

Abstract
The protective effect of dietary saturated fatty acids against the development of alcoholic liver disease has long been known, but the underlying mechanism is incompletely understood. Here we examined the involvement of the adipocyte hormone adiponectin. Circulating adiponectin levels were significantly elevated by chronic ethanol administration to mice consuming a high saturated fat diet. The increase in circulating adiponectin was associated with the activation a set of hepatic signaling pathways mediated through AMPK, PPARα, and PGC-1α, which in turn led to markedly increased rates of fatty acid oxidation, prevention of hepatic steatosis, and alleviation of liver enzyme changes. Furthermore, treatment of rat 3T3-L1 adipocytes with saturated fatty acids (palmitic or stearic acids) in the presence of ethanol increased secretion of adiponectin and enhanced activity of a mouse adiponectin promoter. In conclusion, the protective action of saturated fat against the development of alcoholic fatty liver in mice is partially mediated through induction of adiponectin. The present findings suggest a novel paradigm for dietary fatty acids in the pathogenesis of alcoholic liver disease, and provide a promising therapeutic strategy, which is nutritional modulation of adiponectin, in treating human alcoholic fatty liver disease.
Keywords: adipose tissue, hormone, signal transduction, AMP-activated kinase, liver steatosis
Abbreviations used in the paper: AMPK, AMP-activated protein kinase; ACC, acetyl-CoA carboxylase; CPT I, carnitine palmitoyltransferase I; PPARα, peroxisome proliferator-activated receptor α; PPARγ, peroxisome proliferator-activated receptor γ; PGC-1α, peroxisome proliferator-activated receptor γ co-activator-alpha; AOX, acetyl-CoA oxidase; PPRE, PPAR response element; β-OHB, β-hydroxybutyrate; FFA, free fatty acids; ALT ,alanine aminotransferase; RT-PCR, reverse transcription-polymerase chain reaction
Introduction
Accumulation of fat in the liver in response to chronic alcohol consumption can lead to more severe forms of liver injury (1, 2). Development of specific pharmacological agents to reverse steatosis is at an early stage. While abstinence from alcohol is the long-term goal of management of all forms of alcoholic liver disease, nutritional modulation of alcoholic hepatic steatosis is an attractive approach.
Dietary fat content and composition have been long implicated in the pathogenesis of alcoholic liver disease. By using either the Lieber-DeCarli liquid diet or the intragastric ethanol-fed animal model, several studies have demonstrated that diets enriched in saturated fatty acids or medium chain triglyceride protect against alcoholic liver injury while diets containing polyunsaturated fatty acids promote liver injury (3–7). More strikingly, administration of saturated fat has been shown to reverse established alcoholic liver injury in rats and improve nearly all liver pathological changes despite continued ethanol administration (8, 9). While there is evidence that down-regulation of lipid peroxidation, reduced levels of endotoxin and TNFα, and inhibition of activation of NF-κB pathway might contribute to the therapeutic effects of saturated fat, the underlying mechanisms remain incompletely understood (9–11).
Recently, a number of novel mechanisms involved in the development of alcoholic fatty liver have emerged, providing important therapeutic leads. Chronic ethanol administration in mice is associated with inhibition of AMP-activated kinase (AMPK) and peroxisome proliferator activated receptor α (PPARα), two critical signaling molecules controlling the pathways of hepatic fatty acid oxidation (12–15). These molecules are the major mediators of the metabolic effect of adiponectin (16, 17). Adiponectin is exclusively expressed and secreted from adipose tissue. Circulating adiponectin concentrations are account for ~0.01% of total plasma protein (18). Two adiponectin receptors (AdipoR1 and –R2) have been cloned (16). AdipoR2 is predominantly expressed in the liver, where it mainly serves as the transducer of adiponectin-mediated activation of AMPK and PPARα, leading to increased fatty acid oxidation and reduced fat accumulation (16, 17).
Altered expression of adiponectin may be associated with alcohol-induced liver injury. Chronic ethanol consumption significantly decreased circulating concentrations of adiponectin in mice (19). Delivery of recombinant full-length adiponectin into these mice alleviated liver steatosis and injury (19). Furthermore, pioglitazone, a PPARγ agonist, prevents alcohol-induced liver injury in rats and mice (20–22). While several potential mechanisms have been proposed in these studies, up-regulation of adiponectin by pioglitazone through activation of PPARγ could contribute to the hepatic protective effects (23). Regulation of adiponectin by ethanol was further suggested by human studies which demonstrated that moderate alcohol consumption significantly increased plasma adiponectin levels in both healthy and insulin resistant middle-aged men (24–26). Therefore, the effect of ethanol on adiponectin physiology appears to depend on the amount of alcohol consumed, the dietary context, and nutritional status. In the present study, we examined the role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice.
Materials and Methods
Materials
Most chemicals were purchased from Sigma. PGC-1α antibody was obtained from Calbiochem (San Diego, CA). Full-length recombinant adiponectin was purchased from BioVendor Laboratory Medicine, Inc (Czech Republic). PGC-1α reporter was constructed as described previously (27). PPRE3-tk-luciferase reporter and the expression plasmids for murine PPARα, and -γ were the kind gifts of Dr. Ronald Evans (Salk Institute). Mouse adiponectin promoter reporter plasmid was a kind gift of Dr. Jae Bum Kim (Seoul National University).
