

Abstract
Background
The functions, actions, and regulation of tissue metabolism affected by the consumption of long chain polyunsaturated fatty acids (LC-PUFA) from fish oil and other sources remain poorly understood; particularly how LC-PUFAs affect transcription of genes involved in regulating metabolism. In the present work, mice were fed diets containing fish oil rich in eicosapentaenoic acid and docosahexaenoic acid, fungal oil rich in arachidonic acid, or the combination of both. Liver and hippocampus tissue were then analyzed through a combined gene expression- and lipid- profiling strategy in order to annotate the molecular functions and targets of dietary LC-PUFA.
Results
Using microarray technology, 329 and 356 dietary regulated transcripts were identified in the liver and hippocampus, respectively. All genes selected as differentially expressed were grouped by expression patterns through a combined k-means/hierarchical clustering approach, and annotated using gene ontology classifications. In the liver, groups of genes were linked to the transcription factors PPARα, HNFα, and SREBP-1; transcription factors known to control lipid metabolism. The pattern of differentially regulated genes, further supported with quantitative lipid profiling, suggested that the experimental diets increased hepatic β-oxidation and gluconeogenesis while decreasing fatty acid synthesis. Lastly, novel hippocampal gene changes were identified.
Conclusions
Examining the broad transcriptional effects of LC-PUFAs confirmed previously identified PUFA-mediated gene expression changes and identified novel gene targets. Gene expression profiling displayed a complex and diverse gene pattern underlying the biological response to dietary LC-PUFAs. The results of the studied dietary changes highlighted broad-spectrum effects on the major eukaryotic lipid metabolism transcription factors. Further focused studies, stemming from such transcriptomic data, will need to dissect the transcription factor signaling pathways to fully explain how fish oils and arachidonic acid achieve their specific effects on health.
Keywords: arachidonic acid, docosahexaenoic acid, fatty acid, hippocampus, liver, mice, microarray, PPAR, SREBP, transcription
Background
The presence and abundance of long chain polyunsaturated fatty acids (LC-PUFA) in humans diets have been the basis of scientific investigations for decades. Studies have yet to resolve all the mechanisms of action of LC-PUFA, in particular the remarkable phenotypic differences between populations that consume different quantities of fish and fish oil in their diets [1-5]. Dietary LC-PUFA affect processes including: growth, neurological development, lean and fat mass accretion, reproduction, innate and acquired immunity, infectious pathologies of viruses, bacteria and parasites; and the incidence and severity of virtually all chronic and degenerative diseases including cancer, atherosclerosis, stroke, arthritis, diabetes, osteoporosis, and neurodegenerative, inflammatory, and skin diseases [6-11]. Due to lack of mechanistic knowledge, predictions for optimal intake of nutritionally important LC-PUFA remain vague and dietary recommendations simply relate to prevention of overt deficiency. There is not even scientific consensus on whether LC-PUFA present in human milk should be included in infant formulas [12].
The most often cited action of n3 LC-PUFA is that they induce alterations in eicosanoids by antagonizing the primary substrate of eicosanoid metabolism: arachidonic acid (AA) [13]. Certainly, n3 LC-PUFA replace AA in cellular membranes to some extent and alter eicosanoid products, but is this the primary mechanism underlying the benefits of consuming fish? The n3 LC-PUFA alter postprandial lipoprotein production and clearance, and lower circulating triglyceride-rich lipoproteins [14]. N3 LC-PUFA induce alterations in membrane properties including ion transport, trafficking and vesicular transport, and alter the transcription of genes encoding proteins in lipid biosynthesis, lipid desaturation, and lipid oxidation [15-19].
Fish oil contains, among others, docosahexaenoic acid (DHA; 22:6n3) and eicosapentaenoic acid (EPA; 20:5n3) as important components. The concentration of DHA in the retina and neurological membranes suggests a specific role for DHA in the brain. However, neither the mechanism of uptake nor the precise role of DHA in the brain is currently well understood [20]. Rigorous elimination of DHA and its precursor 18:3n3 from animal diets leads to limited, but significant, phenotypic effects [21]. The basic biochemical pathway of DHA biosynthesis from its precursor 18:3n3 is still being resolved [22]. The EPA component of fish oil is linked to anti-inflammatory, anti-thrombotic, and generally, eicosanoid-mediated effects by antagonizing AA metabolism.
