

Abstract
Chromosomal translocation t(9;11)(p22;q23) in acute myeloid leukemia fuses the MLL and AF9 genes. We have inactivated the murine homologue of AF9 to elucidate its normal role. No effect on hematopoiesis was observed in mice with a null mutation of Af9. However, an Af9 null mutation caused perinatal lethality, and homozygous mice exhibited anomalies of the axial skeleton. Both the cervical and thoracic regions were affected by anterior homeotic transformation. Strikingly, mice lacking functional Af9 exhibited a grossly deformed atlas and an extra cervical vertebra. To determine the molecular mediators of this phenotype, analysis of Hox gene expression by in situ hybridization showed that Af9 null embryos have posterior changes in Hoxd4 gene expression. We conclude that the Af9 gene is required for normal embryogenesis in mice by controlling pattern formation, apparently via control of Hox gene regulation. This is analogous to the role of Mll, the murine homolog of human MLL, to which the Af9 gene fuses in acute myeloid leukemias.
The consequence of chromosomal translocations in human cancer is to cause enforced expression of proto-oncogenes or the creation of tumor-specific fusion genes (reviewed in reference 52). Chromosomal translocations often involve transcription regulators (9) which function as master regulators of cell fate in their normal situations of expression (53). Human AF9 was identified as one of the most common fusion partners of the mixed-lineage leukemia protein (MLL, also called HTRX and ALL-1) (14, 20, 63, 70) and is most usually associated with acute myeloid leukemias (AML). MLL fusion proteins are present in approximately 10% of acute lymphoid leukemias and myeloid leukemias (42) and in up to 80% of leukemias in infants (33, 51), as well as in 85% of cases of secondary leukemias developing after treatment of a primary tumor with alkylating agents or topoisomerase II inhibitors (3, 8, 61). In an individual leukemia, the fusion protein results from one of a variety of reciprocal chromosomal translocations, causing the association of upstream exons of the MLL gene on chromosome 11, band q23, with downstream exons of the partner gene, generally on a separate chromosome. MLL is a human homologue of Drosophila melanogaster trithorax (trx), a master homeotic gene regulator essential for normal patterning during embryo development (2, 43). Gene-targeting studies have demonstrated a similar role for MLL in mammals (17, 21, 67, 69).
More than 30 different MLL fusions have been identified (28), and these code for a structurally and functionally heterogeneous group of proteins (reviewed in references 1 and 10). However, different MLL fusion proteins are consistently associated with hematopoietic tumors of different lineages. Thus, whereas MLL-AF9 is mainly found in AML, the translocation involving the AF4 gene (16, 20, 47) occurs almost exclusively in tumors of the B-cell lineage. This suggests that the fusion partner plays a role in determining disease phenotype, either because it influences the site and timing of the translocation itself or because the mutation has a cell-autonomous effect on a specific subpopulation. The active role of the fusion partner has been further confirmed by experiments demonstrating that mice carrying a knock-in allele of Mll-AF9, in which human AF9 sequences were fused to one allele of mouse Mll, have been shown to develop AML similar to those occurring in human patients (12, 15). Moreover, the product of the analogous t(11;19) translocation, MLL-ENL (48, 55, 63, 68), when used in retroviral transduction (39), transforms murine bone marrow cells, and both the transcriptional transactivation activity of ENL and the DNA-binding motifs of MLL are required for the oncogenic properties of the fusion protein (59). Recently, the putative role in determining tumor phenotype was reinforced by the generation of an Af4 null mutant mouse, which revealed that the Af4 gene, associated with lymphoid malignancy following chromosomal translocation in humans, plays a role in early lymphoid development (30).
The function of the AF9 protein is unknown. It contains a serine- and proline-rich domain, as well as a nuclear localization signal, consistent with a role as a transcription factor (29). AF9 also contains a sequence possessing transcriptional activation properties, which is consistently retained by MLL-AF9 fusion proteins. Moreover, AF9 has regions of extensive homology to both ENL and yeast ANC1 (65). Interestingly, ANC1 has been shown to be involved in the yeast RNA polymerase II transcription complex, as well as the SWI/SNF chromatin-remodeling complex, a multisubunit complex believed to facilitate transcription activator access by disrupting nucleosomes in an ATP-dependent manner (7, 13, 50). The homology of AF9 and ENL to ANC1 has led to the proposal that these proteins may interact with a human SWI/SNF complex, and the fusion proteins may retain these features (5, 6).
We have investigated the function of the Af9 gene by homologous recombination in the mouse. The Af9 null mutation was found to have an effect on the formation of the axial skeleton, suggesting that Af9 plays a role in embryo-patterning processes. While heterozygous Af9 mutant mice were normal and fertile and exhibited no segment anomalies, homozygous gene disruption led to anterior transformation of the cervical and thoracic regions and to early postnatal lethality. The presence of an eighth cervical vertebra generally compensated for the lack of a thoracic vertebra. Thus Af9 is required for normal segmentation in embryo development.
MATERIALS AND METHODS
Gene targeting and generation of chimeric mice.
For the construction of targeting vector pAf9LZ, a 6.5-kb BglII fragment containing the first two coding exons of mouse Af9 (designated exons 1 and 2; see Fig. 1) was isolated from a 129/Sv/E library in λ phage 2001. This fragment was subcloned into the linker HindIII site of pMC1-TK (62) by blunt-end ligation. A 5-kb BamHI fragment from vector pBS-TAG3/IRESlacZ/lox/MC1neoPA/lox (gift from Karen Douglas and Andrew Smith) was subsequently inserted into a HindIII site in Af9 exon 2 by blunt-end ligation. This cassette contained the encephalomyocarditis virus internal ribosome entry site (IRES), followed by the lacZ and neo sequences, the latter flanked by loxP sites. Embryonic stem (ES) cells were transfected as described previously (64). CCB ES cells were grown on neomycin-resistant mouse embryonic feeders during selection. Af9LZ ES cells were transfected with expression vector pPGKCrebpA (gift from Andrew McKenzie) for the removal of the neo cassette by transient expression of Cre recombinase to generate Af9NO clones and grown on mouse embryonic feeders without selection.
FIG. 1.