Animal Studies
Male C57BL/6J (6-8 week-old) mice were obtained from Jackson Laboratory (Bar Harbor, ME). Mice were housed individually in a room with controlled temperature (20–22°C), humidity (55%–65%) and lighting (on at 6 am and off at 6 pm). Liquid diets were based upon the Lieber-DeCarli formulation (28) and provide 1 kcal/ml (prepared by Dyets, Inc., Bethlehem, Philadelphia). Protein content was constant at 18% of total calories and each diet had identical mineral and vitamin content and contained safflower oil (4% of calories) to provide essential fatty acids. The mice were divided into four dietary groups: (a) polyunsaturated fat pair-fed control diet (PUFA, 40% of calories from fat, primarily from corn oil); (b) ethanol-containing polyunsaturated fat diet [PUFA+E, identical to the control PUFA diet but with ethanol added to account for 27.5% of total calories and the caloric equivalent of carbohydrate (maltose-dextrin) removed]; (c) high saturated fat pair-fed control diet [HSF, 40% of calories from fat, primarily from cocoa butter]; (d) ethanol-containing high saturated fat diet [HSF+E, identical to the control HSF diet but with ethanol added to account for 27.5% of total calories]. The dietary and nutritional intake of control mice were matched to those of the ethanol-fed mice by pair-feeding the same volume of isocaloric liquid diet for 4 weeks. For animals on an ethanol-containing diet, animal cages were placed on heating pads to maintain body temperature since ethanol consumption can induce hypothermia. The protocols of animal studies were approved by the Indiana University School of Medicine Animal Care and Use Committee and the Veterans Affairs Animals Use Subcommittee.
Measurement of Hepatic Histology and Lipid
Frozen sections of the liver were stained with Oil Red O (Sigma) as described (14). Total hepatic cholesterol and triglyceride levels were measured as described previously (14).
Assays from Mouse Serum
Serum levels of alanine aminotransferase (ALT) were determined using a kit from Sigma-Aldrich (St. Louis, Missouri). Plasma β-hydroxybutyrate (β-OHB) was measured using a KetoSite kit (GDS Diagnostics, Elkhart, IN). Plasma total homocysteine was analyzed using the IMx System (Abbott Laboratories, Mississauga, Ontario, Canada). Serum leptin was analyzed using an ELISA kit from R&D system (Minneapolis, MN). Circulating levels of TNFα were quantified using commercial ELISA kits from Chemicon International (Temecula, CA). Plasma triglyceride and cholesterol were determined using the Sigma Diagnostics Triglyceride and Infinity Cholesterol Reagent.
Measurement of Adiponectin Levels Using ELISA and Western Blot Analysis for Adiponectin Expression
Serum levels of adiponectin were determined using a commercial ELISA kit from R&D System (Minneapolis, MN). For Western blots, 2 μl of mouse serum were separated by 10% SDS-PAGE, transferred to a nitrocellulose membrane, and probed with anti-mouse adiponectin antibody (CHEMICON® International).
Total RNA Isolation and Relative Quantitative RT-PCR Analysis
Total RNA was prepared from mouse liver using an RNAeasyTM Total RNA kit (QIAGEN Inc.). Quantitative RT-PCR was performed as described (14). The mRNA levels were quantified using a QuantumRNA 18S internal standard kit (Ambion, Austin, TX) according to the manufacturer’s instructions. The primers used are as follow: PGC-1α; (5′aatgcagcggtcttagcact3′)(5′tttctgtgggtttggtgtga3′); acyl-CoA oxidase (AOX); (5′cttgttcgcgcaagtgagg3′) (5′caggatccgactgtttacc3′).
Measurements of Phosphorylation Levels of Hepatic ACC, AMPK and Malonyl CoA Content
At the time of sacrifice, the liver samples were immediately freeze-clamped with the tongs pre-cooled to the temperature of liquid nitrogen. Tissues were ground to a powder under liquid nitrogen and were then homogenized. The liver homogenate was fractionated by electrophoresis in a 10% or 8 % SDS-polyacrylamide gel, and transferred to nitrocellulose filters. AMPKα, phospho-AMPKα, phospho-ACC were visualized using antibodies (Cell Signaling Technology). Hepatic malonyl-CoA concentrations were measured in neutralized perchloric acid liver extracts at the Mouse Metabolic Phenotyping Center (Yale University School of Medicine) (12).
Analysis of Hepatic Palmitate Oxidation
The rates of fatty acid oxidation of liver homogenates were analyzed using [1-14C]-palmitate (Amersham Radiolabeled Chemicals, St. Louis, MO) as described previously (14). Fatty acid β-oxidation activity was expressed as pmol[14C]-palmitate oxidized/g liver/min.
Studies with Rat Hepatoma Cells and Isolated Hepatocytes
The stock fatty acid:BSA complexes were prepared as described (29). Cell transfection assays were carried out as described previously (13). Primary rat hepatocytes were isolated from fed male Sprague-Dawley rats by collagenase digestion (12). The rate of oxidation of [1-14C]-palmitic acid to carbon dioxide was performed as described (12).
Maintenance and Differentiation of 3T3-L1 Cells
The 3T3-L1 cells were maintained as subconfluent cultures in DMEM supplemented with 10% fetal bovine serum. For differentiation, post-confluent cells were induced by incubation with 0.25 μM dexamethasone, 0.5 mM 3-isobutyl-methylxanthine, and 10μg/ml insulin for 2 d. This was followed by incubation with 10 μg/ml insulin for 2 d. The cells were then maintained in DMEM with 10% fetal bovine serum for another 4 d. Staining with Oil red O revealed that more than 90% of cells exhibited typical morphology of adipocytes. At 80-90% confluence, the cells were incubated for 16 h in serum-free DMEM. The cells were then treated with serum-free DMEM containing fatty acid:BSA complex and ethanol.
Data Analysis
All data are presented as the mean ± S.D. Statistical significance was calculated with the Student’s t test, or one-way ANOVA, as appropriate.