Arachidonic acid (AA; 20:4n6), either formed from n6 precursors in mammals or obtained directly from the diet, is believed to exert most of its biological activity via conversion to various eicosanoids. N6 LC-PUFA, and especially AA, are also now recognized to elicit significant physiological effects when consumed in the diet. The lack of significant commercial sources of AA has limited the study of this fatty acid (FA) as a dietary ingredient until relatively recently. Due to the commercialization of fungal biomass production to produce very high levels of AA as a storage oil, it is now possible to study this FA.
Until recently, LC-PUFA effects on gene transcription were thought primarily to be mediated by a single subfamily of orphan nuclear receptors – peroxisome proliferator activated receptors (PPARs); however, it is now becoming evident that FAs can affect many different genes either via direct interactions or indirectly through additional transcription factors including hepatic nuclear-4α (HNF-4α), nuclear factor κβ (NF-κβ), retinoid X receptor α (RXRα), sterol regulatory element binding protein-1c (SREBP-1c), and liver X receptors (LXRα and LXRβ) [17]. Indeed, the enthusiasm for uncovering the biological pathways underlying the beneficial actions of LC-PUFA has revealed a story that is increasingly more complex than originally supposed [23], thereby making microarray technology an ideal platform to further decipher the many roles of these nutritional lipids.
In the present work, mice were fed control diets adequate in 18:2n6 and 18:3n3 but lacking LC-PUFA; or a diet enriched in fungal oil (FUNG) enriched in AA, the n6 elongation and desaturation product of 18:2n6; or a diet containing fish oil (FISH) enriched in 22:6n3 and 20:5n3, the major n3 LC-PUFA elongation and desaturation products of 18:3n3; or a diet containing both fungal oil and fish oil (FUNG+FISH). Thereafter, gene expression profiling was performed on two organs in parallel with quantitative metabolic profiling of a broad spectrum of liver FAs. Tissues of interest were liver, the major lipid metabolizing tissue; and brain, the major neurological tissue and principal site for accumulation and functionality of DHA (and other fish oil components). The hippocampus brain region was specifically chosen because of its importance in memory and learning [24]. Liver and brain are metabolically connected organs as both AA and DHA cross the blood brain barrier via several possible carriers. Diets were fed for 57 days to study chronic and sustained alterations in control of gene expression rather than acute effects.
Results and discussion
Body weight, feed intake, and incorporation of dietary LC-PUFA into tissue membranes
Mice were fed relatively low fat diets to minimize the accretion of body fat and to avoid the confounding effects that LC-PUFA -rich diets are known to exert on the progression of obesity. Body weight and the change in body weight (d58 minus d1) were not affected by dietary FA modulation (p = 0.17). Feed intake data showed no significant differences amongst groups (classic ANOVA considering the cages and diets as cofactors; p > 0.05). There were also no obvious differences in brain and liver weights (p > 0.05). By these criteria, differences in gene expression and metabolic profiles described herein were judged to be linked to the FAs present in FUNG and FISH oils on healthy, normal weight animals.
The incorporation of 20:4n6 and 22:4n6 following feeding of the AA-rich FUNG diet, and the incorporation of 20:5n3, 22:5n3, and 22:6n3 following feeding of the EPA/DHA-rich FISH diet, into whole brain, hippocampal, and hepatic phospholipid (PL) pools (Table 1) was further evidence the diets were consumed, resulting in significant changes to PL acyl composition. Fish oil feeding is well established to increase n3 LC-PUFA in hepatic PL as well as whole brain PL [18]; whereas deficiency of n3 LC-PUFA can decrease 22:6n-3 in hippocampal PL [25]. DHA present in the brain can be formed from precursors in the liver or astrocytes, and intact DHA passes the blood brain barrier via selective transport mechanisms [26]. These data confirm that diets did result in actual increases in the brain of the appropriate FAs and set the stage for interpreting transcriptional responses observed in each organ.
Table 1.
