

Abstract
Several misfolding diseases commence when a secreted folded protein encounters a partially denaturing microenvironment, enabling its self assembly into amyloid. Although amyloidosis is modulated by numerous environmental and genetic factors, single point mutations within the amyloidogenic protein can dramatically influence disease phenotype. Mutations that destabilize the native state predispose an individual to disease; however, thermodynamic stability alone does not reliably predict disease severity. Here we show that the rate of transthyretin (TTR) tetramer dissociation required for amyloid formation is strongly influenced by mutation (V30M, L55P, T119M, V122I), with rapid rates exacerbating and slow rates reducing amyloidogenicity. Although these rates are difficult to predict a priori, they notably influence disease penetrance and age of onset. L55P TTR exhibits severe pathology because the tetramer both dissociates quickly and is highly destabilized. Even though V30M and L55P TTR are similarly destabilized, the V30M disease phenotype is milder because V30M dissociates more slowly, even slower than wild type (WT). Although WT and V122I TTR have nearly equivalent tetramer stabilities, V122I cardiomyopathy, unlike WT cardiomyopathy, has nearly complete penetrance—presumably because of its 2-fold increase in dissociation rate. We show that the T119M homotetramer exhibits kinetic stabilization and therefore dissociates exceedingly slowly, likely explaining how it functions to protect V30M/T119M compound heterozygotes from disease. An understanding of how mutations influence both the kinetics and thermodynamics of misfolding allows us to rationalize the phenotypic diversity of amyloid diseases, especially when considered in concert with other genetic and environmental data.
Amyloid diseases are a large group of an even larger collection of misfolding disorders, the former including greater than 80 familial transthyretin (TTR)-based pathologies (1–11). The TTR missense mutations associated with familial amyloid disease display a wide range of diversity in age of disease onset, penetrance, etc. In diseases resembling and including the TTR amyloidoses, normally folded secreted proteins must first undergo partial denaturation to assemble into amyloid (7–10, 12). Although amyloidosis is modulated by numerous environmental and genetic factors (13), it is known that mutations that destabilize the native state predispose an individual to disease (7, 8, 14). However, thermodynamic stability alone does not reliably predict disease severity (15).
Tetramer dissociation is required for TTR amyloid fibril formation (12, 16, 17). However, the resulting normally folded monomer cannot form amyloid without undergoing partial denaturation (12, 16, 17), yielding the so-called monomeric amyloidogenic intermediate composed of a three-stranded antiparallel β-sheet structure (18). Herein we use chaotropic denaturation studies in an attempt to understand energetic differences between single site variants of TTR, all of which are tetrameric under physiological conditions. These studies demonstrate that kinetic and thermodynamic data, considered together, nicely rationalize why certain mutations lead to severe pathology, whereas others protect against disease or lead to mild pathology.
Methods
Variant TTR Production and Purification.
Recombinant WT TTR and variants thereof were expressed in BL21/DE3 Epicurian gold Escherichia coli (Stratagene) transformed with pmmHα plasmid containing the TTR and ampicillin-resistance genes. Expression and purification were performed as described in detail previously (19). Recombinant expression of WT, L55P, V30M, V122I, and T119M TTR homotetramers in E. coli at 37°C each provide 30–50 mg/liter of purified native tetrameric protein.
TTR Fibril Formation Kinetics.
TTR was buffer exchanged into 10 mM phosphate buffer (pH 7.2) containing 100 mM KCl, 1 mM EDTA, and 1 mM DTT. Solutions of TTR (0.40 mg/ml) were mixed with an equal volume of 200 mM acetate buffer (pH 4.3) containing 100 mM KCl, 1 mM EDTA to yield a final pH of 4.4. The samples were incubated at 37°C without stirring. The turbidity at 400 nm was measured as a function of time up to 360 h. The self-assembly reaction was also followed by thioflavin T (ThT) binding as described previously (20). In fibril formation facilitated by MeOH-mediated dielectric constant lowering, TTR was first buffer-exchanged into 50 mM Tris⋅HCl (pH 7.0) containing 100 mM KCl, 1 mM EDTA, and 1 mM DTT (Tris buffer). Two hundred microliters of this TTR solution (1.5 mg/ml) was then added to 2.8 ml of a methanol/Tris buffer solution at 25°C with constant stirring to yield TTR (0.10 mg/ml) solvated in 50% (vol/vol) MeOH in Tris buffer (pH 7.0). The turbidity at 330 and 400 nm was continuously monitored over the course of 3,600 s. The rate of fibril formation was evaluated by the initial slope of the turbidity at 400 nm (Fig. 4b).
