by boiling in SDS buffer and were analyzed by Western blot analysis as developed with MAbC6, a monoclonal antibody directed against the P/rds C-terminus[12]. No significant increase in sensitivity was observed after treatment with 4M urea or 100 mM NaCarbonate. (B) Hypotonically lysed ROS membranes at 0.4 mg/ml were pretreated with 10mM DTT or were left untreated prior to limited digestion with Proteinase K at 37°C for the times indicated. Reactions were analyzed as described above. Pretreatment with DTT also had no significant effects on C-terminal Proteinase K sensitivity. Results suggest a lack of tertiary structure in the C-terminus.
Expression and purification of a soluble P/rds C-terminal domain (CTER). Structural determinations of integral membrane proteins continue to present numerous technical challenges, particularly with respect to characterization of regions of flexibility. We therefore designed a soluble version of the P/rds C-terminal domain as a cleavable GST fusion protein, based on a previous study [18]. The new expression vector (designated PGEX6CTER) takes advantage of a PreScission protease cleavage site and offers the advantage of facile protease removal using a glutathione affinity matrix. The fusion protein itself, GST6-CTER, is highly expressed in E. coli strain BL21 and yields of approximately 30mgs per liter of LB culture were obtained routinely. Fig. 3A presents a Coomassie blue stained Tricine gel containing fractions from the fusion protein purification procedure. The initial affinity purification from the bacterial lysate yielded a single major band of apparent MW ∼32kDa, with a secondary band of ∼25Da, likely a GST truncation product. Cleavage of GST6-CTER with PreScission protease yielded the expected fragments for GST alone ∼25kDa and the predicted CTER domain ∼7 kDa (Fig. 3B). A second (negative) affinity purification to remove GST, uncleaved GST6-CTER, and the PreScission protease, results in a single major band of the predicted ∼7 kDa molecular weight.
Figure 3.
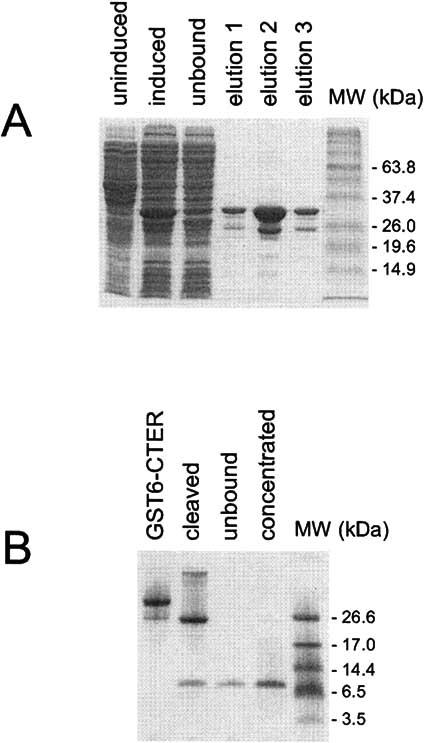
Expression and purification of a soluble form of the P/rds C-terminus analyzed by SDS-PAGE. (A) IPTG induction of a pGEX6CTER transformed BL21 culture resulted in the appearance of a new protein band at ∼33 kDa. GST affinity purification yielded both the ∼33kDa full-length GST6-CTER, and a ∼26kDa GST truncation product. (B) Cleavage of GST6-CTER with PreScission protease released a ∼7kDa fragment (CTER) that did not bind the glutathione-agarose affinity media and is present as a single major product on a Coomassie blue stained Tricine gel. The purified and concentrated CTER was used for further study.