Results
A High Saturated Fat Diet Attenuated Hepatic Steatosis and Reversed Alanine Aminotransferase Abnormalities Caused by Chronic Ethanol Consumption in Mice
Male C57BL/6J mice were fed modified Lieber-DeCarli liquid diet with high polyunsaturated fat (PUFA) or high saturated fat (HSF) diets in the presence or absence of ethanol (27.5% of the total calories) using a pair-feeding protocol for 4 weeks. Ethanol intake had no apparent effect on the health status of the animals, and mice gained a body weight of 1-2 g over 4 weeks. While the mouse body weights did not vary among the groups, the liver/body weight ratio was significantly increased by the PUFA+E diet, whereas it was unchanged by the HSF+E (Fig. 1A). On a PUFA diet, hepatic triglycerides were significantly elevated ~1.9-fold by ethanol feeding as compared with the pair-fed control. Ethanol did not affect liver triglycerides when added to the HSF diet (Fig. 1B). There were no significant differences in hepatic cholesterol content among the various groups (data not shown). Histological analysis of the livers corresponded with the hepatic triglyceride levels. While there was prominent accumulation of lipid droplets in the livers of mice fed the PUFA+E diet, lipid droplets were rare in the livers of mice fed the HSF+E diet (Fig. 1C). Moreover, the PUFA+E diet significantly increased ALT by ~4.5-fold, while there was no increase in the HSF+E diet (Fig. 1D). These results demonstrate that a HSF diet protects against development of alcoholic fatty liver in mice.
Fig. 1. Protective effects of a high saturated fat diet against alcoholic fatty liver.

Liver/body weight ratio (A), hepatic triacylglycerol content (B), Oil red O staining of liver sections (C), and plasma ALT level (D) of mice fed a high polyunsaturated fat diet (PUFA), a high saturated fat diet (HSF) with (PUFA+E, HSF+E) or without ethanol. The mouse body and liver weights were: HSF(body: 22±0.7g, liver: 1.0±0.1g); HSF+E(body: 22±0.9g; liver: 1.0±0.2g); PUFA (body: 22±0.9g; liver: 1.0±0.1g); PUFA+E (body: 22±1.7g; liver: 1.2±0.1g). All data are expressed as the mean ± S.D. n = 8 animals. *p< 0.05, **p< 0.001. aSignificantly different compared with PUFA pair-fed control.
A High Saturated Fat Diet Increased Circulating Adiponectin Level in the Chronic Ethanol-Fed Mice
We next tested the hypothesis that adiponectin might play a role in the prevention of alcoholic fatty liver by the HSF diet. Plasma adiponectin concentration was quantified using both Western blot and ELISA. Plasma adiponectin was significantly decreased ~15% by the PUFA+E diet compared with the PUFA control diet (Fig 2A). However, circulating adiponectin concentration significantly increased nearly 1.8-fold on the HSF+E diet compared with control mice receiving the HSF diet, and 2-fold as compared with the PUFA+E diet.
Fig. 2. A high saturated fat diet increased circulating adiponectin in the chronic ethanol fed-mice.
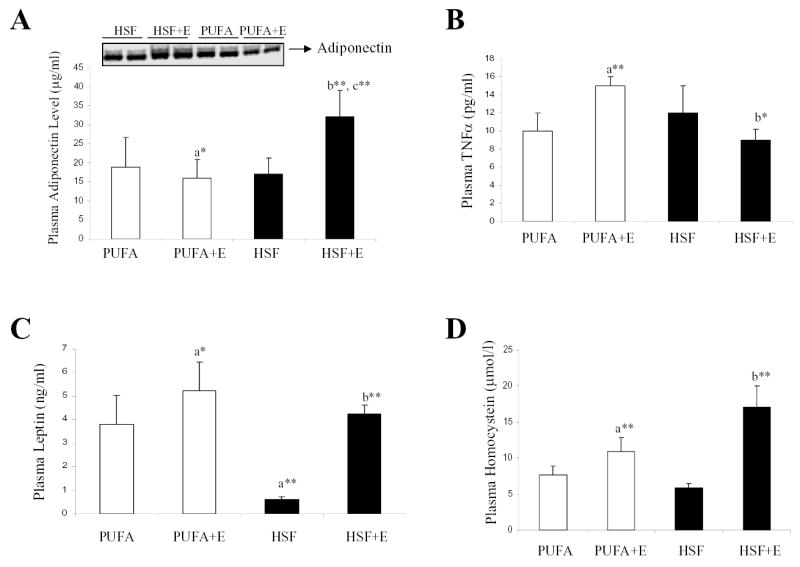
Plasma adiponectin concentrations (A), plasma TNFα (B), plasma leptin (C), and plasma homocysteine (D) of mice fed a high polyunsaturated fat diet (PUFA), a high saturated fat diet (HSF) with (PUFA+E, HSF+E) or without ethanol. The levels of adiponectin were quantified by using ELISA and Western blot analysis (mean ± S.D. n = 4–8 animals). *p< 0.05, **p< 0.001. aSignificantly different compared with PUFA pair-fed control. bSignificantly different compared HSF pair-fed control. cSignificantly different compared with ethanol-PUFA-fed animals.
We observed an inverse relationship between circulating concentrations of adiponectin and TNFα among the various groups (Fig. 2B). Hepatic TNFα mRNA levels were unchanged in all of the groups, indicating that the altered circulating TNFα was not related to changes in production of TNFα from Kupffer cells, hepatocytes, or stellate cells (data not shown). In addition, neither the HSF+E or PUFA+E diets caused a significant increase of plasma endotoxin compared with the control diet, indicating that alcohol-induced endotoxemia may not be the major factor by which the fat composition of the diet modulates the response to ethanol (data not shown).
Feeding the HSF diet dramatically reduced the plasma leptin concentration (Fig 2C). However, in comparison with the control PUFA diet, the PUFA+E or HSF+E diets increased circulating leptin by ~1.7- and 1.6-fold, respectively. The results appear in agreement with a previous report that circulating leptin levels are increased in chronic alcoholism regardless of nutritional status or the presence of liver disease (30).
Ethanol-induced hyperhomocysteinemia has been proposed to play a role in alcoholic liver injury (31, 32). As shown in Fig. 2D, PUFA+E feeding increased plasma homocysteine by ~1.4-fold over PUFA alone, and the HSF+E diet increased homocysteine ~2.9-fold with the HSF-fed control animals.
Taken together, all the ethanol-fed mice had a similar increase in circulating leptin and homocysteine levels despite differences in liver steatosis. The only circulating factor which correlated with the protective effect of the HSF diet was adiponectin.