Figure 4.

TTR fibril formation time courses detected by turbidity at 400 nm (OD400). (a) Amyloid fibril formation mediated by partial acid denaturation of TTR (0.20 mg/ml) in acetate buffer (pH 4.4, 37°C, Fig. 2 symbols apply). (b) TTR (0.10 mg/ml) amyloid fibril formation enabled by MeOH-induced denaturation [50% (vol/vol) in Tris buffer (pH 7.0, 25°C)].
Urea-Mediated TTR Dissociation Measured by Resveratrol Binding.
Resveratrol displays a large increase in its fluorescence quantum yield and a blue shift on binding to tetrameric TTR but does not bind to the TTR monomer (X.J., P.H., and A. Sawkar, unpublished results). Resveratrol-binding curves to quantify the concentration of TTR tetramer were recorded for each TTR variant (0–0.12 mg/ml; 0–2.18 μMtetramer) by using 18 μM resveratrol. The fluorescence intensity at 394 nm (I394) was plotted versus the concentration of TTR, exhibiting a linear fit as expected. Standard curves are provided in Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org. The resveratrol probe was added to TTR subjected to urea denaturation just before the fluorescence measurement in order not to shift the equilibrium toward tetramer significantly. Fortunately, this ligand does not noticeably perturb the reversible concentration-dependent tetramer/monomer equilibrium in urea based on quantification of the tetramer using crosslinking studies (concentration dependence and reversibility data are unpublished). Resveratrol fluorescence was recorded from 350 to 550 nm after excitation at 320 nm. Samples containing TTR (0.10 mg/ml/1.8 μMtetramer) were incubated as a function of urea concentration, buffered with 50 mM phosphate (pH 7.0) containing 100 mM KCl, 1 mM EDTA, and 1 mM DTT. After incubation (96 h), 3.6 μl of resveratrol from a 2.5 mM stock solution in DMSO was added to a 500-μl protein sample just before the measurement yielding a final concentration of 18 μM resveratrol.
Urea Unfolding of TTR Measured by Tryptophan Fluorescence.
Samples containing TTR (0.10 mg/ml) were incubated (25°C) in varying concentrations of urea buffered with 50 mM phosphate (pH 7.0) containing 100 mM KCl, 1 mM EDTA, and 1 mM DTT. Tryptophan fluorescence spectra were recorded between 310 and 410 nm with excitation at 295 nm. Equilibrium data were recorded after a 96-h incubation period sufficient to reach equilibrium in all cases except T119M. The fluorescence ratio at 355 and 335 nm was used as a structural probe as described previously (21).
Kinetics of Monomer Unfolding and Tetramer Dissociation as a Function of Urea.
Unfolding of monomeric TTR (M-TTR; F87M/L110M) (17) was monitored by stopped-flow fluorescence (355 nm, 4°C) using a 1:10 dilution of M-TTR (50 μM) in 50 mM phosphate buffer containing 100 mM KCl, 1 mM EDTA, and 1 mM DTT into 5.55 M urea (Aviv Associates, Lakewood, NJ) ATF-105 spectrometer]. The unfolding rate of tetrameric WT TTR was also measured at 4°C [which is faster than at 25°C, due to lower stability (21)]. Because the monomer unfolds five to six orders of magnitude faster than tetrameric TTR dissociates, the rate-determining dissociation step was measured by purposefully linking the quaternary structural changes to tertiary structural changes (measured by tryptophan fluorescence) mediated by urea concentrations in the unfolding posttransition region. Time courses (25°C) were recorded for WT, V30M, L55P, and V122I TTR (27 s = dead time for manual mixing) up to 250–300 h. The final data points for the T119M variant were recorded after 480 h. No burst phase was observed for any TTR sequence. The kinetic data fit well to a single exponential function: I355/335 = I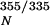 + A(1−e−kdisst), where I
+ A(1−e−kdisst), where I is the native protein fluorescence intensity ratio (355/335 nm), A is the amplitude difference, kdiss is the tetramer dissociation rate constant, and t is time in hours.
is the native protein fluorescence intensity ratio (355/335 nm), A is the amplitude difference, kdiss is the tetramer dissociation rate constant, and t is time in hours.
TTR Reassembly Kinetics.