Self association of CTER. Previous studies have demonstrated that full-length P/rds from ROS membranes is detergent solubilized as a non-covalent tetramer under reducing conditions [32]. Moreover, specific determinants within the large extracellular/intradiskal loop region, EC2, are used to guide protein folding and subunit assembly [12]. Although no evidence has yet been presented that C-terminal defects can alter protein stoichiometry, we thought it worthwhile to directly test the possibility that this domain may self-associate - particularly since a 15 amino acid synthetic peptide corresponding to sequence within this region is reported to form dimers and tetramers [17]. We asked whether we could detect a similar self-association of CTER using a hydrodynamic assay. Low pressure size exclusion chromatography (SEC) was performed in a moderate ionic strength buffer system at neutral pH, using protein concentrations from 0.1 to 1.5 mg/ml on a column of Sephacryl S-100HR. Fig. 4 shows a typical SEC elution profile of purified CTER as measured by A280. A single symmetrical peak, with an elution time of 118.1 min is observed - corresponding to an apparent MW of approximately 25kDa. The left inset figure shows that an identical profile was observed when TCA-precipitated SEC fractions were analyzed by Tricine-buffered SDS-PAGE in conjunction with Coomassie blue staining. The CTER elution position was unaffected by initial protein concentration (0.05 to 1.5 mg/ml), low ionic strength, or phospholipid vesicles (not shown). Collection, concentration and re-injection of the CTER peak resulted in an essentially identical elution profile (not shown). These results suggested that the purified CTER was present as a single non-equilibrating species of ∼25 kDa apparent MW. Given its predicted monomer mass of 7.2 kDa, the SEC data suggested that CTER could self-associate as a non-equilibrating trimer or tetramer. This was an intriguing outcome, given our previous findings that: 1) tetrameric P/rds subunit assembly is not affected by large insertional mutations in its C-terminus [12], and 2) strong denaturants do not increase accessibility of the C-terminus to proteases (Fig. 2). However, since molecular mass determinations by SEC assume globularity and can be affected by binding interactions with chromatographic media [26,33], we decided to consider an alternative interpretation to self-association and test the possibility that CTER is non-globular.
Figure 4.
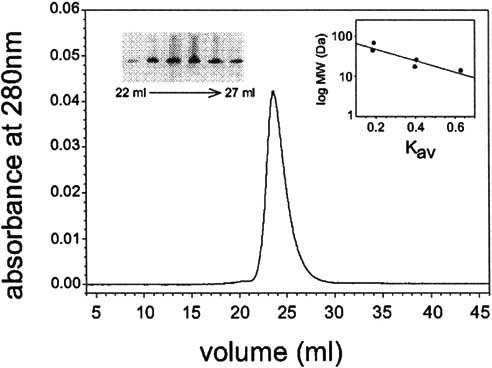
CTER apparent MW under non-denaturing conditions was assessed by SEC. Purified CTER was chromatographed in 50mM TRIS-HCl, 150mM NaCl, 1 mM TCEP, pH 7.5, on a calibrated column of Sephacryl S-100HR at 0.2 ml/min. The protein eluted as a single major peak with Kav = 0.396. CTER elution position was confirmed by SDS-PAGE and coomassie blue analysis of the collected fractions (left inset). An apparent MW of ∼ 25 kDa was determined using a 1st order regression analysis of known standards (right inset).
Analytical ultracentrifugation (AUC) was employed to differentiate between self-association and non-globularity and rigorously define CTER stoichiometry. CTER sedimentation velocity was assessed for protein concentrations ranging from 0.1 to 1.0 mg/ml and the data were analyzed using a model of continuous distribution, c(s), of sedimentation coefficients [25]. The results are presented in a scaled and offset form in Fig. 5. Peak positions and ratios remain essentially constant; these findings indicate that CTER does not participate in a reversible mass-action equilibrium at the concentrations measured. The weight-average s-values (Sw) presented in Table 1 were obtained by a second-moment integration of the sedimentation profiles. The similar values confirm that Sw remains constant over the range of protein concentration examined. A model of non-interacting discrete species as applied to the 1 mg/ml velocity sedimentation data suggest that CTER is present as a single major species, with < 10% of a higher MW contaminant present.
Figure 5.