Targeting strategy for the generation of Af9 null alleles. (A) A partial genomic map of the mouse Af9 gene around the two exons encoding the first 64 amino acids of mouse Af9 (designated exons 1 and 2) is shown in the top line. Targeting vector pAf9LZ (second line) contained a 6.5-kb genomic BglII fragment, with a 5-kb lacZ-neo cassette inserted into a HindIII site in exon 2. The IRES sequence allowed independent translation of the Escherichia coli lacZ gene, which served as an Af9 gene expression marker. The neo sequence conferred G-418 resistance, allowing positive selection of targeted ES cells. This sequence was flanked by loxP sites to allow subsequent excision by transient expression of Cre recombinase (the starting vector was a gift from K. Douglas and A. Smith). Upstream of the targeting fragment, the herpes simplex virus thymidine kinase gene (TK) cassette allowed negative selection of nonhomologous integration events (62). The regions of homology with the endogenous Af9 gene are indicated. The mutant Af9 allele (Af9LZ) resulting from homologous recombination is illustrated in the third line. The Af9NO allele, resulting from transient expression of Cre recombinase, is illustrated in the fourth line. H, HindIII; B, BamHI; Bg, BglII; R, EcoRI; RV, EcoRV; Xh, XhoI. Blue boxes, exons; red arrowheads, LoxP sites. (B) Filter hybridization analysis of ES clones. A 2-kb nonrepetitive EcoRI-BglII fragment 5′ of the targeted region (A) detected a 10-kb EcoRV fragment in the wild-type (wt) genomic DNA, reduced to 7.5 kb in the Af9LZ allele (left). A nonrepetitive 3′ probe consisting of a 1-kb BglII-BamHI fragment (A) detected a 6.5-kb wild-type HindIII band, increased to 11 kb in Af9LZ and then reduced to 10 kb in Af9NO (center). The same 11-kb Af9LZ band hybridized with a probe specific for the inserted neo cassette (right); a single neo insertion was observed. This band was not visible in the Af9NO clones after removal of the neo sequence by expression of Cre recombinase. (C) RT-PCR analysis of RNA from a representative litter of E11.5 Af9NO embryos was used to detect the presence of the Af9 transcript. PCRs were analyzed on agarose gels. PCR carried out with no DNA template (H2O) served as a negative control. A 207-bp band amplified from the Af9 mRNA was generated from all samples, except those from embryos 1, 3, and 4, which were null Af9 mutants. Actin gene-specific RT-PCR, indicating that all cDNA populations analyzed were of comparable quality and concentration, is shown below. Embryo numbers are indicated above the gel. Embryos 1, 3, and 4 were Af9NO−/−, while the remaining embryos were either Af9NO+/− (2, 5, and 7 to 9) or wild type (6 and 10).
DNA from ES cells and from mouse tissues was prepared with Puregene (Gentra Systems). Ten micrograms of DNA was digested with a restriction enzyme and resolved on 0.8% agarose gels. After electrophoresis, the DNA was transferred to nylon membranes and hybridized to radioactive probes as described previously (41). Homologous recombinant Af9LZ clones were identified by using a nonrepetitive, 2-kb EcoRI-BglII genomic fragment as an external probe (5′ probe). Positive clones were confirmed by using an external 3′ probe consisting of a 1-kb BglII-BamHI fragment and by hybridization with an internal neo probe to exclude multiple insertions. Af9NO genomic DNA was analyzed with the 3′ probe and the internal neo fragment.
Clones Af9LZ and Af9NO were injected into C57BL/6 blastocysts. High-percentage male chimeras were mated with wild-type C57BL/6 females, and the resulting offspring were assayed for germ line transmission of the Af9LZ and Af9NO alleles by filter hybridization of tail DNA with the 5′ and 3′ probes, respectively.
RNA extraction and RT-PCR analysis.
Extraction of total RNA from whole mouse embryos was carried out with Trizol (Gibco-BRL). cDNA was generated with SuperRT reverse transcriptase (RT; HT Biotechnology). cDNA from 5 μg of total RNA was diluted to 100 μl. One microliter of cDNA in a reaction volume of 25 μl was subjected to 35 cycles of PCR amplification with Af9 primers 5′-GGCTAGCTCGTGTTCCG-3′ and 5′-GGTGGATCTTTGCACAC-3′. Temperature cycling was 95°C for 1 min, 65°C for 1 min, and 72°C for 1 min.
Immunophenotyping of hematopoietic cells in Af9 null mice.
Single-cell suspensions were prepared from bone marrow, spleens, and thymuses of Af9LZ mice and wild-type controls and stained with fluorescent antibodies followed by fluorescence-activated cell sorter analysis. The following antibodies were used: B220, Gr-1 (spleen cells), CD4, CD8 (thymus cells), CD44, Ter119, Gr-1, Mac-1, and c-Kit (bone marrow cells).
β-Galactosidase staining of whole embryos.
β-Galactosidase staining of mouse embryos was carried out according to published protocols (23). Pregnant females were sacrificed by lethal injection, and embryos were dissected from the uterine horns. Embryonic day 10.5 (E10.5) and E11.5 embryos were prefixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 30 min at room temperature. E12.5 and E14.5 embryos were prefixed in 4% PFA in PBS supplemented with 0.25% Nonidet P-40 for 45 min and 1 h, respectively. Embryos were subsequently washed three times with PBS for 10 min each before addition of 2 ml of freshly prepared X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining solution (0.2% X-Gal, 2 mM MgCl2, 5 mM K3Fe[CN]6). After incubation at 37°C for 24 h, X-Gal was removed by repeated rinsing in PBS, and embryos were postfixed in 4% PFA at room temperature for 48 h. Embryos were rinsed in PBS and equilibrated in 30% sucrose in PBS supplemented with 0.05% azide for long-term storage at 4°C.
Skeletal preparations of newborn mice.
Skeletons of newborn mice were stained for cartilage with Alcian blue and for bone with Alizarin red as described previously (23). Newborn mice were sacrificed by lethal injection. Specimens were skinned, eviscerated, and fixed in 95% ethanol for 3 to 5 days and then incubated at room temperature for 24 h in Alcian blue stain (15 mg of Alcian blue in 80 ml of 95% ethanol-20 ml of glacial acetic acid). Samples were rinsed twice in 95% ethanol for 24 h each. Specimens were cleared by being placed in 1% KOH for approximately 6 h and counterstained overnight with Alizarin red stain (50 mg of Alizarin red per liter of 2% KOH). Finally, samples were cleared by being placed in 2% KOH solutions of decreasing strengths. Initially, specimens were placed in 2% KOH for 2 to 3 days, and the clearing process was completed in solutions having the following ratios of 2% KOH to glycerol for 24 h each: 80:20, 40:60, and 20:80. Samples were stored indefinitely in 2% KOH-glycerol (20:80).
Whole-mount RNA in situ hybridization.
Mouse embryos were obtained by crossing heterozygous Af9NO mice and genotyped by filter hybridization of yolk sac DNA. They were fixed overnight in 4% PFA-PBS and whole-mount in situ hybridization was carried out as described previously (66). Hox gene riboprobes were made by using cDNA fragments previously described and labeled with digoxigenin (46). Cdx-I has been described (60), and the Cdx-4 probe was derived from a 520-bp cDNA fragment encoding the 5′ part of the translated sequence and finishing just before the homeobox domain. This fragment was obtained by RT-PCR on mouse E8.5 embryo total RNA with primers having the following sequences (26): 5′-ATGTACGGAAGCTGTCTTTTGGAG-3′ and 5′-CTTTTGTCCTGGTTTTCCCCGTCAC-3′.
RESULTS
Generation of Af9 null mutant mice.