WT and T119M TTR were unfolded by incubation in 8 M urea at 4°C for 7 days [TTR is destabilized at low temperatures, facilitating denaturation (21)]. The proteins were confirmed to be unfolded by Trp fluorescence and the lack of resveratrol binding. The reassembly reaction was initiated by diluting unfolded TTR 10-fold (to 1.8 μMtetramer) with phosphate buffer to achieve the desired final urea concentration in the presence of 18 μM resveratrol. Resveratrol fluorescence was monitored at 390 nm after excitation at 320 nm as a function of time. The TTR reassembly reaction was biphasic and the data best fit to a double exponential function. Under these conditions, the amplitudes of the two phases were equal for WT and depended on the urea concentration in the case of T119M. The final yield of reassembly for both variants is 90%, demonstrating that the process is reversible.
Results and Discussion
Relative Thermodynamic Stability of the TTR Variants.
In this study, we used TTR tetramers composed of identical monomer subunits to elucidate the effects of human mutations (Fig. 1) on thermodynamic stability, rate of tetramer dissociation, and rate of amyloid fibril formation. TTR tetramers do not denature in urea; hence dissociation to monomers is required for urea-induced tertiary structural changes detected by tryptophan fluorescence (Fig. 2b) (15, 21).
Figure 1.
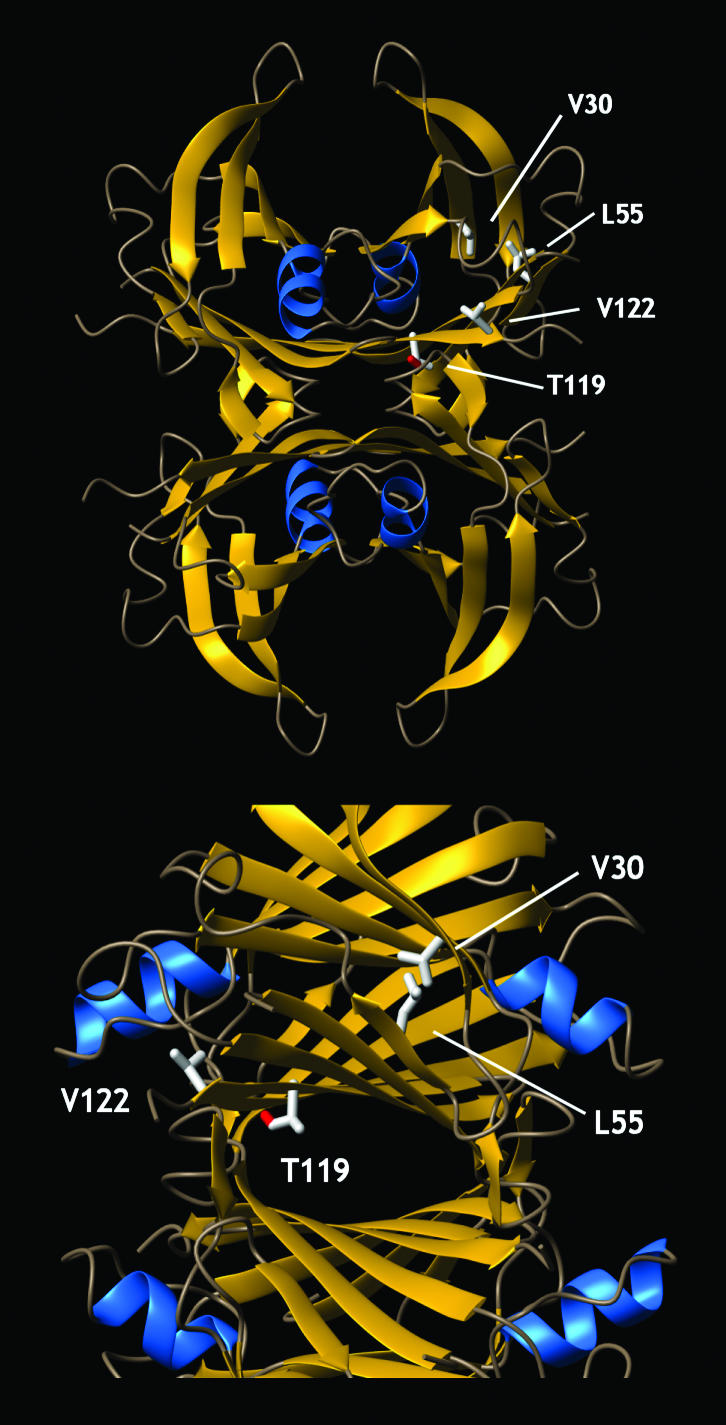
The structure of tetrameric TTR, showing the location of the point mutations that change the kinetics and/or thermodynamics of partial denaturation and influence amyloidogenicity and disease phenotype (mutations identified in only one subunit). The V30M TTR mutation is located in the hydrophobic core, whereas the L55P substitution is located in β-strand D. The V122I TTR mutation is close to the C terminus and the dimer/dimer interface region of TTR. T119M contributes to the dimer/dimer interface and comprises the thyroxine-binding sites. The T119M TTR mutation appears to stabilize the quarternary structure and dramatically slows the rate of tetramer dissociation, enabling it to function as a transsuppressor of misfolding (20), apparently by increasing the hydrophobic surface area buried at the dimer/dimer interface (34).