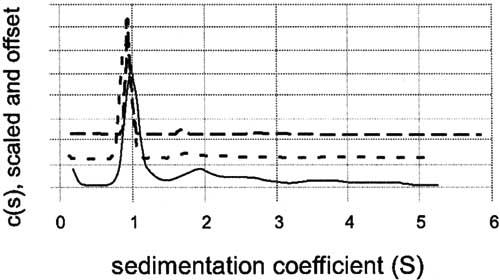
Analytical ultracentrifugation of purified CTER by velocity sedimentation. CTER was prepared in 50mM Tris-HCl, 150mM NaCl, 1 mM TCEP, pH 7.5 at concentrations of 0.1, 0.3, and 1.0 mg/ml. After reaching temperature equilibrium, the samples were centrifuged at 50,000rpm, and scanned at one minute intervals for 7h. The raw data were analyzed with the Sedfit (version 8.5) program using the model of a continuous distribution of sedimentation coefficients [25]. Distributions for 0.1 mg/ml (solid line), 0.3 mg/ml (short dashed line), and 1.0 mg/ml (long dashed line), are plotted together using an arbitrary y-offset. The data indicate that CTER is monomeric and not undergoing a reversible mass-action equilibrium.
Table I.
Weight-average S-values for samples of purified CTER at concentrations from 0.1 to 1.0 mg/ml obtained from second order integration of the respective velocity sedimentation profiles. No change is observed over the concentration range examined.
| Concentration | sw (calc) | C0 (calc) |
|---|---|---|
| 0.1 mg/ml | 1.17 S | 0.62 fringes |
| 0.3 mg/ml | 0.98 S | 1.89 fringes |
| 1.0 mg/ml | 1.06 S | 3.8 fringes |
CTER was subsequently subjected to sedimentation equilibrium analysis at three protein concentrations ranging from 0.11 to 1.0 mg/ml. Data sets (six in total) collected for each concentration at both 28k and 40k rpm were analyzed with nonlinear least-squares techniques using the WinNonlin program. As was expected from sedimentation velocity experiments, fits using only a single ideal species were poor, and large systematic errors indicated an incorrect model. Various association models were tried, and the best fit was obtained assuming two non-interacting discrete species. The results from this fit, using the known value for CTER of 7.21 kDa is shown in Table 2. The deviations are essentially random and close to the noise level inherent in the optical system. The values of the fitted K’s indicate a higher order MW contaminant present in the CTER sample at approximately 5-10%. In sum, the combined AUC measurements indicate that the purified CTER is monomeric and that the sample also contains <10% of a higher molecular weight contaminant. The rigorous determination that CTER is monomeric (by AUC) combined with a substantially retarded hydrodynamic elution position (observed by SEC) suggests that CTER possesses an extended conformation and may have a weak affinity for the SEC matrix.
Table II.
Parameters obtained from fitting sedimentation equilibrium data obtained at concentrations of CTER from 0.11 to 1.0 mg/ml at 28,000 and 40,000 rpm. Fits using a single ideal species were poor, and the heterogeneous model used here indicates that CTER is predominantly monomeric, and less than 10% of a higher order molecular weight contaminant is present.
| Speed (rpm) | C0 (mg/ml) | MW (kDa) | LnK1,4 (K is in terms of fringes) | rms (fringes) |
|---|---|---|---|---|
| 28K | 0.11 | 7.21 (fixed) | 1.3 | 0.0118 |
| 0.33 | −3.2 | |||
| 1.0 | −6.3 | |||
| 40K | 0.11 | −0.72 | ||
| 0.33 | −4.3 | |||
| 1.0 | −6.7 |
Tryptophan solvent exposure. We wished to test further the hypothesis that CTER possesses an extended conformation; this model predicts that the two tryptophan residues present should be readily accessible to solvent. We measured intrinsic tryptophan fluorescence of CTER under a variety of solution conditions and found this to be so. Fig. 6A (solid line) shows an emission spectrum of CTER, using an excitation wavelength of 295 nm. The characteristic broad peak at 348 nm suggested to us that the two tryptophans contained within this domain were rather solvent exposed, as had been predicted by the SEC and AUC results. Variation of ionic strength and divalent ion concentration had no appreciable effect on these results (not shown). The idea that CTER possesses an extended conformation was supported further by the finding that the addition of a strong denaturant, 6M GuCl, altered the emission spectrum peak only marginally (Fig. 6A, dashed line). Protein denaturation and increase of tryptophan solvent exposure is typically accompanied by a significant red shift; however in this instance, the small shift observed likely results solely from an altered solvent environment. Finally, we assessed tryptophan accessibility by adding increasing amounts of the commonly used fluorescence quencher, acrylamide. As seen in Fig. 6B, acrylamide quenching of CTER intrinsic tryptophan fluorescence is highly effective and shows a steep dependence similar to that seen for random coil peptides [34]. The combined data clearly indicate a high solvent accessibility for both of the tryptophan residues in CTER, and these observations are most consistent with an extended protein conformation.