The physiological role of endogenous Af9 was investigated in mice with a null mutation of Af9. A targeting vector (pAF9LZ) was constructed in which the second coding exon of mouse Af9 was disrupted by inserting an IRES-β-galactosidase gene (IRES-lacZ) cassette as an expression marker and a neomycin resistance cassette (neo) as a positive selection marker (Fig. 1A). ES cells were transfected with pAF9LZ, and two independent, homologous recombinant clones were identified by filter hybridization (Fig. 1B). One of these was used to generate chimeric mice and in turn germ line heterozygous carriers of the null Af9 mutation (designated Af9LZ mice). Furthermore, to eliminate any possible interference of the neo gene enhancer with lacZ expression patterns, the Cre-loxP system of phage P1 (22) was used to remove the neo sequence (flanked in the targeting cassette by loxP sites) from the second of these targeted Af9LZ ES clones to generate the Af9NO clone (Fig. 1A). High-percentage chimeric mice were generated from this clone, and the transmission of the disrupted alleles to their offspring was verified by filter hybridization.
Heterozygous Af9 mutant mice from the two independent Af9-targeted ES clones (i.e., Af9LZ and Af9NO) appeared normal and fertile and were bred to generate homozygous mice. Twenty-five percent of mice observed at birth were of the Af9−/− genotype, showing that the null mutants survive embryogenesis. The absence of functional Af9 in these animals was verified by RT-PCR, confirming the lack of the Af9 transcript in Af9−/− mutants (Fig. 1C). However, no homozygous mutant animals survived to weaning age. Approximately 50% of Af9−/− mice identified were found dead at birth or within hours thereafter. The remaining homozygous mutants generally became severely runted and died in the first 2 weeks. No obvious external malformations or internal changes to organs were observed. Moreover, hematopoiesis appeared normal in newborn Af9−/− animals, as analyzed by histology and flow cytometry (data not shown). AF9 mRNA transcripts are mainly detected in cell lines of megakaryocytic and erythroid origins, which does not correlate with the myeloid specificity of MLL-AF9-induced tumors (29). Together with the apparently unaffected hematopoiesis of the Af9−/− mice, this suggests that the primary function of Af9 is not to regulate genes that are normally active in myeloid cells.
Af9 is highly expressed in the developing skeleton during embryogenesis.
Our mutant Af9LZ allele contained an IRES-lacZ reporter, allowing assessment of Af9 expression patterns during embryogenesis. Figure 2 shows β-galactosidase staining of heterozygous Af9LZ embryos taken at E10.5, E11.5, E12.5, and E14.5, with wild-type littermates as negative controls. X-Gal staining was observed in the developing skeletal system of limb buds, prevertebrae, rib anlagen, developing jaw, nose, and skull. Staining was also observed in the vibrissae, gut, and forming genital organs, as well as parts of the developing neural system, in particular in the caudal hindbrain and at the midbrain-hindbrain border. Identical staining patterns were obtained with the Af9LZ and Af9NO strains, the latter lacking the neo gene, indicating that the neo enhancer had no effect on lacZ or Af9 expression (Fig. 3A) and confirming the concordance of expression in the two independent lines. Hence, the β-galactosidase activity observed reflects normal Af9 gene expression.
FIG. 2.

Af9 expression in embryogenesis detected by β-galactosidase staining of Af9LZ mouse embryos. Af9LZ heterozygous mice were mated with C57BL/6 mice, and staged embryos were removed from the uterus, prefixed in 4% PFA, and stained with X-Gal solution overnight. Wild-type embryos (+/+) and heterozygous littermates (+/−) were examined at E10.5, E11.5, E12.5, and E14.5. Activity of the Af9 gene, leading to expression of the β-galactosidase gene in the Af9LZ allele, is visible as blue staining. No staining by endogenous β-galactosidase was observed in the wild-type controls. Areas of precartilage primordium, such as the developing jaw, nose, and skull, as well as the limb buds, developing ribs, and vertebrae, exhibited Af9 expression, particularly at the later stages of development. At E14.5, the external ear and the vibrissae also exhibited strong Af9 expression. Strong staining was also observed in neural tissues, especially the caudal hindbrain, the midbrain-hindbrain junction, and the sympathetic ganglia, as well as in the heart tube, at E10.5.
FIG. 3.

Effect of homozygous Af9 mutation on embryonic development. Heterozygous Af9LZ or Af9NO mice were mated, and embryos were removed at E12.5 (A) or E14.5 (B). All embryos were prefixed in 4% PFA and stained with X-Gal solution overnight. (A) Comparison of β-galactosidase staining patterns of Af9LZ and Af9NO heterozygous embryos at E12.5. Similar staining patterns were observed with both lines. (B) Comparison of β-galactosidase staining patterns of heterozygous and homozygous Af9NO embryos. An Af9NO+/− embryo (top) is compared with an Af9NO−/− embryo (bottom; E14.5). A dorsal view of each embryo is shown on the left, with an enlargement of the cervical region on the right. Staining along the axial skeleton extended to a more anterior limit in the homozygous Af9 mutants. In addition, the structures exhibiting Af9 expression appeared to spread over a broader lateral area in this region.
β-Galactosidase staining patterns of homozygous Af9 mutants were also investigated and compared with those of heterozygous littermates (Fig. 3B). A subtle difference could be observed in the cervical region at E14.5, a stage of development in which cartilage and bone formation has begun. In Af9−/− embryos, expression along the developing axial skeleton appeared to extend to a more anterior limit than in Af9+/− specimens, as well as appearing slightly broader. Otherwise staining patterns appeared similar in Af9−/− and Af9+/− specimens, and no obvious malformations were observed.
Af9 homozygous mutants exhibit homeotic transformations of the axial skeleton.
Since β-galactosidase staining results indicated significant Af9 expression in developing bony structures, skeletons of Af9 mutants were analyzed for possible anomalies. Long bone structures of Af9−/− animals appeared normal, as assessed by histology analysis (data not shown). However, whole-skeleton preparations of newborn animals, stained for cartilage and bone with Alcian blue and Alizarin red, revealed abnormalities of the axial skeletons of homozygous mutants (Fig. 4). While heterozygous Af9LZ and Af9NO specimens appeared normal, the homozygous knockout mice were generally lacking a pair of floating ribs and the associated vertebra, having a total number of 12 instead of 13 pairs of ribs and vertebrae (Fig. 4). Only 1 out of 13 skeletons from Af9−/− pups had a 13th thoracic vertebra, but this carried very small rib anlagen. Moreover, the ribs of Af9−/− pups were not attached to the sternum in pairs but rather were staggered randomly (Fig. 4). Consequently, sternums were deformed, consisting mainly of bone rather than of segments of bone separated by cartilage, as observed in normal newborn mice.
FIG.4.

Skeletal development in Af9 mutant mice. Skeletons of newborn Af9LZ pups were cleared in 2% KOH and stained for bone with Alizarin red and for cartilage with Alcian blue. A wild-type control mouse (+/+) is shown together with two homozygous Af9 mutant specimens (−/− 1 and 2). Ventral and dorsal views are shown. Among the anomalies observed in the homozygous mutant mice was a lack of the third pair of floating ribs (fr) and malformations of the first and second cervical vertebrae (C1 and C2). Moreover, articulated ribs did not join the sternum (s) pairwise but rather in a staggered, disorganized fashion, resulting in sternum malformations of various levels of severity (see in particular specimen −/− 2). C1, atlas; C2, axis; L1 and L6, first and last lumbar vertebrae, respectively.