Figure 2.

Evaluation of the stability of TTR sequences as a function of urea concentration. WT, filled triangles; V30M, open squares; L55P, filled circles; V122I, open circles; T119M, open triangles. (a) Tetramer dissociation curve measured by resveratrol binding (96-h incubation). (b) Tertiary structure unfolding curve measured by intrinsic tryptophan fluorescence changes.
In principle, TTR quaternary and tertiary structural changes can be unlinked by conducting biophysical experiments at low TTR concentrations. Under these conditions, the tetramer-folded monomer equilibrium is concentration dependent, whereas the folded–unfolded monomer equilibrium should be concentration independent. Over the physiologically relevant range of concentrations studied thus far (0.1−0.5 mg/ml), the unfolding transitions detected by Trp fluorescence changes exhibit a TTR concentration dependence, strongly suggesting that the quaternary and tertiary structural transitions are linked for V30M, L55P, WT, V122I, and T119M TTR. Further evidence that these transitions are linked includes the nearly coincident quaternary and tertiary structural transitions mediated by increasing urea concentrations (Fig. 7, which is published as supporting information on the PNAS web site).
The fraction of TTR tetramer retained as a function of urea concentration (quaternary structural transition) was evaluated by using the small molecule resveratrol, which fluoresces when bound to at least one of the two thyroid-binding sites in the tetramer (Fig. 2a). The dissociation of TTR into monomeric subunits is not associated with intrinsic fluorescence or circular dichroism changes; therefore, ligand binding was used to probe the tetramer–monomer equilibrium (17). Although it is reasonable to hypothesize that this method may overestimate the fraction tetramer as a function of denaturant by shifting the equilibrium toward tetramer (Le Chatelier's principle), this was not noticeable when the resveratrol data were compared with analytical ultracentrifugation and glutaraldehyde crosslinking data (unpublished data). The dissociation curves depicted in Fig. 2a require a 96-h incubation period due to the high kinetic barrier associated with tetramer dissociation (see below). Trp fluorescence as a function of urea concentration is used to follow TTR tertiary structural changes (unfolding; Fig. 2b).
The tetramers composed exclusively of mutated subunits associated with disease are clearly less stable than WT based on their denaturation midpoints for dissociation or unfolding. Furthermore, all of the TTR variants have similar sensitivity to urea concentration, allowing direct comparisons. The stability of V30M and L55P is very similar and lower than WT, which is similar to V122I. The T119M suppressor tetramer appears to be the most stable (Fig. 2a); however, predictions about relative thermodynamic stability have to be made with caution because of its extremely slow dissociation rate (it does not reach equilibrium within 96 h). An identical rank ordering of stabilities is exhibited by comparing unfolding transitions evaluated by Trp fluorescence (Fig. 2b), consistent with linked tetramer dissociation and unfolding equilibria.
The Influence of Mutations on TTR Tetramer Dissociation Kinetics.
It is perplexing that the disease-associated variants L55P and V30M, which are similarly destabilized relative to WT TTR, exhibit dramatically different clinical features, including disease penetrance and age of onset (22, 23). To understand this discrepancy, we explored the influence of mutations on the rate of tetramer dissociation and amyloid fibril formation. Given that the WT tetramer cannot undergo tertiary structural changes before it dissociates, one can detect the rate of quaternary structural changes by linking tetramer dissociation to TTR tertiary structural changes, provided that the tertiary structural changes are much faster. The rate of monomeric TTR (17) denaturation (t1/2 = 69 ms; in 5.0 M urea, 4°C, Fig. 3a Inset) is ≈5 × 105-fold faster than the dissociation rate of WT TTR (t1/2 = 9.6 h; in 5.0 M urea, 4°C), demonstrating this to be the case. Using urea concentrations in the posttransition region for tertiary structural changes directly links the slow TTR quaternary structural changes to the rapid tertiary structural changes and renders unfolding irreversible. Dissociation time courses for the TTR variants described above fit well to a single exponential over a range of urea concentrations. Representative 6.0 M urea dissociation time courses are depicted in Fig. 3a.