A High Saturated Fat Diet Stimulated AMPKα and ACC Phosphorylation and Reduced Malonyl CoA Content in the Livers of Chronic Ethanol-Administrated Mice
Stimulation of fatty acid oxidation by adiponectin occurs through activation of AMPK, diminished ACC activity, and reduction of malonyl CoA content in the liver (16–18). The phosphorylation of Thr172 in the α subunit of AMPK is essential for AMPK activity, and the phosphorylation of ACC at Ser79 by AMPK is associated with reduced activity (12). To determine whether the changes in serum adiponectin concentrations resulted in altered activities of AMPK and ACC in the livers of ethanol-fed mice, the phosphorylation states of AMPKα and ACC were analyzed. Compared to the respective control diets, the HSF+E diet led to nearly 180% and 200% increases in the phosphorylation of AMPKα and ACC, respectively. Conversely, the PUFA+E diet resulted in modest 42% and 20% decreases in the phosphorylation of AMPKα and ACC, respectively (Fig. 3A).
Fig. 3. A high saturated fat diet caused increased phosphorylation levels of AMPK and ACC and enhanced PGC-1α mRNA and protein levels.

(A) Western blots were performed using anti-P-AMPKα, anti-AMPKα, and anti-P-ACC antibodies from liver extracts of mice fed a high polyunsaturated fat diet (PUFA), a high saturated fat diet (HSF) with (PUFA+E, HSF+E) or without ethanol. (B) Western blots of PGC-1α protein levels. (C) Relative hepatic levels of PGC-1α mRNA. (D) Relative hepatic levels of AOX mRNA. Western blots were quantified by a PhosphorImager and ImageQuant software analysis. All data are expressed as the mean ± S.D. n = 3–5 animals. *p< 0.05, **p< 0.001. aSignificantly different compared with PUFA pair-fed control. bSignificantly different compared with HSF pair-fed control.
Malonyl CoA, the product of ACC, is a potent inhibitor of carnitine palmitoyltransferase I (CPT I), the rate-limiting step for mitochondrial fatty acid oxidation. The HSF+E diet reduced the concentration of malonyl CoA to 54% of the HSF diet control. The malonyl CoA content was significantly enhanced ~1.4-fold by the PUFA+E diet compared with control (Table I).
Table I. Interaction of diets and ethanol feeding in mice on selected parameters.
The male C57BL/6J mice were divided into four dietary groups: (a) polyunsaturated fat pair-fed control diet (PUFA, 40% of calories from corn oil); (b) ethanol-containing polyunsaturated fat diet [PUFA+E, identical to the control PUFA diet but with ethanol added to account for 27.5% of total calories and the caloric equivalent of carbohydrate (maltose-dextrin) removed]; (c) high saturated fat pair-fed control diet [HSF, 40% of calories from cocoa butter]; (d) ethanol-containing high saturated fat diet [HSF+E, identical to the control HSF diet but with ethanol added to account for 27.5% of total calories]. Protein content was constant at 18% of total calories and each diet had identical mineral and vitamin content and contained safflower oil (4% of calories) to provide essential fatty acids. The animals were pair-fed for 4 weeks. Results are expressed as means ± SD of 4–8 animals.
| Parameters | PUFA | PUFA+E | HSF | HSF+E |
|---|---|---|---|---|
| Plasma triglycerides (mg/dl) | 142±60 | 131±30 | 74±38 | 196±30b* |
| Plasma cholesterol (mg/dl) | 161±22 | 162±17 | 103± 54 | 162±17 |
| Plasma β-hydrobutyrate (mg/dl) | 0.5± 0.2 | 1.0±0.3a* | 0.3±0.1 | 2.4±0.6b** |
| Liver malonyl CoA (nmol/g liver) | 4.8±1.3 | 6.6±1.4a* | 5.5±2.9 | 3.0±1.5b* |
| Liver palmitate oxidation (pmol/g liver/min) | 11.8±6.1 | 8.6±4.0 | 10.9±2.4 | 29.4±10b** |
Significant difference from the PUFA pair-fed group.
Significant difference from the HSF pair-fed group.
p<0.05
p<0.001.
A High Saturated Fat Diet Increased the mRNA and Protein Expression of PGC-1α in the Livers of Ethanol-Fed Mice
Ethanol treatment impairs hepatic fatty acid oxidation by inhibiting the transcriptional activities of PPARα (13). PGC-1α is a coactivator of PPARα (33, 34); therefore, we examined the mRNA and protein levels of PGC-1α. As shown in Fig.3B and C, both mRNA and protein expression of PGC-1α were significantly increased nearly 2-fold in the livers of HSF+E fed mice, and the hepatic mRNA level of acyl-CoA oxidase (AOX), a target of both PPARα and PGC-1α, was elevated ~2.5-fold as compared to the HSF diet control (Fig. 3D). Interestingly, the HSF diet alone significantly reduced the mRNA expression level of PGC-1α compared with PUFA diet-fed mice (Fig.3C).
Increased Rate of Hepatic Fatty Acid β-Oxidation and Elevated Plasma β-Hydroxybutyrate (β-OHB) Level Induced by a High Saturated Fat Diet Plus Ethanol Consumption
To further investigate whether increased AMPKα, diminished ACC activity, reduced malonyl CoA content, and up-regulated PGC-1α expression induced by the HSF+E diet led to increased fatty acid oxidation, we assessed the total hepatic fatty acid β-oxidation capacity. It was robustly increased ~3-fold by the HSF+E diet while the PUFA+E diet modestly suppressed the fatty acid β-oxidation activity (Table I). Consistent with this finding, the HSF+E diet markedly increased (~ 8-fold) the plasma β-hydroxybutyate (β-OHB) levels (Table I).
Collectively, in the setting of a HSF diet, consumption of ethanol was associated with a dramatically increased capacity for hepatic fatty acid oxidation and plasma ketone body production. These changes indicate that the livers of HSF+E fed mice handle fatty acid loads much more effectively than livers from animals fed other diets.