Figure 6.
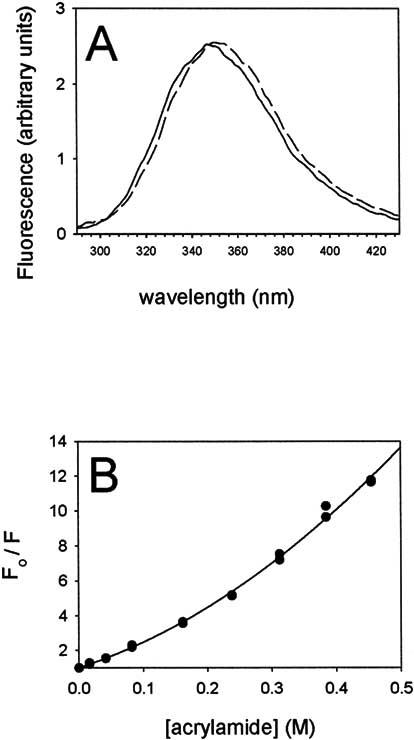
Hydrophobic residue exposure to solvent as assessed by intrinsic tryptophan fluorescence. Intrinsic tryptophan fluorescence was measured in response to 295nm excitation of a 5 μg/ml solution of CTER in 50 mM Tris-HCL, 150 mM NaCl, 1 mM TCEP, pH 7.5 at 25°C. Buffer background subtracted spectra are presented. (A) Fluorescence emission spectra for purified CTER are shown in the presence (dashed line) and absence (solid line) of 6M GuCl. Little difference is observed. (B) Quenching of CTER tryptophan fluorescence by acrylamide shows a steep dependence, similar to that reported for ACTH, a random coil peptide [34].
Far UV Circular dichroism. Since findings from SEC, AUC and intrinsic tryptophan fluorescence all pointed to an extended conformation and lack of globular structure, far UV CD was employed to help assay the degree of structure present in CTER. A previous investigation using far UV CD concluded that CTER displays considerable secondary structure in aqueous solution, but did not address whether tertiary structure is present [18]. Fig. 7A shows averaged far UV-CD spectra measured for CTER at 10°C (solid line) and 20°C (dotted line). They are similar and characterized by roughly 10% α-helix, in agreement with prior measurements. As expected from SEC, AUC, intrinsic tryptophan fluorescence and previous CD measurements, the spectra also indicate strong contributions from random coil elements (estimated here at ∼36%). Far UV CD was subsequently applied to ask whether CTER displays significant tertiary structure. A thermal denaturation of CTER (from 10°C to 60°C) was performed while monitoring CD at 220 nm, a routine diagnostic for loss of α-helical and tertiary structure. Contrary to expectations for a folded protein, Fig. 7B illustrates a small increase in CTER α-helix with elevated temperature. The combined results suggest that CTER largely lacks fixed tertiary structure and possesses limited secondary structure.
Figure 7.