Figure 3.

TTR tetramer dissociation rates as a function of urea concentration (Fig. 2 symbols apply). (a) Unfolding time course measured by tryptophan fluorescence provides the rate of TTR tetramer dissociation in 6.0 M urea. Inset shows the rapid unfolding of monomeric TTR in 5.0 M urea measured by stopped-flow fluorescence. (b) The logarithm of the rate of tetramer dissociation, lnkdiss (kdiss in h−1) plotted as a function of urea concentration. The lnkdiss vs. urea concentration plot is linear, allowing extrapolation to 0 M urea.
Tetramer Dissociation Kinetics.
It is clear that single amino acid changes in the TTR sequence can have a significant and not easily predicted influence on the rate of tetramer dissociation (Table 1). The logarithm of the rate constant (lnkdiss) varies linearly with urea concentration (Fig. 3b), allowing extrapolations to more physiological conditions (0 M urea). The dissociation half life (t1/2) of WT TTR is 42 h (0 M urea, 25°C). The most pathogenic familial amyloid polyneuropathy variant (L55P) dissociates 10-fold faster (t1/2 = 4.4 h), whereas the V122I cardiac variant dissociates 2.2-fold faster than WT under identical conditions. Similar relative rates are exhibited under denaturing conditions (Fig. 3b). The L55P and V122I mutations lower the activation barrier for dissociation apparently by destabilizing the tetrameric ground state more than the transition state associated with dissociation. Amyloidogenic lysozyme mutations also destabilize the structure and increase the rate of denaturation relative to WT, suggesting that the rate of denaturation could be generally important in amyloidoses (24, 25). Furthermore, it has been shown that thermodynamic destabilization of the prion protein by familial point mutations is unlikely to be the determinant for disease phenotype (26).
Table 1.
TTR homotetramer dissociation rates derived from time courses as a function of urea concentration extrapolated to 0 M urea concentration
| Sequence | kdiss*, h−1 | mkin*, M−1 |
|---|---|---|
| WT | 1.68 ± 0.15⋅10−2 | 0.094 ± 0.013 |
| V30M | 1.02 ± 0.37⋅10−2 | 0.121 ± 0.066 |
| L55P | 15.7 ± 0.58⋅10−2 | 0.179 ± 0.008 |
| V122I | 3.64 ± 0.38⋅10−2 | 0.100 ± 0.02 |
| T119M | 4.52 ± 1.18⋅10−4 | 0.303 ± 0.03 |
kdiss is the tetramer dissociation rate constant, and mkin is the urea dependence of the tetramer dissociation rate constant.
In stark contrast to the disease-associated TTR sequences, the T119M TTR variant exhibits an exceedingly slow tetramer dissociation rate (t1/2 of 1,534 h), demonstrating that this suppressor protects against amyloid disease by effectively precluding tetramer dissociation on a biologically relevant time scale. Hence, T119M misfolding is prevented by kinetic stabilization, because the activation barrier for dissociation is insurmountable (27, 28). The very slow dissociation rate exhibited by the T119M suppressor homotetramer is consistent with previous experiments that were unable to detect dissociation by subunit exchange (20, 29). The V30M mutant dissociates slightly slower than WT TTR (Fig. 3b), demonstrating that there are mutations that destabilize TTR significantly without increasing its dissociation rate (apparently V30M nearly equally destabilizes the ground and transition states of dissociation or changes the pathway for denaturation). That the V30M and WT tetramer dissociation rates are similar under physiological conditions is also supported by subunit exchange rates (20, 29). Even though the V30M tetramer is slightly more destabilized than L55P, the V30M disease phenotype is milder because V30M dissociates slowly, even more slowly than WT. For this and related arguments to be relevant to disease, tetramer dissociation rates have to correlate with amyloid formation rates, as demonstrated below.
Fibril Formation Rates Are Predicted by Tetramer Dissociation Rates.
TTR will form amyloid fibrils in vitro under partially denaturing conditions imposed by lowering either the pH (simulating the endocytic pathway) or the dielectric constant of the aqueous medium (12, 20). The mechanism of TTR amyloid fibril formation is not yet fully understood; however, it is clear that it lacks a lag phase and is not seedable (J. White and J.W.K., unpublished data). The initial rates of TTR amyloid fibril formation for all of the variants shown in Fig. 4a (37°C, 0.20 mg/ml of TTR, pH 4.4) fit to single exponentials. This observation and others (15, 17) provide strong evidence that the rate-determining step for TTR amyloid formation is tetramer dissociation. In fact, the fibril formation rates of the TTR variants displayed in Fig. 4a are predictable from the tetramer dissociation rates, suggesting that the relative denaturation energy landscapes in two different denaturants (urea and acid) are similar. The most pathogenic variant, L55P, forms fibrils 9.2-fold faster that WT TTR, similar to the V122I variant, which is 3.6-fold faster than WT (each characterized by 100% disease penetrance), whereas V30M is slightly slower than WT (V30M and WT TTR show incomplete disease penetrance). The T119M suppressor homotetramer forms fibrils 3,000-fold slower than WT, explaining how it likely protects against pathology in compound heterozygotes.