When the forelimbs and shoulder blades were removed, we observed that 11 out of 13 skeletons from Af9−/− mice also presented eight cervical vertebrae, compensating for the missing thoracic vertebra, instead of the normal seven, common to almost all mammals (Fig. 5B and D). In the remaining two Af9−/− samples, the second vertebra or axis had the appearance of two vertebrae partially fused dorsally (Fig. 5C). One of these was the same specimen that presented a 13th pair of ribs, raising the possibility that this represented a milder penetrance of the phenotype. Furthermore, vertebra C2 was malformed to various degrees even in the specimens where it was not fused to the third vertebra (Fig. 5).
FIG. 5.

Cervical skeletons of Af9 mutant mice. The cervical regions of skeletons of newborn Af9LZ mice, stained with Alizarin red and Alcian blue, were examined after removal of the forelimbs and shoulder blades. (A) Wild-type (+/+) control mouse; (B to D) homozygous Af9 mutant specimens. The atlases (C1) of Af9 knockout mice were severely deformed; each had the appearance of two vertebrae partially fused, with a single vertebral body (vb) extending along both substructures. Moreover, most Af9 mutants exhibited a complete, supplementary cervical vertebra, bringing the total number of cervical vertebrae to eight, as opposed to the normal seven (B and D). Occasionally, the third vertebra (C3) was partially fused to C2, but with a vertebral body of its own (C). In addition, Af9 knockout mice generally exhibited small protruding rib anlagen (ra) on vertebra C8, whereas wild-type controls usually had a pair of cervical ribs on C6. Furthermore, the spinous process (sp) normally found on the second thoracic vertebra was observed on T3 instead. C1 to C8, cervical vertebrae; T1 to T3, first thoracic vertebrae.
In addition to the presence, complete or partial, of the extra cervical vertebra, the first vertebra or atlas also had the appearance of two partially fused vertebrae (Fig. 5). In all 13 Af9−/− specimens analyzed, the atlas was approximately twice as high as in wild-type pups, with lateral gaps. The vertebral body extended the whole height of the vertebra, making it substantially larger than those in the wild-type controls. Hence, Af9 knockout mice appeared to possess eight cervical vertebrae, as well as an additional, partially duplicated structure. This unprecedented situation makes the numbering and nomenclature of vertebrae in the Af9−/− pups somewhat subjective. For descriptive purposes, the misshapen double atlas was labeled C1, the following structure, resembling a normal axis in most Af9−/− specimens, was designated C2, and the following cervical vertebrae were designated C3 to C8. Thus, in two of the Af9−/− skeletons, vertebrae C2 and C3 were considered partially fused.
In most wild-type mice, vertebra C6 carried a pair of cervical ribs, joined to the first pair of thoracic ribs, themselves joined to the sternum (Fig. 5). This was not observed in homozygous Af9 knockout mice; however, vertebra C8 usually carried a pair of rib anlagen (Fig. 5), which were not attached to the sternum or to the ribs on the first thoracic vertebra. Moreover, the spinous process normally present on thoracic vertebra T2 was usually observed on T3 (Fig. 5). Finally, in the C3-to-C8 region, the cervical spine was noticeably compressed, causing an S-shaped deformation of the neck and a forward tilt of the head in comparison with the Af9+/+ samples. This may contribute to the early postnatal death of the homozygous mutant pups, due to respiratory and/or feeding difficulties as well as possible neurological problems.
Individually dissected, stained vertebrae of an Af9LZ−/− newborn pup and a wild-type control littermate are shown in Fig. 6 (all cervical vertebrae as well as T1 are shown). The atlas and the axis (C1 and C2) of Af9−/− animals had cross-section shapes similar to those of the controls. However, in normal mouse vertebrae C3, C4, and C5 have practically identical morphologies. In the Af9−/− specimen, the third vertebra was more similar to C2 than to C4, with a round rather than an oval shape, and instead C4, C5, and C6 appeared almost indistinguishable. Thus, an anterior shift in identity was observed from the third vertebra onwards. The appearance of cervical vertebrae C7 and C8 in the Af9−/− pups was consistent with this observation as, like those of C6 and C7 in the wild-type pups, the shapes of C7 and C8 in the Af9−/− mutants progressed toward that of a thoracic vertebra, with C8 even carrying small rib anlagen.
FIG. 6.
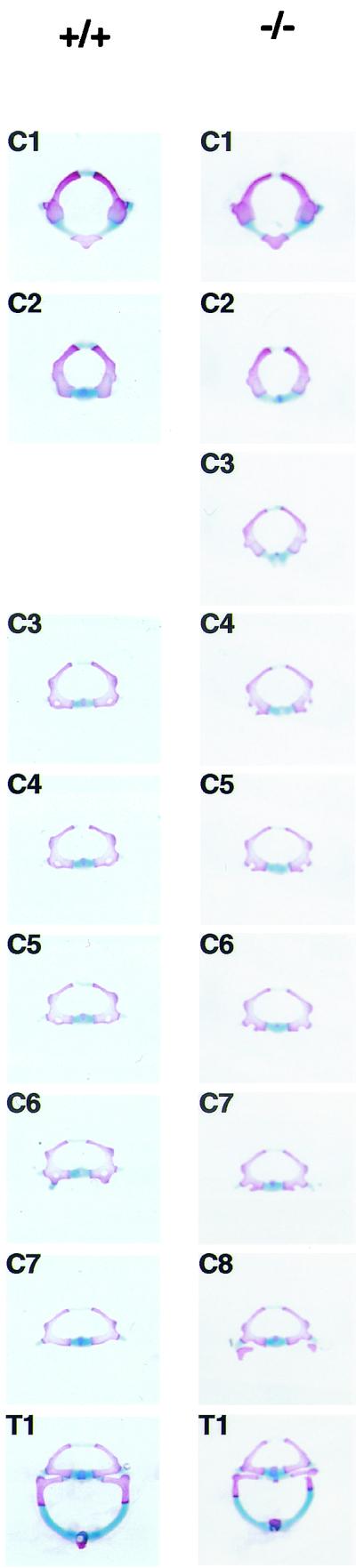
Morphology of individual vertebrae of Af9 mutant mice. Vertebrae from the cervical and thoracic regions of stained skeletons of newborn Af9LZ mice were dissected and examined under a microscope. Samples from a homozygous Af9 knockout animal (−/−) and the corresponding segments from a wild-type control mouse (+/+) are shown. All cervical vertebrae (C1 to C8) and the first thoracic vertebra (T1) are shown. The third vertebra of Af9 mutants exhibited C2 characteristics, while C4 to C8 resembled C3 to C7 in the wild type.