N6/N3 LC-PUFA ratios [(20:4n6 + 22:4n6)/(20:5n3+22:5n3+22:6n3)] in whole brain, hippocampus, and hepatic phospholipid pools. Typically, the above ratios were highest following feeding of the AA-enriched diet, lowest following feeding of the FISH diet, and intermediary following feeding of the combination diet. Abbreviations: SPN, sphingomyelin; PC, phosphatidylcholine; PS/PI, mixture of phosphatidylserine and phosphatidylinositol; PE, phosphatidylethanolamine; CL, cardiolipin. 20:4, 20:4n6; 22:4, 22:4n6; 20:5, 20:5n3; 22:5, 22:5n3; 22:6, 22:6n3.
| Dietary Group | Tissue | (20:4+22:4)/(20:5+22:5+22:6) ratio in various phospholipids | ||||
| SPN | PC | PS/PI | PE | CL | ||
| Control | Whole Brain | - | 1.31 | 0.53 | 0.75 | 1.18 |
| FUNG | Whole Brain | - | 2.33 | 0.68 | 0.94 | 0.73 |
| FISH | Whole Brain | - | 0.99 | 0.41 | 0.50 | 0.36 |
| FUNG +FISH | Whole Brain | - | 1.20 | 0.50 | 0.67 | 1.08 |
| Control | Hippocampus | - | 2.96 | 0.50 | 0.79 | 1.30 |
| FUNG | Hippocampus | - | 4.18 | 0.67 | 0.96 | 1.09 |
| FISH | Hippocampus | - | 1.86 | 0.44 | 0.51 | 0.76 |
| FUNG +FISH | Hippocampus | - | 2.45 | 0.52 | 0.62 | 0.92 |
| Control | Liver | 0.83 | 2.14 | 8.02 | 2.00 | 0.69 |
| FUNG | Liver | 1.91 | 9.73 | 12.51 | 3.53 | 1.97 |
| FISH | Liver | 0.32 | 0.32 | 1.52 | 0.35 | 0.16 |
| FUNG +FISH | Liver | 0.90 | 1.48 | 2.93 | 0.62 | 0.39 |
Importantly, our lipid profiling data revealed that FUNG and FISH FAs were not identically incorporated into individual PL in the two organs studied (partly shown in Table 1), and this may affect subsequent transcription factor signaling. For example, there was consistently more AA accumulating in various hepatic PL than in the hippocampus following AA feeding; and more DHA accumulated in hepatic cardiolipin (CL) than in hippocampal CL, whereas the reverse was true for phosphatidylserine/phosphatidylinositol (PS/PI). Thus, there are several explanations for why FUNG, FISH, and FUNG+FISH feeding resulted in different transcriptional profiles in liver and hippocampus, as discussed subsequently.
Gene Expression analysis
Differentially expressed genes induced by the dietary treatments were selected using a limit fold change (LFC) model, in addition to a more conventional confidence interval test [27]. Genes that satisfied the 5% LFC model and which lay above the 99.9% confidence interval of pooled replicates were selected as differentially expressed. It should also be noted that for the liver data, inter-individual variation was examined. All genes selected as differentially expressed using pooled data in the liver also laid outside the 99.0% confidence interval when compared to individual mouse variability, thereby lending additional confidence to the selection method used with both the liver and hippocampus data. According to these criteria, which involves examining all possible pair-wise comparisons between the four diets, 329 and 356 differentially expressed genes were selected in the liver and hippocampus, respectively. In the present document, genes that are discussed are followed by parentheses indicating fold changes according to the following convention: FUNG versus control diet, FISH versus control diet, and FUNG+FISH versus control diet, e.g. G6pc (1.1, 1.6*, -1.9*). Those fold changes that are significant by the 5% LFC model are further identified with an asterisk (*). It is important to note that the nature of the LFC model allows differentially expressed genes to be selected based on differences between the treatments, and therefore explains why some genes discussed in the text have no fold changes marked with an asterisk (as significance was observed between treatments and not versus the control diet). All liver genes discussed in the present manuscript are found in Figure 1 (green dots) and lie within the range of validation data (black squares). The high degree of concordance between RT-PCR and microarray data (86%- reported in detail elsewhere [27]) establishes a certain level of confidence in the results discussed throughout this manuscript.
Figure 1.