Far UV circular dichroism spectroscopy of purified CTER. (A) CD spectra of 0.31 mg/ml CTER taken in 50 mM Tris-HCl, 150 mM NaCl, 1mM TCEP, pH 7.5 at 10°C (solid line) and 20°C (dotted line) as described in Methods. The spectra are similar and each is characterized by about 10% helix and 25% sheet. (B) Thermal melting of 0.31 mg/ml CTER performed in 50 mM Tris-HCl, 150 mM NaCl, 1mM TCEP, pH 7.5 was followed at 220nm from 10°C to 60 °C, using a scan rate of 20°C/h. A very slight increase in helical content is indicated. (C) Effect of a membrane mimetic environment on CTER secondary structure. Background subtracted CD spectra collected from 0.082 mg/ml CTER in 20 mM Tris-HCl, 0.5mM TCEP, pH 7.5 with the indicated concentrations of TFE. Estimates of helical content (assayed at 220nm) more than tripled as TFE content increased from 0 to 50%.
Since evidence suggests that the high macromolecule concentrations in cellular environments can promote compact, vs. extended protein conformations [35], we examined whether macromolecular crowding effects could induce additional structure in CTER. As well, given the fact that the native P/rds C-terminus is membrane anchored, exists in a membrane-rich environment, and can interact with liposomes [3,16], we tested the effect of a membrane mimetic environment on CTER structure. The presence of 320 mg/ml dextran (40,000), a commonly used macromolecular crowding agent [35], did not have a appreciable effect on the far UV CD spectrum of CTER; an increase in helical content of roughly 3% was estimated. In contrast, increasing concentrations of trifluoroethanol (TFE), a cosolvent that is documented to stabilize functionally important protein structures [36], had a significant effect on increasing α-helical content in CTER. Fig. 7C shows that signals at 220 nm rise with TFE concentration. CTER helical content is estimated to increase more than threefold (10% to 32%), in the presence of 50% TFE. Overall these results suggest that CTER structure, like that in other disordered domains is not affected by macromolecular crowding effects [22], but is appreciably increased in the presence of a hydrophobic environment.
DISCUSSION
Studies described in this report suggest that structure plays quite a different role for the C-terminus of P/rds than it does for the previously characterized EC2 domain. The latter has fulfilled usual expectations for a hydrophilic extracellular domain; it contains a high degree of predicted secondary structure, numerous disulfide linkages, several regions shown to be essential for protein folding, and discrete determinants for oligomeric subunit assembly [12]. In contrast, we show here that the cytoplasmic C-terminus displays far less fixed structure and our results suggest that this domain is extended and possesses an inherent conformational flexibility. In retrospect, the high antigenicity documented previously for this region may have provided a clue that a substantial degree of C-terminal disorder was present [37]. Increasingly frequent reports from a variety of laboratories suggest that many proteins include sizable regions that lack fixed tertiary structure, and further, that these regions (or molecules) often perform critical functions within eukaryotic cells [21-23]. The presence of a non-globular C-terminal domain in P/rds raises the possibility that intrinsic disorder plays a regulatory role for its function, and potentially for tetraspanins more generally.
We report here that modeling programs predict a high degree of conformational flexibility for this region and we present four lines of evidence with regard to domain structure. Firstly, we used limited proteolysis in conjunction with denaturing agents, a common approach for probing domain structure, since denaturation increases protease sensitivity of globular domains. To perform the assays we utilized Proteinase K, a highly active endopepetidase that does not exhibit pronounced cleavage site specificity or trypsin, an enzyme that exhibits specificity for cleavage at basic residues, in combination with MabC6, an antibody that binds the C-terminus of P/rds. Pretreatments that denature proteins and strip rod OS membranes of peripheral proteins, including 4M urea and 100mM sodium carbonate, pH 11 [38], had no effect on digestion rates. We were unable to assay higher denaturant concentrations (i.e. 8M urea, 6M GuCl), as they induced irreversible leakiness in the membrane vesicles. Treatment with 10mM DTT, a condition that disrupts P/rds binding to GARP-related proteins [31], also had no measurable effects on digestion rates. Secondly, we utilized AUC, performed in both velocity and equilibrium modes to show that a soluble version of the C-terminus, CTER, behaves as a ∼7kDa monomer in aqueous solution and shows no tendency to self-associate. Combined with the AUC result, the elution of CTER at the equivalent of ∼25kDa by SEC is a strong indication that it possesses a highly extended and non-globular