Thus, it appeared that an anterior homeotic transformation of the axial skeleton had occurred throughout most of the cervical region and the entire thoracic region, resulting in the shift of C3→C2 through to T13→T12, with the third cervical vertebra possibly resulting from a duplication of the C2 segment. In addition, the gross deformation of C1 seemed to indicate the presence of another extra vertebral segment fused to the atlas. In contrast, the number and morphology of all the lumbar and caudal vertebrae as well as the appendicular skeleton, were apparently normal.
Af9 null mutation affects Hoxd-4 gene expression.
The homeotic transformation observed in Af9 null mice suggests a role for Af9 as a master gene controlling expression of downstream effector genes such as Hox and Cdx genes. Such a role has been shown for Mll (69), the common fusion partner of Af9 in leukemia-associated chromosomal translocations. Accordingly expression patterns of specific Hox genes in wild-type, heterozygous, and Af9−/− embryos were compared by whole-mount in situ hybridization. We examined Hoxa2, -a3, -a4, -b2, and -b3 as well as Cdx1 and Cdx4. A caudal shift in the anterior expression boundary of Hoxd4 was detected in E9.5 Af9−/− embryos compared with wild-type or heterozygous specimens (Fig. 7 ). At this stage, Hoxd4 expression normally reaches an anterior limit localized between rhombomeric segments r6 and r7 (19, 27), but in Af9−/− embryos the anterior limit of Hoxd4 expression was localized between r7 and r8 (Fig. 7). In both wild-type and heterozygous embryos (the latter not shown), the Hoxd4 staining extended to the normal expression boundary localized between rhombomeric segments r6 and r7. In the Af9−/− embryos, a posterior shift of the anterior expression limit was observed, corresponding to approximately one rhombomeric segment. We did not find evidence for alterations of the other Hox genes analyzed or for alterations of Cdx1 or Cdx4 expression.
FIG. 7.

Hoxd4 gene expression in Af9 null mutant embryos. Hoxd4 expression was analyzed by whole-mount in situ hybridization (66) of wild type (A to C) and Af9−/− (D to F) E9.5 embryos. A Hoxd4 riboprobe was made from a cDNA fragment and labeled with digoxigenin (46). (A and D) Dorsal views; (B, C, E, and F) lateral views. (C and F) Magnification (×2.5) of the specimens from panels B and E. In wild-type embryos, the Hoxd4 staining extended to a boundary localized between rhombomeres r6 and r7. In the Af9−/− embryos, a posterior shift of the anterior expression limit, corresponding to approximately one rhombomere, was observed. Ov, otic vesicle.
DISCUSSION
Af9 may be an upstream master regulator of genes involved in embryo patterning.
The skeletal abnormalities exhibited by Af9 null mutant mice suggest a role in embryo patterning. The transformations of homeotic character observed in individual vertebrae are reminiscent of, but distinct from, phenotypes obtained with aberrant expression of genes such as members of the vertebrate HOX family of homeobox genes (for a review see reference 38). Experimental alteration of Hox gene expression in transgenic or knockout mice (44) or by maternal exposure to retinoic acid (34, 36) results in homeotic transformations of the axial skeleton. For instance, several Hox gene null mutants exhibit an effect that mirrors the Af9 null mutation, namely, the posterior transformation of C7 into T1, complete with a pair of ribs. This effect is observed in Hoxa4, Hoxa5, and Hoxa6 mutants and at low frequency in Hoxd4−/− mutants (24, 25, 31, 37), suggesting that the C7-T1 junction may be particularly susceptible to perturbations in Hox gene expression. The first two vertebrae, the atlas and the axis, are also frequent targets of homeotic transformation following Hox gene deregulation. Loss of either Hoxb4 or Hoxd4 function leads to a partial posterior homeotic transformation of C1 to C2 (25, 54), whereas disruption of Hoxd3 causes anterior transformations of these two vertebrae (11). Interestingly, overexpression of Hoxa7 also results in the atlas and the axis acquiring the characteristics of more-posterior vertebrae, as well as, in the presence of an additional vertebra, the proatlas (35). In wild-type mice, the proatlas anlage is a temporary embryonic structure which subsequently contributes cells to the basioccipital bone, but, following Hoxa7 overexpression, this structure appears to undergo a partial transformation to a vertebral fate.
Unlike the phenotypes observed with aberrant expression of individual Hox genes, the homeotic transformations exhibited by Af9 null mutant mice were spread along the embryo axis from the first cervical vertebra to the last thoracic segment. This suggests that Af9 may play a role as a master regulator, perhaps of Hox genes. Indeed, in many respects the characteristics of Af9−/− mice were found to be remarkably similar to those of mice carrying null mutations of Hox gene regulator Cdx1 (60). Cdx1 expression is established at E7.5 and subsequently regresses in a temporally controlled fashion along the entire embryo (45, 60). Homozygous disruption of Cdx1 leads to axial skeletal abnormalities with anterior homeotic transformation, and in situ hybridization analysis of various Hox genes has demonstrated a posterior shift of their expression domains by one segment in the Cdx1 null mutants (60).
The similarities between the Af9 and Cdx1 mutant phenotypes suggest that they may have a regulatory effect on related groups of target genes, presumably Hox genes. Nonetheless, important differences were observed, and in particular the Af9 phenotype appeared more severe than that of Cdx1. First, disruption of Af9 was lethal shortly after birth, whereas Cdx1−/− mice are viable and fertile. Moreover, the homeotic transformation in the Af9 knockout mice extended throughout the thoracic region, with the last pair of ribs missing, whereas no morphological changes are apparent posterior to T9 in Cdx1−/− animals. In addition, while Cdx1 mutants exhibit an incomplete first vertebra, partly fused to the basioccipital bone, the atlases of Af9 mutants showed a grossly deformed, duplicated structure. This was presumably due either to a fusion of the atlas with a persisting proatlas or to a duplication of the developing C1 structure in embryogenesis.
In view of the homeotic transformation phenotype of Af9 null mutant mice it seems likely that Af9 plays a role in the regulation of genes involved in the complex processes of embryo patterning. Comparison with the effects observed following aberrant expression of Hox genes suggests that Af9 may affect the activity of these genes, directly or indirectly. In particular, striking parallels between the Af9 phenotype and the effects of null mutations of Hox gene members of the paralogous group 4 (notably Hoxa4, Hoxb4, and Hoxd4) suggested a possible effect on the expression of these genes (24, 25, 37, 54). Indeed, the in situ hybridization experiments illustrated in Fig. 7 demonstrated a posterior shift in the anterior expression limits of Hoxd4 in E9.5 Af9−/− embryos. This effect is similar to that observed with expression in Cdx1 null mutant embryos (60) and confirms that Af9 affects Hox gene expression, directly or indirectly.
A link between Af9 and Mll genes, embryo patterning, and leukemia.