Adiponectin Increased Activities of a Mouse PGC-1α Promoter Reporter and a PPARα Responsive Reporter and Caused stimulation of AMPK and the Rate of Fatty Acid Oxidation in Rat H4IIEC3 Cells and Rat Hepatocytes
To determine whether PGC-1α is a direct target for adiponectin action, the effect of adiponectin on a mouse PGC-1α promoter was tested in rat H4IIEC3 hepatoma cells. As shown in Fig. 4A, PGC-1α promoter activity was significantly induced in a dose-dependent manner by adiponectin treatment, as well as by a known inducer of this promoter, forskolin, acting via cAMP (33).
Fig. 4. Adiponectin increased activities of a mouse PGC-1α promoter reporter and a PPARα responsive reporter and stimulated AMPK activity and fatty acid oxidation in rat H4IIEC3 cells and rat hepatocytes.
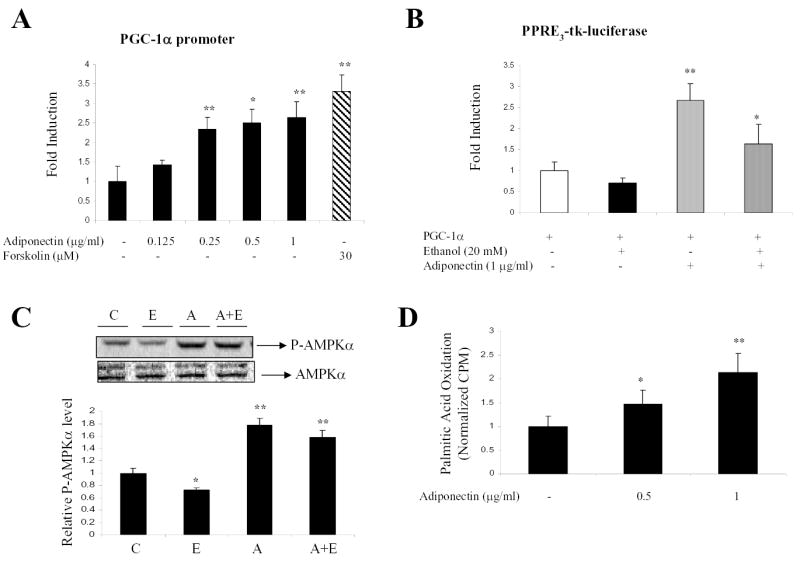
(A) H4IIEC3 cells were plated in MEM (low glucose) supplemented with 10% FBS and transfected with a mouse PGC-1α promoter-luciferase reporter, PPARα expression plasmid, and internal control (pSV2CAT) for 5 h. Cells were then incubated with adiponectin at the concentrations as indicated for 48 h. The adenylate cyclase activator, forskolin (30 μM), was used as a positive control. (B) H4IIEC3 cells were transfected with PPRE3-tk-luciferase reporter, PPARα and PGC-1α expression plasmids and internal control (pSV2CAT). After 24 h, cells were then incubated with adiponectin in the absence or presence of ethanol at concentrations as indicated. Forty-eight hours after transfection, the cells were harvested for assay of the reporter enzymes. Normalized luciferase activities are shown as mean ± SE. (C) Rat hepatocytes were grown in DMEM containing 10% fetal bovine serum, 100 nM insulin, 100 nM dexamethasone, and 5 μg/ml transferrin for 4 hours. Cells were then cultured in serum-free DMEM for 16 hours followed by treatment. C, control; E, ethanol (100 mM); A, adiponectin (1 μg/ml); A+E, ethanol (100 mM) plus adiponectin (1 μg/ml). Western blots were quantified by a PhosphorImager and ImageQuant software analysis. (D) Rat hepatocytes were treated with adiponectin at concentrations as indicated for 4 h. Palmitic acid oxidation assays were performed as described under “Methods”. The data represent at least three replications. *p < 0.05; **p < 0.01 compared with control group by one-way ANOVA.
We further examine the direct effect of adiponectin on activities of AMPK and PPARα. The effect of adiponectin on PPARα activity was examined by using a PPARα responsive reporter plasmid transfected into H4IIEC3 cells. Since the levels of endogenous PPARα and PGC-1α were low in H4IIEC3 cells (13), PPARα and PGC-1α expression plasmids were co-transfected into cells. As shown in Fig. 4B, adiponectin, at 1 μg/ml increased the reporter activity by over 3-fold. Ethanol inhibited activity of PPRE3-tk-luciferase as reported (13), and co-incubation of adiponectin (1 μg/ml) partially restored the ethanol-inhibited reporter activity. More interestingly, the activation of the PPARα-responsive promoter by adiponectin requires co-expression of PGC-1α, suggesting that adiponectin-induced PPARα activity may be mediated through up-regulation of PGC-1α expression.
We next examined the effects of adiponectin on the phosphorylation state of AMPKα in the absence or presence of ethanol in rat hepatocytes. The inhibition of AMPKα phosphorylation by ethanol is reversed by the presence of adiponectin (Fig. 4C). Furthermore, adiponectin treatment increased rates of palmitic acid oxidation in a dose dependent manner in freshly isolated rat hepatocytes (Fig. 4D).
Effects of Dietary Fatty Acids on Adiponectin Production Mediated by Ethanol in 3T3-L1 Adipocytes
To examine the direct effects of fatty acids and ethanol on adiponectin secretion, differentiated 3T3-L1 adipocytes were incubated with various fatty acid:BSA complexes, i.e., palmitic and stearic acids (the major saturated fatty acids in cocoa butter) or linoleic and oleic acids (the major unsaturated fatty acids in corn oil), in the presence or absence of ethanol for 24 h. Adiponectin concentrations in the conditioned medium were significantly increased by either palmitic or stearic acids in the presence of ethanol (Fig. 5A). There was only a small, non-significant decrease in adiponectin production by adipocytes treated with ethanol in the presence of either oleic or linoleic acids (Fig. 5A). Ethanol (100 mM) treatment alone for 24 h had no significant effect on adiponectin secretion.
Fig. 5. Effects of dietary fatty acids and ethanol on adiponectin production in 3T3-L1 adipocytes.