Lowering the dielectric constant of the aqueous medium solvating TTR by adding MeOH (50% vol/vol) also leads to partial denaturation and amyloid fibril formation (20). The fibril formation rate in aqueous MeOH is dramatically faster than that mediated by partial acid denaturation (compare Fig. 4 b to a). Nonetheless, the relative TTR tetramer dissociation rates still predict amyloid fibril formation velocity, implying that tetramer dissociation remains rate limiting. The L55P familial amyloid polyneuropathy (FAP) variant forms fibrils with a relative rate of 3.4, similar to the V122I familial amyloid cardiomyopathy variant (3.1), whereas the rate of the V30M FAP variant (0.92) is similar to WT (1), in stark contrast to T119M (0.048), which forms amyloid 20-fold slower than WT TTR.
The T119M Transsuppressor Exhibits High Kinetic Stability.
The 40-fold slower dissociation rate of the T119M homotetramer relative to WT is consistent with a high kinetic barrier of dissociation (Table 1, Fig. 3). To provide further evidence for an increase in barrier height relative to WT, we monitored the reassembly kinetics of T119M and compared them to WT. The reassembly rate of T119M is 90- or 200-fold slower than WT, depending on which of the two phases are compared (Fig. 5). This demonstrates that the barriers for T119M dissociation and reassociation are both considerably higher than those characterizing WT. The increased T119M barriers lead to a tetramer exhibiting very high kinetic stability (27, 30) under amyloidogenic conditions. Preventing tetramer dissociation by increasing the kinetic barrier should be a very effective strategy to confer stability on a protein that can adopt a lower free energy amyloid state under denaturing conditions (28). The transsuppressor efficacy exhibited by inclusion of T119M subunits in V30M/T119M hybrid tetramers reported previously (20) is likely to be mediated by barrier height tuning, although this has not been directly demonstrated.
Figure 5.

The kinetics of WT (filled triangles) and T119M TTR (open triangles) reconstitution (folding and reassembly) monitored by resveratrol fluorescence (monitors tetramer formation). A 10-fold dilution of urea unfolded TTR (8 M) to a final TTR concentration of 1.8 μMtetramer initiates reconstitution (final urea concentration = 1.0 M). Inset shows the complete trace for the very slow T119M reassembly time course. The rate constants (extrapolated to 0 M urea) for the fast phase are 0.294 ± 0.014 s−1 (WT) and 3.16⋅10−3 ± 1.65⋅10−3⋅s−1 (T119M). For the slow phase, the rate constants are 6.24⋅10−2 ± 0.62⋅10−2⋅s−1 (WT) and 2.76⋅10−4 ± 2.3⋅10−4⋅s−1 (T119M). Therefore, the reassembly rate of T119M (extrapolated to 0 M urea) is 93-fold slower (fast phase) and 226-fold slower (slow phase) relative to WT.
Sequence-Dependent Energetics Significantly Contribute to Amyloid Disease Diversity.
The sequence-dependent variation in TTR tetramer dissociation rates (rate-determining step for amyloid fibril formation), when considered in combination with the apparent thermodynamic stability of the tetramer (as judged by susceptibility to urea denaturation, a valid comparison due to the similarity in the m values presented by the denaturation curves in Fig. 2 a and b), nicely rationalizes the clinical data associated with the five TTR sequences studied herein. Although thermodynamic stability dictates whether amyloid formation is possible in a given denaturing environment, the rate at which the tetramer dissociates to the partially unfolded monomeric amyloidogenic intermediate governs the rate of fibril formation and therefore contributes significantly to disease severity. L55P TTR exhibits severe pathology, because the tetramer both dissociates rapidly and is highly destabilized, explaining why this mutation confers 100% disease penetrance with the earliest age of disease onset (15–25 years). Even though the V30M tetramer is slightly more destabilized than L55P, the disease phenotype is milder because V30M dissociates even more slowly than WT. Therefore, the V30M monomeric amyloidogenic intermediate cannot form to the extent prescribed by thermodynamics because the slow tetramer dissociation rate limits its steady-state concentration—suggesting why penetrance of V30M disease can be as low as 2% (23, 31). Even though the V122I tetramer is similar in stability to WT TTR, the tetramer dissociates >2-fold faster than WT, causing cardiac amyloid disease with near 100% penetrance, unlike the <25% penetrance exhibited by WT cardiac disease (32). Compound heterozygotes expressing both T119M and V30M TTR develop a mild late-onset pathology—if at all (20). This may be explained by extending the slow dissociation rates exhibited by the T119M homotetramers studied within to the mixed V30M/T119M tetramers showing dramatically lower amyloidogenicity in vitro (20). The amyloidogenicity of TTR and disease phenotype is also likely influenced by the stoichiometry of TTR-binding partners and other factors whose concentrations are dictated by genetic background (13).