AF9 is a common partner of MLL in AML. Interestingly, the skeletal phenotype of the Af9 knockout mouse is reminiscent of that observed in mice with a disruption of the Mll gene (69), indicating that both genes play a crucial role in embryo-patterning processes. Homozygous Mll inactivation is lethal by E11; however, Mll+/− mice are viable, though they exhibit growth retardation and hypofertility. Heterozygous Mll mutants are haploinsufficient and display segment abnormalities, with disordered identity observed in the cervical, thoracic, and lumbar regions. Anterior transformations are present in the cervical and upper thoracic regions, while posterior transformations are observed in the lower thoracic, lumbar, and sacral regions. All specimens lack either a thoracic or a lumbar vertebra, and, furthermore, C7 has C6 characteristics and the spinous process characteristic of T2 is shifted to T3 or is present on both vertebrae. These effects presumably reflect the caudal shifts observed in the anterior expression boundaries of Hox genes in Mll+/− embryos. Interestingly, Hoxa7 and Hoxc9 transcripts cannot be detected in homozygous Mll null mutant embryos, despite the presence of structures normally exhibiting Hox expression, such as somites, spinal ganglia, limb buds, and the neural tube (69).
There are further apparent links between human embryo development and cancer, such as the rapidly expanding evidence for the involvement of Hox genes in the regulation of hematopoiesis and leukemia (4, 40, 58). Experimental dysregulation of Hox gene expression can lead to altered characteristics of blood cells or disturbance of blood cell development. For example, overexpression of Hoxa7 and Hoxa9 following retroviral gene transfer leads to myeloid leukemia in mice (49). Moreover, in cases of early childhood cancer, including AML and lymphoblastic leukemia, the incidence of a cervical rib (a posterior homeotic transformation of a cervical vertebra toward a thoracic-type vertebra) appears to be increased to about 25%, compared to approximately 2% in the general human population (57). This intriguing observation poses some important questions about the link between embryo-patterning processes and cancer and about the status of genes such as AF9 in children with leukemia.
The molecular mechanism by which the MLL-AF9 fusion causes leukemia is obscure. The association of the C-terminal portion of the AF9 protein with N-terminal MLL sequences may target the fusion protein to sites not normally affected by AF9 through the DNA-binding properties of MLL, creating a novel Hox gene regulator. The induction of a chimeric MLL-AF9 protein in 32Dcl3 cells inhibits the up-regulation of Hoxa7, Hoxb7, and Hoxc9 normally observed in the presence of granulocyte colony-stimulating factor when it substitutes for interleukin 3 (32). Similarly, MLL-ENL has been shown to transactivate the promoter of Hoxa7, an effect dependent on the DNA-binding properties of MLL and the C-terminal portion of ENL (56). Moreover, a recent study has revealed that the AF9 and ENL portions of MLL-AF9 and MLL-ENL, respectively, recruit HPC3, a member of the human Polycomb group of proteins (18), a group which regulates Hox gene expression and axial skeleton development. Further, the expression of an Mll-AF9 fusion gene in mice disrupts myelopoiesis (15). These findings suggest that the two related fusion proteins, MLL-AF9 and MLL-ENL, may affect hematopoietic cell differentiation and proliferation through dysregulation of Hox genes, thereby leading to leukemogenesis.
Acknowledgments
E.C.C. was a recipient of an LRF Gordon Pillar Studentship, and A.A. is the recipient of a Lady Tata Fellowship. This work was supported by the Medical Research Council.
We are indebted to Rob Krumlauf for invaluable help and advice with the in situ hybridization technique, for the Hox probes, and for his advice on embryo patterning. We also thank John O'Brien and Bill Wisden for interpretation of lacZ expression patterns. We thank Karen Douglas and Andrew Smith for pBS-TAG3/IRESlacZ/lox/MC1neoPA/lox, Andrew McKenzie for PGKCre-pA, Vasanta Subramanian for the Cdx-1 probe, and Alan Ashworth for Cdx4 cDNA sequences.
REFERENCES
- 1.Ayton, P. M., and M. L. Cleary. 2001. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene Rev. 20:5695-5707. [DOI] [PubMed] [Google Scholar]
- 2.Breen, T. R., and P. J. Harte. 1993. Trithorax regulates multiple homeotic genes in the bithorax and Antennapedia complexes and exerts different tissue-specific, parasegment-specific and promoter-specific effects on each. Development 117:119-134. [DOI] [PubMed] [Google Scholar]
- 3.Broeker, P. L., H. G. Super, M. J. Thirman, H. Pomykala, Y. Yonebayashi, S. Tanabe, N. Zeleznik-Le, and J. D. Rowley. 1996. Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood 87:1912-1922. [PubMed] [Google Scholar]
- 4.Buske, C., and R. K. Humphries. 2000. Homeobox genes in leukemogenesis. Int. J. Hematol. 71:301-308. [PubMed] [Google Scholar]
- 5.Cairns, B. R., N. L. Henry, and R. D. Kornberg. 1996. TFG3/TAF30/ANC1, a component of the yeast SW1/SNF complex that is similar to the leukemogenic proteins ENL and AF-9. Mol. Cell. Biol. 16:3308-3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cairns, B. R., Y. J. Kim, M. H. Sayre, B. C. Laurent, and R. D. Kornberg. 1994. A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5, and SNF6 gene products isolated from yeast. Proc. Natl. Acad. Sci. USA 91:1950-1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlson, M., and B. C. Laurent. 1994. The SNF/SWI family of global transcriptional activators. Curr. Opin. Cell Biol. 6:396-402. [DOI] [PubMed] [Google Scholar]
- 8.Cimino, C., M. C. Rapanotti, A. Biondi, L. Elia, F. Lo Coco, C. Price, V. Rossi, A. Rivolta, E. Canaani, C. M. Croce, F. Mandelli, and M. Greaves. 1997. Infant acute leukemias show the same biased distribution of ALL1 gene breaks as topoisomerase II related secondary acute leukemias. Cancer Res. 57:2879-2883. [PubMed] [Google Scholar]
- 9.Cleary, M. L. 1991. Oncogenic conversion of transcription factors by chromosomal translocations. Cell 66:619-622. [DOI] [PubMed] [Google Scholar]
- 10.Collins, E. C., and T. H. Rabbitts. 2002. The promiscuous MLL gene links chromosomal translocations to cellular differentiation and tumour tropism. Trends Mol. Med. 8:436-442. [DOI] [PubMed]