(A) The differentiated 3T3-L1 adipocytes were incubated for 16 h in serum-free DMEM and then treated with various fatty acid:BSA complexes in the absence or presence of ethanol as indicated. Analysis of adiponectin concentrations in the culture media was performed using an ELISA-based assay. (B) Hela cells were transfected with a mouse adiponectin promoter-luciferase reporter and expression plasmids for PPARγ and RXRα. Following transfection, cells were treated with various fatty acid:BSA complexes in the absence or presence of ethanol. Rosiglitazone (10 μM) was used as a positive control. Normalized luciferase activities are shown as mean ± SE from at least three experiments performed in duplicate. *p < 0.05; **p < 0.01 in comparison with controls.
We further examined the ability of various dietary fatty acids and ethanol to transactivate a mouse adiponectin promoter reporter. This study utilized Hela cells since the 3T3-L1 adipocytes are difficult to transfect. The cells were co-transfected with PPARγ and retinoid X receptor (RXRα) expression plasmids, which activated the adiponectin reporter, and rosiglitazone, a PPARγ agonist, further enhanced promoter activity by ~2.5-fold (Fig. 5B). The promoter activity was not significantly changed by either ethanol alone or any of the dietary fatty acids tested in the absence of ethanol. However, the mouse adiponectin promoter activity was significantly increased by addition of ethanol in the presence of the saturated fatty acids, palmitic or stearic acids. Conversely, treatment with unsaturated fatty acids (linoleic or oleic acids) with ethanol suppressed adiponectin promoter activity by 20–30% (Fig. 5B).
Discussion
The present study provided evidence that adiponectin plays a role in the ability of dietary saturated fat to protect against the development of alcoholic fatty liver in mice. Circulating adiponectin levels were significantly elevated by chronic ethanol administration in the setting of the HSF diet. In parallel, a HSF+E diet stimulated the phosphorylation of AMPKα and ACC, reduced malonyl CoA contents, and up-regulated PGC-1α mRNA and protein levels in the liver. Treatment of rat 3T3-L1 adipocytes with saturated fatty acids in the presence of ethanol increased secretion of adiponectin and enhanced activity of an adiponectin promoter to approximately the same extent. Thus, dietary saturated fat alleviated alcoholic fatty liver partially through up-regulation of adiponectin expression and production in adipose tissue. The increased circulating adiponectin is likely to coordinate multiple signaling pathways in the liver (Fig. 6), leading to enhanced oxidation of fatty acids, prevention of hepatic steatosis, and alleviation of liver injury.
Fig. 6.
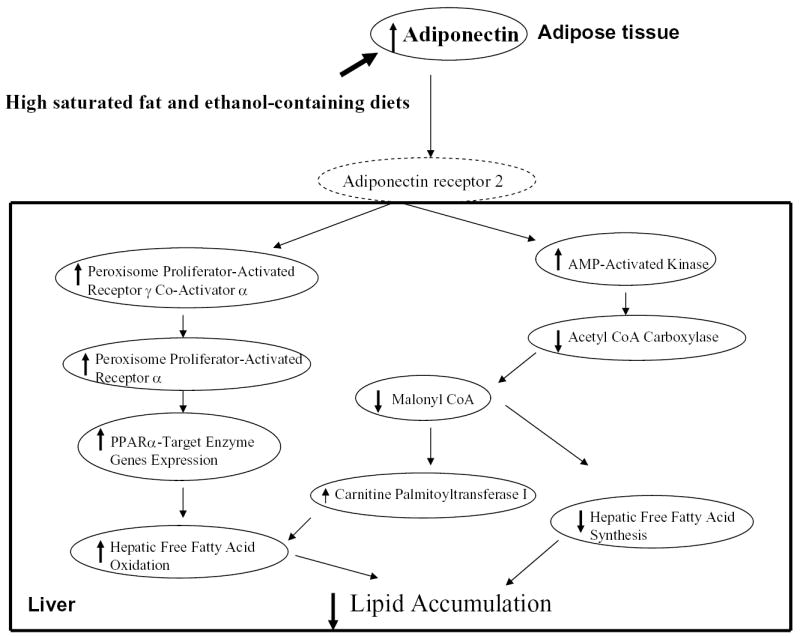
Schematic diagram of the potential mechanisms that underlie the protective action of dietary saturated fatty acids against alcoholic fatty liver.
The precise mechanism by which dietary fatty acids plus ethanol affect adiponectin expression and its secretion remains to be determined. Adiponectin and TNFα regulate each other’s expression. Down-regulation of TNFα has been proposed as a mechanism for the protective effects of saturated fatty acids (10, 35). Thus, it is possible that the HSF+E diet reduced TNFα concentrations and thereby enhanced adiponectin expression. Indeed, we found that plasma adiponectin levels were inversely related to serum TNFα concentrations. However, secretion of adiponectin by 3T3-L1 adipocytes and the response of the adiponectin promoter suggested that dietary fatty acids plus ethanol could also directly regulate adiponectin gene expression. In this scenario, the increased levels of adiponectin in animals fed the HSF+E diet might have led to reduction of TNFα production. A recent study demonstrates that moderate alcohol consumption significantly enhanced circulating adiponectin concentrations without affecting the plasma TNFα levels, suggesting that regulation of adiponectin by ethanol could be independent of the TNFα levels (24).
The mouse adiponectin promoter contains a binding site for PPARγ and its cofactor, retinoid X receptor (RXR) (23). Previously, we reported that ethanol treatment slightly but significantly reduced both basal and clofibrate-stimulated PPARγ-dependent transcriptional activities, and the mRNA and protein levels of RXRα were decreased in chronic ethanol-fed mice (13, 14). Therefore, dietary fatty acids plus ethanol may regulate the adiponectin promoter activity through directly modulating PPARγ function.