The 80 different TTR familial amyloid disease mutations coupled with the availability of clinical data will allow us to further scrutinize the hypothesis that it is necessary to consider both thermodynamics and the kinetics of partial denaturation to rationalize the spectrum of amyloid disease phenotypes. Interestingly, disease-associated mutations are also proposed to accelerate the conversion of the monomeric prion protein to a misfolded state possibly associated with prion pathology by analogous alterations in the free energy landscape (26, 33). From the data now available, it is clear that TTR mutations influencing amyloidogenicity can change thermodynamic stability without significant changes in the rate of partial denaturation or vice versa. Alternatively, mutations can change both the thermodynamics and the kinetics of partial denaturation. Mutations that destabilize the structure and increase the rate of monomer accumulation lead to severe amyloid diseases (e.g., L55P). Single amino acid sequence changes that increase the denaturation rate without significant structural destabilization (e.g., V122I) lead to highly penetrant diseases of intermediate severity. Mutations that destabilize without increasing the denaturation rate (e.g., V30M) lead to diseases with incomplete penetrance and intermediate severity, whereas mutations that apparently stabilize the homotetramer and slow the rate of partial denaturation (e.g., T119M) protect against amyloid disease onset in the context of compound heterozygotes.
How and where amyloid fibrils form in a human being are not yet established. That many amyloidogenic proteins and peptides form fibrils under acidic conditions suggests this may be an intracellular process, perhaps occurring within an organelle such as a lysosome. Despite significant effort, however, we have not been able to demonstrate intracellular TTR amyloidosis. The slow tetramer dissociation process required for amyloid fibril formation revealed within may provide some clues regarding how and where amyloidosis occurs in a human. The time scale of dissociation may be surprising if one assumes that the rate-limiting step of TTR amyloidosis is the formation of a high-energy multimeric intermediate referred to as a nucleus. Nucleated polymerizations are common for amyloidogenic peptides; however, TTR amyloidogenesis is different and very efficient in that it proceeds by a downhill polymerization mechanism not requiring high-energy multimeric nucleus formation (J. White and J.W.K., unpublished data). The monomeric misfolded form of TTR appears to be polymerization competent, consistent with the inability of TTR amyloidosis to be accelerated by seeding with preformed fibrils or protofilaments (fibril precursors). The slow dissociation process and the efficiency of TTR amyloidosis may provide useful constraints for discerning how and where amyloid fibrils form in mammals.
Supplementary Material
Acknowledgments
We thank Ted Foss for the preparation of Fig. 1. Support from the National Institutes of Health (NIH) (DK 46335), the Skaggs Institute of Chemical Biology, and the Lita Annenberg Hazen Foundation is appreciated. Postdoctoral fellowships to P.H. from the Wenner–Gren Foundation and to A.R.H. from NIH National Research Service Award (AG00080) are also valued.
Abbreviation
- TTR
transthyretin
Footnotes
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Self-Perpetuating Structural States in Biology, Disease, and Genetics,” held March 22–24, 2002, at the National Academy of Sciences in Washington, DC.