structure. Thirdly, the intrinsic tryptophan fluorescence spectrum of CTER was essentially unaffected by a strong denaturant (6M GuCl) and showed a steep dependence of quenching by acrylamide. These findings indicate that neither of the tryptophans present are protected within a hydrophobic core, but instead are exposed to solvent - consistent with a polypeptide that lacks tertiary structure. Since the fluorescence spectrum was not affected significantly by divalent cations, or CTER phosphorylation by casein kinase II or protein kinase C, we also conclude these factors also do not significantly affect its structure (results not shown). Finally, observations from far UV CD studies, here and elsewhere [18], predict that a substantial portion of the CTER sequence is present as random coil elements. Our present report also demonstrates that CTER displays no measurable loss of helical content with heat denaturation; this result is most consistent with minimal long-range interactions between polypeptide segments. Overall, the combination of structural predictions and experimental evidence suggests that the bulk of the P/rds C-terminus is intrinsically disordered. We consider it unlikely, but cannot formally exclude the possibility that loss of membrane tethering and/or bacterial expression contributed to the high degree of disorder we measured for CTER. We note that the recombinant CTER is not merely misfolded, as it is both highly expressed and soluble to >10 mg/ml in a purified form. In addition, CTER fused to green fluorescent protein possesses targeting and localization activity in X. laevis photoreceptors [19,20], while CTER fusions with GST display fusogenic activity [18,20]; these results indicate that soluble versions of the P/rds C-terminus domain can function autonomously, at least in part.
Intrinsically disordered regions (also called: intrinsically unstructured, natively unstructured, and natively unfolded regions), are portions of proteins or entire proteins without fixed tertiary structure. Although disordered regions lack a defined 3-D structure in their native states, they are involved in a variety of cellular activities and frequently undergo disorder-to-order transitions upon binding to partners [21-23]. This conformational flexibility has been suggested to act as a regulatory mechanism that allows a single domain the adaptability required to engage in multiple interactions [21,22]. Disordered regions have been documented in several photoreceptor proteins and flexible switch regions appear to be important elements for the regulation of the phototrandsduction cascade [39-41]. We hypothesize that C-terminal flexibility in P/rds likewise may be important for regulating its function; albeit that that function remains uncertain. Although clearly required for OS formation, P/rds mechanism of action at the molecular level is still controversial. The C-terminus is important for P/rds OS targeting/localization and fusogenic activity, and is potentially involved in binding to GARP-related proteins as well [16,19,31]. We speculate that intrinsic disorder may allow this region to participate in, and switch between some or all of these activities; additional studies are required to address this possibility. Increases in CTER structure induced by phospholipid vesicles [18], and by TFE, the alcohol co-solvent used here (Fig. 7C), suggest the possibility that such regulation might involve an induced conformational change.
We note that although there is significant divergence between tetraspanin C-terminal sequences, family members generally have in common very petite C-termini - as few as five amino acids in some instances, [42]. We consider it likely that intrinsic disorder is an important aspect of function for these domains, since sequences of this length are generally unable to overcome entropic forces and maintain regular secondary structures in solution [43]. Although the functional importance of tetraspanin C-termini is not known generally, reports for two tetraspanins, CD151 and CD63 suggest that their C-termini are essential for protein function. An investigation of CD151, an integrin-binding member of the tetraspanins proposed to regulate adhesion-dependent signaling, identified a critical role for its predicted 9 amino acid C-terminus for activity toward Ras [13]. A more recent study concluded that the CD151 C-terminus is critically important for regulating α6β1 integrin adhesion strengthening [14]. As well, the predicted 11 amino acid C-terminus of CD63 has been shown important for subcellular targeting [15]. These findings suggest that tetraspanins other than P/rds also possess disordered, yet functionally active C-termini. In accord with a previous proposal for the general significance of intrinsically disordered domains [21], we speculate that conformational flexibility in tetraspanin C-termini may allow these minimal sequences to interact with multiple intracellular signaling partners.