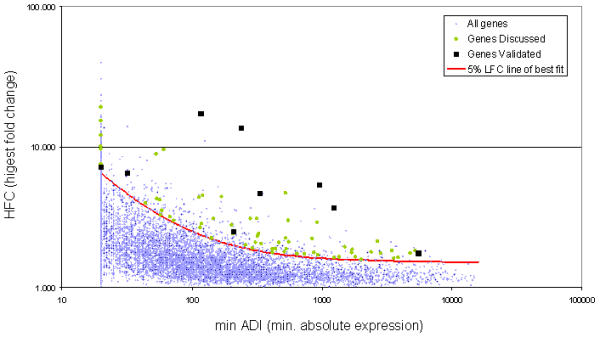
Graphical representation of the trend and spread of the hepatic microarray data. Blue dots indicate the highest fold change for each probe set on the Affymetrix Mu11K chip, where the highest fold change was identified by examining all fold changes in every possible pairwise comparison between diets. Those probe sets that were identified as absent (by Affymetrix software) across all dietary treatments were treated as unchanged and therefore not considered in the limit fold change (LFC) selection model. The trend within this dataset indicates that as absolute expression increases (along the x-axis), fold changes decrease, i.e. the chance of seeing large fold changes with highly expressed genes is minimal. The 5% LFC line of best fit (red line) takes this observed trend into account and identifies the upper 5% of those probe sets with the greatest fold changes within predefined bins (model parameters fully explained in [27]). The 329 probe sets that lie above the 5% LFC line are those genes defined as differentially regulated. Green circles represent the hepatic genes discussed in the current manuscript and black squares represent those genes validated by real time PCR. When examining the spread of the genes discussed and validated, it is apparent that genes were selected across the entire range of absolute values and not concentrated in one area. Furthermore, as the validation data (which had 86% concordance with microarray data; see [27]) lies across the entire range of absolute expression levels, a high degree of confidence is established for the genes discussed within the present manuscript.
To understand the trends in transcriptional response within and between the two organs studied, genes were annotated using GO classifications; a dynamic, structured, yet precisely defined system which characterizes genes based on defined categories of molecular function, biological process, and cellular component [28]. Since the GO classifications contain several thousand descriptors, many differentially expressed genes had a unique annotation. Record counts for most GO identifiers were not high, although a number of significant trends were observable. Venn diagrams were constructed for the top 10 GO descriptors for each category, according to record count, to visualize broad transcriptional responses induced by dietary LC-PUFA (Figure 2). The interpretation of transcriptomic data, especially that stemming from a nutritional study such as this, benefits greatly from such a classification system. The diverse whole-body effects of LC-PUFA render individual gene interpretation difficult; however, considering groups of genes reveals preliminary information concerning the biological mechanisms responding to the addition of dietary LC-PUFA.
Figure 2.
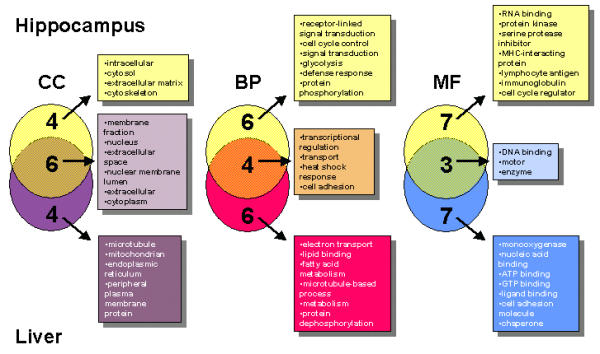
Venn diagrams depicting the top 10 GO functional categories in the hippocampus and liver. Examining the top 10 GO classifications for cellular component (CC), biological process (BP), and molecular function (MF) indicated that functional commonalties (overlapping circles) in response to LC-PUFAs exist between the two organs, implying that molecular responses to dietary LC-PUFA occurs via common biological pathways rather than through identical genes. Additional organ-specific functions are also indicated (individual circles).
The GO analysis and specific gene expression patterns were interpreted to indicate that LC-PUFA-enriched oils affected liver metabolism through the regulation of genes directly involved in FA metabolism and lipid binding/transport (Figure 2), as previously reported in rats [18]. When examining GO classifications for the hippocampus, such descriptors as cell signaling and cell cycle control are seen in response to dietary LC-PUFA. This result was expected, as the two organs have very different biochemical roles and import dietary FA via distinctly different means. The tissue functions were reflected in the transcriptomic profile. Although there were relatively few overlapping functions between the two organs, the most prominent overlapping function indicated that LC-PUFA was affecting genes related to transcription and DNA/RNA-binding.