PGC-1α was initially discovered as a coactivator for PPARγ, and has subsequently been shown to serve as a coactivator of PPARα (33), and is therefore involved in the transcriptional control of the genes encoding enzymes of fatty acid oxidation (34). Our data suggest a close relationship between PGC-1α expression and the action of adiponectin as demonstrated in cultured hepatoma cells and animal livers. The ability of adiponectin to enhance PPARα responsive reporter activity has been shown in primary hepatocytes (16). Our studies showed that adiponectin-mediated activation of a PPARα-responsive reporter required PGC-1α co-expression, indicating that adiponectin may increase the transcriptional activity of PPARα through its up-regulation of PGC-1α expression. We speculate that PGC-1α may provide a critical signaling link between adiponectin and PPARα.
While our findings strongly suggest that adiponectin acts as an important mediator of the protective action of saturated fatty acids, our data fall short of establishing a cause and effect relationship. However, Xu et al. reported that delivery of recombinant full-length adiponectin into chronic ethanol-fed mice dramatically alleviated fatty liver and ALT abnormalities, partially through adiponectin-mediated induction of hepatic fatty acid oxidation and inhibition of hepatic fatty acid synthesis (19). More definitive demonstration of the hypothesis that the protective effect of dietary fatty acids is mediated by adiponectin would be provided using either adiponectin knockout or hepatic adiponectin receptor 2 null mice in the future.
The possibility that dietary saturated fatty acids plus ethanol directly regulate AMPK, PPARα and PGC-1α can not be excluded. Saturated fatty acids such as palmitic acid stimulated AMPK activity in the presence of ethanol in cardiomyocytes (36). We also observed that PGC-1α promoter activity in vitro is modified by various fatty acids only in the presence of ethanol (unpublished data). These direct effects could be, in part, due to the differences in the metabolism of PUFA and HSF in the presence or absence of ethanol (37). Additional studies addressing these potential multiple mechanisms are required and currently under investigation in our laboratory.
The translation of our current findings to human beings will need to be further studied. The HSF-rich diet has well known health risks. The lower PPARα expression levels in human than in rodent liver may also limit clinical applications that depend on PPARα actions (38, 39). However, our study suggests that nutritional modulation of adiponectin levels may be therapeutic for human alcoholic fatty liver disease.
Acknowledgments
We are grateful to Drs. Michinaga Matsumoto, Won Kyoo Cho, Izabela Cyganek, Mark Deeg, and Mrs. Ruth Ann Ross for the outstanding technical and intellectual contributions.
Footnotes
Financial Support: This work was supported by grant AA013623 (to M.Y.), grant AA015070 (to D.W.C.) by the National Institute on Alcohol Abuse and Alcoholism, and grant P50-AA-07611 from the Alcohol Research Center (to D.W.C.).
References
- 1.Diehl AM. Nonalcoholic fatty liver disease: implications for alcoholic liver disease pathogenesis. Alcohol Clin Exp Res. 2001;25:8S–14S. doi: 10.1097/00000374-200105051-00004. [DOI] [PubMed] [Google Scholar]
- 2.Yang S, Lin H, Diehl AM. Fatty liver vulnerability to endotoxin-induced damage despite NF-kappaB induction and inhibited caspase 3 activation. Am J Physiol Gastrointest Liver Physiol. 2001;281:G382–392. doi: 10.1152/ajpgi.2001.281.2.G382. [DOI] [PubMed] [Google Scholar]
- 3.Nanji AA, Mendenhall CL, French SW. Beef fat prevents alcoholic liver disease in the rat. Alcohol Clin Exp Res. 1989;13:15–19. doi: 10.1111/j.1530-0277.1989.tb00276.x. [DOI] [PubMed] [Google Scholar]
- 4.Mezey E. Dietary fat and alcoholic liver disease. Hepatology. 1998;28:901–905. doi: 10.1002/hep.510280401. [DOI] [PubMed] [Google Scholar]
- 5.Nanji AA, Sadrzadeh SM, Yang EK, Fogt F, Meydani M, Dannenberg AJ. Dietary saturated fatty acids: a novel treatment for alcoholic liver disease. Gastroenterology. 1995;109:547–554. doi: 10.1016/0016-5085(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 6.Nanji AA, Satoh S, Uetake S, Ohata M, Nakajima H, Yamauchi M. Effect of type of dietary fat and ethanol on hepatic leukotriene level in experimental alcoholic liver disease. Nihon Arukoru Yakubutsu Igakkai Zasshi. 2003;38:350–363. [PubMed] [Google Scholar]
- 7.Ronis MJ, Korourian S, Zipperman M, Hakkak R, Badger TM. Dietary saturated fat reduces alcoholic hepatotoxicity in rats by altering fatty acid metabolism and membrane composition. J Nutr. 2004;134:904–912. doi: 10.1093/jn/134.4.904. [DOI] [PubMed] [Google Scholar]
- 8.Nanji AA, Yang EK, Fogt F, Sadrzadeh SM, Dannenberg AJ. Medium chain triglycerides and vitamin E reduce the severity of established experimental alcoholic liver disease. J Pharmacol Exp Ther. 1996;277:1694–1700. [PubMed] [Google Scholar]
- 9.Nanji AA, Jokelainen K, Tipoe GL, Rahemtulla A, Dannenberg AJ. Dietary saturated fatty acids reverse inflammatory and fibrotic changes in rat liver despite continued ethanol administration. J Pharmacol Exp Ther. 2001;299:638–644. [PubMed] [Google Scholar]
- 10.Zakim D, Rahemtulla A, Daly T, Miao L, Zhao S, Khwaja S, et al. Dietary saturated fatty acids down-regulate cyclooxygenase-2 and tumor necrosis factor alpha and reverse fibrosis in alcohol-induced liver disease in the rat. Hepatology. 1997;26:1538–1545. doi: 10.1002/hep.510260622. [DOI] [PubMed] [Google Scholar]