This report introduces the notion that intrinsic disorder is an important feature of the C-terminal domain of photoreceptor P/rds. In agreement with secondary structure and disorder predictions, we show that the native C-terminus in situ is readily accessible to proteases and that denaturing conditions do not alter its accessibility. In addition, four independent lines of evidence demonstrate that a soluble form of this domain largely lacks tertiary structure and possesses limited secondary structure. Overall, the combination of structural predictions and experimental evidence suggests the P/rds C-terminal domain is intrinsically disordered. These findings raise the possibility that conformational flexibility might play a role in regulating P/rds activity, a proposal that may apply to other tetraspanins as well. Our continuing studies should help determine whether and under what circumstances intrinsic disorder in the C-terminus of P/rds plays a regulatory role in photoreceptor OS architecture and tetraspanin structure/function more generally.
ACKNOWLEDGEMENTS
We thank Ms. Jinny Johnson and Drs. James Cole and Frank Giblin for technical assistance, and Drs. John Philo, Sitaramayya Ari, and Krzysztof Palczewski for helpful discussions.
ABBREVIATIONS
- AUC
analytical ultracentrifugation
- β-ME
β-mercaptoethanol
- bp
basepair
- DTT
dithiothreitol
- EC2
extracellular loop 2
- GST
glutathione S-transferase
- IPTG
isopropylthio-β-galactosidase
- MAb
monoclonal antibody
- PBS
phosphate-buffered saline
- PONDR
predictor of naturally disordered regions
- OS
outer segment
- P/rds
peripherin/rds
- SEC
size exclusion chromatography
- TBS
Tris-buffered saline
- TCEP
tris(carboxyethyl) phosphine
- TFE
trifluoroethanol
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
Supported by a grant (EY013246) to A.F.X.G. from the National Eye Institute.
REFERENCES
- Connell G, Bascom R, Molday L, Reid D, McInnes RR, Molday RS. Proc. Natl. Acad. Sci. USA. 1991;88:723–726. doi: 10.1073/pnas.88.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis GH, Sutcliffe JG, Bok D. Neuron. 1991;6:61–70. doi: 10.1016/0896-6273(91)90122-g. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Goldberg AF. Int. Rev. Cytol. 2002;217:183–225. doi: 10.1016/s0074-7696(02)17015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl S, Giddings I, Besch D, Apfelstedt-Sylla E, Zrenner E, Wissinger B. Acta Anat. (Basel) 1998;162:75–84. doi: 10.1159/000046471. [DOI] [PubMed] [Google Scholar]
- Travis GH, Hepler JE. Nat. Genet. 1993;3:191–192. doi: 10.1038/ng0393-191. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Todd SC, Levy S. FASEB J. 1997;11:428–442. [PubMed] [Google Scholar]
- Boucheix C, Rubinstein E. Cell Mol. Life Sci. 2001;58:1189–1205. doi: 10.1007/PL00000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneuret M, Delaguillaumie A, Lagaudriere-Gesbert C, Conjeaud H. J. Biol. Chem. 2001;276:40055–40064. doi: 10.1074/jbc.M105557200. [DOI] [PubMed] [Google Scholar]
- Kitadokoro K, Ponassi M, Galli G, Petracca R, Falugi F, Grandi G, Bolognesi M. Biol. Chem. 2002;383:1447–1452. doi: 10.1515/BC.2002.164. [DOI] [PubMed] [Google Scholar]
- Kitadokoro K, Bordo D, Galli G, Petracca R, Falugi F, Abrignani S, Grandi G, Bolognesi M. EMBO J. 2001;20:12–18. doi: 10.1093/emboj/20.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AF, Loewen CJ, Molday RS. Biochemistry. 1998;37:680–685. doi: 10.1021/bi972036i. [DOI] [PubMed] [Google Scholar]