References
- 1.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 2. Dobson, C. M. (1999) Trends Biochem. Sci. 329–332. [DOI] [PubMed]
- 3.Goldberg M S, Lansbury P T., Jr Nat Cell Biol. 2000;2:E115–E119. doi: 10.1038/35017124. [DOI] [PubMed] [Google Scholar]
- 4.Fink A L. Folding Des. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 5.Uemichi T. Rinsho Kagaku (Nippon Rinsho Kagakkai) 1997;26:74–87. [Google Scholar]
- 6.Saraiva M J M. Hum Mutat. 1995;5:191–196. doi: 10.1002/humu.1380050302. [DOI] [PubMed] [Google Scholar]
- 7.Booth D R, Sunde M, Bellotti V, Robinson C V, Hutchinson W L, Fraser P E, Hawkins P N, Dobson C M, Radford S E, Blake C C F, Pepys M B. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 8.Hurle M R, Helms L R, Li L, Chan W, Wetzel R. Proc Natl Acad Sci USA. 1994;91:5446–5450. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McParland V, Kad N, Kalverda A, Brown A, Kirwin-Jones P, Hunter M, Sunde M, Radford S. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- 10.Chiti F, Mangione P, Andreola A, Giorgetti S, Stefani M, Dobson C, Bellotti V, Taddei N. J Mol Biol. 2001;307:379–391. doi: 10.1006/jmbi.2000.4478. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson D R, Pastore R D, Yaghoubian R, Kane I, Gallo G, Buck F S, Buxbaum J N. New Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 12.Colon W, Kelly J W. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 13.White J T, Kelly J W. Proc Natl Acad Sci USA. 2001;98:13019–13024. doi: 10.1073/pnas.241406698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCutchen S L, Lai Z, Miroy G, Kelly J W, Colon W. Biochemistry. 1995;34:13527–13536. doi: 10.1021/bi00041a032. [DOI] [PubMed] [Google Scholar]
- 15.Jiang X, Buxbaum J N, Kelly J W. Proc Natl Acad Sci USA. 2001;98:14943–14948. doi: 10.1073/pnas.261419998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai Z, Colon W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 17.Jiang X, Smith C S, Petrassi H M, Hammarström P, White J T, Sacchettini J C, Kelly J W. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 18.Liu K, Cho H S, Lashuel H A, Kelly J W, Wemmer D E. Nat Struct Biol. 2000;7:754–757. doi: 10.1038/78980. [DOI] [PubMed] [Google Scholar]
- 19.Lashuel H A, Wurth C, Woo L, Kelly J W. Biochemistry. 1999;38:13560–13573. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- 20.Hammarström P, Schneider F, Kelly J W. Science. 2001;293:2459–2461. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 21.Hammarström P, Jiang X, Deechongkit S, Kelly J W. Biochemistry. 2001;40:11453–11459. doi: 10.1021/bi010673+. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson D R, Buxbaum J N. Adv Hum Genet. 1991;20:69–123. doi: 10.1007/978-1-4684-5958-6_2. [DOI] [PubMed] [Google Scholar]
- 23.Coelho T. Curr Opin Neurol. 1996;9:355–359. [PubMed] [Google Scholar]
- 24.Canet D, Sunde M, Last A M, Miranker A, Spencer A, Robinson C V, Dobson C M. Biochemistry. 1999;38:6419–6427. doi: 10.1021/bi983037t. [DOI] [PubMed] [Google Scholar]
- 25.Takano K, Funahashi J, Yutani K. Eur J Biochem. 2001;268:155–159. doi: 10.1046/j.1432-1327.2001.01863.x. [DOI] [PubMed] [Google Scholar]
- 26.Liemann S, Glockshuber R. Biochemistry. 1999;38:3258–3267. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- 27.Baker D, Sohl J L, Agard D A. Nature (London) 1992;356:263–266. doi: 10.1038/356263a0. [DOI] [PubMed] [Google Scholar]
- 28.Jaswal S S, Sohl J L, Davis J H, Agard D A. Nature (London) 2002;415:343–346. doi: 10.1038/415343a. [DOI] [PubMed] [Google Scholar]
- 29.Schneider F, Hammarström P, Kelly J W. Protein Sci. 2001;10:1606–1613. doi: 10.1110/ps.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sohl J L, Jaswal S S, Agard D A. Nature (London) 1998;395:817–819. doi: 10.1038/27470. [DOI] [PubMed] [Google Scholar]
- 31.Holmgren G, Costa P M, Andersson C, Asplund K, Steen L, Bechman L, Nylander P O, Teixeira A, Saraiva M J, Costa P P. J Med Genet. 1994;31:351–354. doi: 10.1136/jmg.31.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westermark P, Sletten K, Johansson B, Cornwell G G. Proc Natl Acad Sci USA. 1990;87:2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baskakov I V, Legname G, Prusiner S B, Cohen F E. J Biol Chem. 2001;276:19687–19690. doi: 10.1074/jbc.C100180200. [DOI] [PubMed] [Google Scholar]
- 34.Sebastiao M P, Lamzin V, Saraiva M J, Damas A M. J Mol Biol. 2001;306:733–744. doi: 10.1006/jmbi.2000.4415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.