- 11.Condie, B. G., and M. R. Capecchi. 1993. Mice homozygous for a targeted disruption of Hoxd-3 (Hox-4.1) exhibit anterior transformations of the first and second cervical vertebrae, the atlas and the axis. Development 119:579-595. [DOI] [PubMed] [Google Scholar]
- 12.Corral, J., I. Lavenir, H. Impey, A. J. Warren, A. Forster, T. A. Larson, S. Bell, A. N. J. McKenzie, G. King, and T. H. Rabbitts. 1996. An Mll-Af9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell 85:853-861. [DOI] [PubMed] [Google Scholar]
- 13.Cote, J., J. Quinn, J. L. Workman, and C. L. Peterson. 1994. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science 265:53-60. [DOI] [PubMed] [Google Scholar]
- 14.Djabali, M., L. Selleri, P. Parry, M. Bower, B. D. Young, and G. A. Evans. 1992. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 2:113-118. [DOI] [PubMed] [Google Scholar]
- 15.Dobson, C. L., A. J. Warren, R. Pannell, A. Forster, I. Lavenir, J. Corral, A. J. H. Smith, and T. H. Rabbitts. 1999. The Mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 18:3564-3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domer, P. H., S. S. Fakharzadeh, C. S. Chen, J. Jockel, L. Johansen, G. A. Silverman, J. H. Kersey, and S. J. Korsmeyer. 1993. Acute mixed-lineage t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc. Natl. Acad. Sci. USA 90:7884-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fidanza, V., P. Melotti, T. Yano, T. Nakamura, A. Bradley, E. Canaani, B. Calabretta, and C. M. Croce. 1996. Double knockout of the ALL-1 gene blocks hematopoietic differentiation in vitro. Cancer Res. 56:1179-1183. [PubMed] [Google Scholar]
- 18.Garcia-Cuellar, M. P., O. Ziller, S. A. Schreiner, M. Birke, T. M. Winkler, and T. M. Slany. 2001. The ENL moiety of the childhood leukemia-associated MLL-ENL oncoprotein recruits human Polycomb 3. Oncogene 20:411-419. [DOI] [PubMed] [Google Scholar]
- 19.Gaunt, S. J., R. Krumlauf, and D. Duboule. 1989. Mouse homeo-genes within a subfamily Hox-1.4, -2.6 and -5.1 display similar anteroposterior domains of expression in the embryo but show stage- and tissue-dependent differences in their regulation. Development 107:131-141. [DOI] [PubMed] [Google Scholar]
- 20.Gu, Y., T. Nakamura, H. Alder, R. Prasad, O. Canaani, G. Cimino, C. M. Croce, and E. Canaani. 1992. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 71:701-708. [DOI] [PubMed] [Google Scholar]
- 21.Hess, J. L., B. D. Yu, B. Li, R. Hanson, and S. J. Korsmeyer. 1997. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood 90:1799-1806. [PubMed] [Google Scholar]
- 22.Hoess, R. H., M. Ziese, and N. Sternberg. 1982. P1 site-specific recombination: nucleotide sequence of the recombining sites. Proc. Natl. Acad. Sci. USA 79:3398-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogan, B., R. Beddington, F. Costantini, and E. Lacy. 1994. Manipulating the mouse embryo: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 24.Horan, G. S., K. Wu, D. J. Wolgemuth, and R. R. Behringer. 1994. Homeotic transformation of cervical vertebrae in Hoxa-4 mutant mice. Proc. Natl. Acad. Sci. USA 91:12644-12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horan, G. S. B., E. N. Kovacs, R. R. Behringer, and M. S. Featherstone. 1995. Mutations in paralogous Hox genes result in overlapping homeotic transformations of the axial skeleton: evidence for unique and redundant function. Dev. Biol. 169:359-372. [DOI] [PubMed] [Google Scholar]
- 26.Horn, J. M., and A. Ashworth. 1995. A member of the caudal family of homeobox genes maps to the X-inactivation centre region of the mouse and human X chromosomes. Hum. Mol. Genet. 4:1041-1047. [DOI] [PubMed] [Google Scholar]
- 27.Hunt, P., M. Gulisano, M. Cook, M. H. Sham, A. Faiella, D. Wilkinson, E. Bonicelli, and R. Krumlauf. 1991. A distinct Hox code for the branchial region of the vertebrate head. Nature 353:861-864. [DOI] [PubMed] [Google Scholar]
- 28.Huret, J. L., P. Dessen, and A. Bernheim. 2001. An atlas of chromosomes in hematological malignancies. Example: 11q23 and MLL. Leukaemia 15:987-999. [DOI] [PubMed] [Google Scholar]
- 29.Iida, S., M. Seto, K. Yamamoto, H. Komatsu, A. Tojo, S. Asano, N. Kamada, Y. Ariyoshi, T. Takahashi, and R. Ueda. 1993. MLLT3 gene on 9p22 in t(9;11) leukemia encodes a serine/proline rich protein homologous to MLLT1 on 19p13. Oncogene 8:3085-3092. [PubMed] [Google Scholar]
- 30.Isnard, P., N. Core, P. Naquet, and M. Djabali. 2000. Altered lymphoid development in mice deficient for the mAF4 proto-oncogene. Blood 96:705-710. [PubMed] [Google Scholar]
- 31.Jeannotte, L., M. Lemieux, J. Charron, F. Poirier, and E. J. Robertson. 1993. Specification of axial identity in the mouse: role of the Hoxa-5 (Hox1.3) gene. Genes Dev. 7:2085-2096. [DOI] [PubMed] [Google Scholar]
- 32.Joh, T., Y. Hosokawa, R. Suzuki, T. Takahashi, and M. Seto. 1999. Establishment of an inducible expression system of chimeric MLL-LTG9 protein and inhibition of Hox a7, Hox b7 and Hox c9 expression by MLL-LTG9 in 32Dcl3 cells. Oncogene 18:1125-1130. [DOI] [PubMed] [Google Scholar]
- 33.Kaneko, Y., N. Maseki, N. Takasaki, M. Sakurai, Y. Hayashi, S. Nakazawa, T. Mori, M. Sakurai, T. Takeda, T. Shikano, and Y. Hiyoshi. 1986. Clinical and hematologic characteristics in acute leukemia with 11q23 translocations. Blood 67:484-491. [PubMed] [Google Scholar]
- 34.Kessel, M. 1992. Respecification of vertebral identities by retinoic acid. Development 115:487-501. [DOI] [PubMed] [Google Scholar]
- 35.Kessel, M., R. Balling, and P. Gruss. 1990. Variations of cervical vertebrae after expression of a Hox-1.1 transgene in mice. Cell 61:301-308. [DOI] [PubMed] [Google Scholar]
- 36.Kessel, M., and P. Gruss. 1991. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell 67:89-104. [DOI] [PubMed] [Google Scholar]
- 37.Kostic, D., and M. R. Capecchi. 1994. Targeted disruptions of the murine Hoxa-4 and Hoxa-6 genes result in homeotic transformations of components of the vertebral column. Mech. Dev. 46:231-247. [DOI] [PubMed] [Google Scholar]
- 38.Krumlauf, R. 1994. Hox genes in vertebrate development. Cell 78:191-201. [DOI] [PubMed] [Google Scholar]
- 39.Lavau, C., S. J. Szilvassy, R. Slany, and M. L. Cleary. 1997. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 16:4226-4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence, H. J., G. Sauvageau, R. K. Humphries, and C. Largman. 1996. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells 14:281-291. [DOI] [PubMed] [Google Scholar]