- Goldberg AF, Fales LM, Hurley JB, Khattree N. J. Biol. Chem. 2001;276:42700–42706. doi: 10.1074/jbc.M107511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada S, Yoshimoto M, Odintsova E, Hotchin NA, Berditchevski F. J. Biol. Chem. 2003;278:26323–26326. doi: 10.1074/jbc.C300210200. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Kazarov AR, Huang H, Lee RT, Hemler ME. Proc. Natl. Acad. Sci. USA, 2003;100:7616–7621. doi: 10.1073/pnas.1337546100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rous BA, Reaves BJ, Ihrke G, Briggs JA, Gray SR, Stephens DJ, Banting G, Luzio JP. Mol. Biol. Cell. 2002;13:1071–1082. doi: 10.1091/mbc.01-08-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Lamba OP, Napoli AA, Jr., Sinha S, Guo Y. Biochemistry. 1998;37:9477–9487. doi: 10.1021/bi980173p. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Stefano FP, Fenner M, Napoli AA., Jr. Biochim. Biophys. Acta. 2000;1463:343–354. doi: 10.1016/s0005-2736(99)00226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Goldberg AF, Dispoto J, Katragadda M, Cesarone G, Albert AD. Exp. Eye Res. 2003;77:505–514. doi: 10.1016/s0014-4835(03)00151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL, Papermaster DS. Mol. Biol. Cell. 2004;15:2027–2037. doi: 10.1091/mbc.E03-09-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter LM, Boesze-Battaglia K, Tam BM, Moritz OL, Khattree N, Chen SC, Goldberg AF. J. Biol. Chem. 2004;279:39958–39967. doi: 10.1074/jbc.M403943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- Tompa P. Trends in Biochem. Scie. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- McDowell JH. Meths. Neurosci. 1993;15:123–130. [Google Scholar]
- Schuck P. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent TC, Killander J. J. Chromatog. 1964;14:317–330. [Google Scholar]
- Bohm G, Muhr R, Jaenicke R. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- Garnier J, Gibrat J-F, Robson B. Meths. Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. Bioinformatics. 1998;14:892–893. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Poetsch A, Molday LL, Molday RS. J. Biol. Chem. 2001;276:48009–48016. doi: 10.1074/jbc.M108941200. [DOI] [PubMed] [Google Scholar]
- Goldberg AF, Molday RS. Biochemistry. 1996;35:6144–6149. doi: 10.1021/bi960259n. [DOI] [PubMed] [Google Scholar]
- Nozaki Y, Schechter NM, Reynolds JA, Tanford C. Biochemistry. 1976;15:3884–3890. doi: 10.1021/bi00662a036. [DOI] [PubMed] [Google Scholar]
- Eftink MR, Ghiron CA. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- Sasahara K, McPhie P, Minton AP. J. Mol. Biol. 2003;326:1227–1237. doi: 10.1016/s0022-2836(02)01443-2. [DOI] [PubMed] [Google Scholar]
- Buck M. Structure (Camb.) 2003;11:735–736. doi: 10.1016/s0969-2126(03)00130-8. [DOI] [PubMed] [Google Scholar]
- Molday RS. Prog. in Reti. and Eye Res. 1994;13:271–299. [Google Scholar]
- Penefsky HS, Tzagoloff A. Meths. Enzymol. 1971;22:204–219. [Google Scholar]
- Langen R, Cai K, Altenbach C, Khorana HG, Hubbell WL. Biochemistry. 1999;38:7918–7924. doi: 10.1021/bi990010g. [DOI] [PubMed] [Google Scholar]
- Kisselev OG, Downs MA. Structure. (Camb.) 2003;11:367–373. doi: 10.1016/s0969-2126(03)00045-5. [DOI] [PubMed] [Google Scholar]
- Kisselev OG, Kao J, Ponder JW, Fann YC, Gautam N, Marshall GR. Proc. Natl. Acad. Sci. USA. 1998;95:4270–4275. doi: 10.1073/pnas.95.8.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipp CS, Kolesnikova TV, Hemler ME. Trends Biochem. Sci. 2003;28:106–112. doi: 10.1016/S0968-0004(02)00014-2. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. FASEB J. 1995;9:37–42. doi: 10.1096/fasebj.9.1.7821757. [DOI] [PubMed] [Google Scholar]