- 11.Nanji AA. Role of different dietary fatty acids in the pathogenesis of experimental alcoholic liver disease. Alcohol. 2004;34:21–25. doi: 10.1016/j.alcohol.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 12.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gasteroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 13.Galli A, Pinaire J, Fischer M, Dorris R, Crabb DW. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J Biol Chem. 2001;276:68–75. doi: 10.1074/jbc.M008791200. [DOI] [PubMed] [Google Scholar]
- 14.Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–8004. doi: 10.1074/jbc.M302140200. [DOI] [PubMed] [Google Scholar]
- 15.Nanji AA, Dannenberg AJ, Jokelainen K, Bass NM. Alcoholic liver injury in the rat is associated with reduced expression of peroxisome proliferator-alpha (PPARalpha)-regulated genes and is ameliorated by PPARalpha activation. J Pharmacol Exp Ther. 2004;310:417–424. doi: 10.1124/jpet.103.064717. [DOI] [PubMed] [Google Scholar]
- 16.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 17.Shklyaev S, Aslanidi G, Tennant M, Prima V, Kohlbrenner E, Kroutov V, et al. Sustained peripheral expression of transgene adiponectin offsets the development of diet-induced obesity in rats. Proc Natl Acad Sci U S A. 2003;100:14217–14222. doi: 10.1073/pnas.2333912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 19.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomita K, Azuma T, Kitamura N, Nishida J, Tamiya G, Oka A, et al. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-Met. Gastroenterology. 2004;126:873–885. doi: 10.1053/j.gastro.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Ohata M, Suzuki H, Sakamoto K, Hashimoto K, Nakajima H, Yamauchi M, et al. Pioglitazone Prevents Acute Liver Injury Induced by Ethanol and Lipopolysaccharide Through the Suppression of Tumor Necrosis Factor-alpha. Alcohol Clin Exp Res. 2004;28:139S–144S. doi: 10.1097/01.alc.0000134412.38510.f7. [DOI] [PubMed] [Google Scholar]
- 22.Enomoto N, Takei Y, Hirose M, Konno A, Shibuya T, Matsuyama S, et al. Prevention of ethanol-induced liver injury in rats by an agonist of peroxisome proliferator-activated receptor-gamma, pioglitazone. J Pharmacol Exp Ther. 2003;306:846–854. doi: 10.1124/jpet.102.047217. [DOI] [PubMed] [Google Scholar]
- 23.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–2099. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 24.Sierksma A, Patel H, Ouchi N, Kihara S, Funahashi T, Heine RJ, et al. Effect of moderate alcohol consumption on adiponectin, tumor necrosis factor-alpha, and insulin sensitivity. Diabetes Care. 2004;27:184–189. doi: 10.2337/diacare.27.1.184. [DOI] [PubMed] [Google Scholar]
- 25.Shai I, Rimm EB, Schulze MB, Rifai N, Stampfer MJ, Hu FB. Moderate alcohol intake and markers of inflammation and endothelial dysfunction among diabetic men. Diabetologia. 2004;47:1760–1767. doi: 10.1007/s00125-004-1526-0. [DOI] [PubMed] [Google Scholar]
- 26.Pischon T, Girman CJ, Rifai N, Hotamisligil GS, Rimm EB. Association between dietary factors and plasma adiponectin concentrations in men. Am J Clin Nutr. 2005;81:780–786. doi: 10.1093/ajcn/81.4.780. [DOI] [PubMed] [Google Scholar]
- 27.Schaeffer PJ, Wende AR, Magee CJ, Neilson JR, Leone TC, Chen F, et al. Calcineurin and calcium/calmodulin dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J Biol Chem. 2004;279:39593–39603. doi: 10.1074/jbc.M403649200. [DOI] [PubMed] [Google Scholar]
- 28.Lieber CS, DeCarli LM, Sorrell MF. Experimental methods of ethanol administration. Hepatology. 1989;10:501–510. doi: 10.1002/hep.1840100417. [DOI] [PubMed] [Google Scholar]
- 29.Van Harken DR, Dixon CW, Heimberg M. Hepatic lipid metabolism in experimental diabetes. V. The effect of concentration of oleate on metabolism of triglycerides and on ketogenesis. J Biol Chem. 1969;244:2278–2285. [PubMed] [Google Scholar]
- 30.Nicolas JM, Fernandez-Sola J, Fatjo F, Casamitjana R, Bataller R, Sacanella E, et al. Increased circulating leptin levels in chronic alcoholism. Alcohol Clin Exp Res. 2001;25:83–88. [PubMed] [Google Scholar]
- 31.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 32.Esfandiari F, Villanueva JA, Wong DH, French SW, Halsted CH. Chronic Ethanol Feeding and Folate Deficiency Activate Hepatic Endoplasmic Reticulum Stress Pathway in Micropigs. Am J Physiol Gastrointest Liver Physiol 2005; in press. [DOI] [PubMed]
- 33.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 34.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin HZ, Yang SQ, Zeldin G, Diehl AM. Chronic ethanol consumption induces the production of tumor necrosis factor-alpha and related cytokines in liver and adipose tissue. Alcohol Clin Exp Res. 1998;22:231S–237S. doi: 10.1097/00000374-199805001-00004. [DOI] [PubMed] [Google Scholar]
- 36.Sparagna GC, Jones CE, Hickson-Bick DL. Attenuation of fatty acid-induced apoptosis by low dose alcohol in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2004;287:H2209–2215. doi: 10.1152/ajpheart.00247.2004. [DOI] [PubMed] [Google Scholar]
- 37.Piquet MA, Roulet M, Nogueira V, Filippi C, Sibille B, Hourmand-Ollivier I, Pilet M, Rouleau V, Leverve XM. Polyunsaturated fatty acid deficiency reverses effects of alcohol on mitochondrial energy metabolism. J Hepatol. 2004;41:721–729. doi: 10.1016/j.jhep.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet. 1999;354:141–148. doi: 10.1016/S0140-6736(98)10364-1. [DOI] [PubMed] [Google Scholar]
- 39.Everett L, Galli A, Crabb D. The role of hepatic peroxisome proliferator-activated receptors (PPARs) in health and disease. Liver. 2000;20:191–199. doi: 10.1034/j.1600-0676.2000.020003191.x. [DOI] [PubMed] [Google Scholar]