- 41.LeFranc, M.-P., A. Forster, R. Baer, M. A. Stinson, and T. H. Rabbitts. 1986. Diversity and rearrangement of the human T cell rearranging γ genes: nine germ-line variable genes belonging to two subgroups. Cell 45:237-246. [DOI] [PubMed] [Google Scholar]
- 42.Look, A. T. 1997. Oncogenic transcription factors in the human acute leukemias. Science 278:1059-1065. [DOI] [PubMed] [Google Scholar]
- 43.Mazo, A. M., D.-H. Huang, B. A. Mozer, and I. B. Dawid. 1990. The trithorax gene, a trans-acting regulator of the bithorax complex in Drosophila, encodes a protein with zinc-finger domains. Proc. Natl. Acad. Sci. USA 87:2112-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGinnis, W., and R. Krumlauf. 1992. Homeobox genes and axial patterning. Cell 68:283-302. [DOI] [PubMed] [Google Scholar]
- 45.Meyer, B. I., and P. Gruss. 1993. Mouse Cdx-1 expression during gastrulation. Development 117:191-203. [DOI] [PubMed] [Google Scholar]
- 46.Morrison, A., L. Ariza-McNaughton, A. Gould, M. Featherstone, and R. Krumlauf. 1997. Hoxd4 and regulation of the group 4 paralogous genes. Development 124:3135-3146. [DOI] [PubMed] [Google Scholar]
- 47.Morrissey, J., D. C. Tkachuk, A. Milatovitch, U. Francke, M. Link, and M. L. Cleary. 1993. A serine/proline-rich protein is fused to HRX in t(4;11) acute leukemias. Blood 81:1124-1131. [PubMed] [Google Scholar]
- 48.Nakamura, T., H. Alder, Y. Gu, R. Prasad, O. Canaani, N. Kamada, R. P. Gale, B. Lange, W. M. Crist, P. C. Nowell, C. M. Croce, and E. Canaani. 1993. Genes on chromosome 4, 9 and 19 involved in 11q23 abnormalities in acute leukemia share homology and/or common motifs. Proc. Natl. Acad. Sci. USA 90:4631-4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura, T., D. A. Largaespada, J. D. Shaughnessy, Jr., N. A. Jenkins, and N. G. Copeland. 1996. Cooperative activation of Hoxa and Pbx1-related genes in murine myeloid leukaemias. Nat. Genet. 12:149-153. [DOI] [PubMed] [Google Scholar]
- 50.Peterson, C. L., A. Dingwall, and M. P. Scott. 1994. Five SWI/SNF gene products are components of a large multisubunit complex required for transcriptional enhancement. Proc. Natl. Acad. Sci. USA 91:2905-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pui, C. H., J. R. Kane, and W. M. Crist. 1995. Biology and treatment of infant leukemias. Leukemia 9:762-769. [PubMed] [Google Scholar]
- 52.Rabbitts, T. H. 1994. Chromosomal translocations in human cancer. Nature 372:143-149. [DOI] [PubMed] [Google Scholar]
- 53.Rabbitts, T. H. 1991. Translocations, master genes, and differences between the origins of acute and chronic leukemias. Cell 67:641-644. [DOI] [PubMed] [Google Scholar]
- 54.Ramirez-Solis, R., H. Zheng, J. Whiting, R. Krumlauf, and A. Bradley. 1993. Hoxb-4 (Hox-2.6) mutant mice show homeotic transformation of a cervical vertebra and defects in the closure of the sternal rudiments. Cell 73:279-294. [DOI] [PubMed] [Google Scholar]
- 55.Rubnitz, J. E., J. Morrissey, P. A. Savage, and M. L. Cleary. 1994. ENL, the gene fused with HRX in t(11;19) leukemias, encodes a nuclear protein with transcriptional activation potential in lymphoid and myeloid cells. Blood 84:1747-1752. [PubMed] [Google Scholar]
- 56.Schreiner, S. A., M. P. Garcia-Cuellar, G. H. Fey, and R. K. Slany. 1999. The leukemogenic fusion of MLL with ENL creates a novel transcriptional transactivator. Leukemia 13:1525-1533. [DOI] [PubMed] [Google Scholar]
- 57.Schumacher, R., A. Mai, and P. Gutjahr. 1992. Association of rib anomalies and malignancy in childhood. Eur. J. Pediatr. 151:432-434. [DOI] [PubMed] [Google Scholar]
- 58.Shen, W. F., K. Detmer, C. H. Mathews, F. M. Hack, D. A. Morgan, C. Largman, and H. J. Lawrence. 1992. Modulation of homeobox gene expression alters the phenotype of human hematopoietic cell lines. EMBO J. 11:983-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slany, R. K., C. Lavau, and M. L. Cleary. 1998. The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol. Cell. Biol. 18:122-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramanian, V., B. I. Meyer, and P. Gruss. 1995. Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell 83:641-653. [DOI] [PubMed] [Google Scholar]
- 61.Super, H. J., N. R. McCabe, M. J. Thirman, R. A. Larson, M. M. Le Beau, J. Pedersen-Bjergaard, P. Philip, M. O. Diaz, and J. D. Rowley. 1993. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 82:3705-3711. [PubMed] [Google Scholar]
- 62.Thomas, K. R., and M. R. Capecchi. 1987. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51:503-512. [DOI] [PubMed] [Google Scholar]
- 63.Tkachuk, D. C., S. Kohler, and M. L. Cleary. 1992. Involvement of a homolog of Drosophila Trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 71:691-700. [DOI] [PubMed] [Google Scholar]
- 64.Warren, A. J., W. H. Colledge, M. B. L. Carlton, M. J. Evans, A. J. H. Smith, and T. H. Rabbitts. 1994. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell 78:45-58. [DOI] [PubMed] [Google Scholar]
- 65.Welch, M. D., and D. G. Drubin. 1994. A nuclear protein with sequence similarity to proteins implicated in human acute leukemias is important for cellular morphogenesis and actin cytoskeletal function in Saccharomyces cerevisiae. Mol. Biol. Cell 5:617-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilkinson, D. G., and M. A. Nieto. 1993. Detection of messenger RNA by in situ hybridisation to tissue sections and whole mounts. Methods Enzymol. 225:361-373. [DOI] [PubMed] [Google Scholar]
- 67.Yagi, H., K. Deguchi, A. Aono, Y. Tani, T. Kishimoto, and T. Komori. 1998. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood 92:108-117. [PubMed] [Google Scholar]
- 68.Yamamoto, K., M. Seto, Y. Akao, S. Iida, S. Nakazawa, M. Oshimura, T. Takahashi, and R. Ueda. 1993. Gene rearrangement and truncated mRNA in cell lines with 11q23 translocation. Oncogene 8:479-485. [PubMed] [Google Scholar]
- 69.Yu, B. D., J. L. Hess, S. E. Horning, G. A. J. Brown, and S. J. Korsmeyer. 1995. Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378:505-508. [DOI] [PubMed] [Google Scholar]
- 70.Ziemin-van der Poel, S., N. R. McCabe, H. J. Gill, R. Espinosa, Y. Patel, A. Harden, P. Rubinelli, S. D. Smith, M. M. LeBeau, J. D. Rowley, and M. O. Diaz. 1991. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 88:10735-10739. [DOI] [PMC free article] [PubMed] [Google Scholar]