Hepatic gene expression analysis and transcription factor signatures
It is difficult to interpret transcriptional profiling data alone, since dimerization, post-translational modifications, relative quantities of co-activators and co-repressors, and the formation of multiple transcriptional complexes binding to common promoter regions ultimately determines the binding activation potential of transcription factors. Nevertheless, clustering analysis of differentially regulated genes revealed numerous genes known to be regulated by specific transcription factors, thereby enabling several transcription factor signatures to be established. These signatures then provide clues concerning the various pathways through which FA may effect gene transcription. Additionally, FUNG and FISH diets regulated some transcription factors not (Table 2) previously associated with these dietary oils, thereby indicating novel LC-PUFA mediated molecular mechanisms of action. These include: Spot14 (Thrsp), transcription factor 7 T-cell specific (Tcf7), transcription factor 2 (Tcf2), nuclear transcription factor-Y β(NF-Y β; Nfyb transcript), D site albumin promoter binding protein (Dbp), general transcription factor II (Gtf2I), activating transcription factor 5 (Atf5), Msx-interacting-zinc finger (Miz1), and pre B-cell leukemia transcription factor 1 (Pbx1). In the present work, the focus will be on transcription factors previously identified as mediators of LC-PUFA actions, such as PPARs, SREBP, and HNFs.
Table 2.
Differentially expressed genes in response to dietary LC-PUFAs in (A) hippocampus and (B) liver. Table includes gene categories discussed in the manuscript. Genes are grouped by functional category, sorted alphabetically, and annotated with NCBI LocusLink. Average difference intensity (ADI) represents gene absolute expression, and was used to calculate fold changes. Differentially expressed genes were clustered with a combined k-means/hierarchical approach. Asterisk (*) indicates ADIs significantly different from control diet, using 5% LFC gene selection model equations (LFChippocampus = 1.36 + (90/min ADI); LFCliver = 1.52+(100/min ADI)).
| (A) | |||||||
| Gene Symbol | Description | Unigene |
ADI (Average Difference Intensity) |
Cluster Coordinates | |||
|
Control |
FUNG |
FISH |
FUNG + FISH |
||||
| Nucleic Acid Binding | |||||||
| Ar | androgen receptor | Mm.4470 | 83 | 209* | 136 | 101 | H1-K2 |
| Atoh4 | atonal homolog 4 (Drosophila) | Mm.42017 | 21 | 97 | 74 | 119* | H1-K2 |
| Btf3 | basic transcription factor 3 | Mm.1538 | 992 | 522* | 475* | 877 | H5-K5 |
| Dbp | D site albumin promoter binding protein | Mm.3459 | 111 | 206 | 84 | 154 | H1-K2 |
| Ddb1 | damage specific DNA binding protein 1 | Mm.29623 | 239 | 299 | 242 | 139 | H4-K4 |
| Dscr1 | Down syndrome critical region homolog 1 (human) | Mm.56 | 304 | 258 | 138* | 130* | H5-K3 |
| E2f3 | E2F transcription factor 3 | Mm.6333 | 110 | 55 | 214 | 80 | H3-K1 |
| Elk1 | ELK1, member of ETS oncogene family | Mm.3064 | 613 | 425 | 690 | 402 | H3-K1 |
| Hdac1 (Rpd3) | high homology to histone deacetylase 1 | Mm.2602 | 393 | 231 | 411 | 353 | H3-K1 |
| Hoxd12 | homeo box D12 | Mm.57124 | 436 | 368 | 403 | 234* | H4-K4 |
| Idb1 | inhibitor of DNA binding 1 | Mm.444 | 148 | 169 | 344* | 238 | H3-K1 |
| Idb3 | inhibitor of DNA binding 3 | Mm.110 | 41 | 117 | 156* | 138 | H1-K2 |
| Nhlh1 | nescient helix loop helix 1 | Mm.2474 | 111 | 198 | 79 | 132 | H1-K2 |
| Pax9 | paired box gene 9 | Mm.5035 | 104 | 20 | 165 | 77 | H3-K1 |
| Pcbp3 | poly(rC) binding protein 3 | Mm.143816 | 187 | 180 | 52* | 184 | H5-K5 |
| Purb | purine rich element binding protein B | Mm.154651 | 391 | 313 | 172* | 246 | H5-K3 |
| Lipoprotein Metabolism | |||||||
| Apod | apolipoprotein D | Mm.2082 | 325 | 423 | 575* | 456 | H3-K1 |
| Miscellaneous Genes | |||||||
| Htr4 | 5 hydroxytryptamine (serotonin) receptor 4 (5-HT) | Mm.20440 | 20 | 130* | 76 | 89 | H1-K2 |
| Pomc | proopiomelanocortin | Mm.21878 | 51 | 304* | 80 | 71 | H1-K2 |
| Prkcd | protein kinase C, delta (PKC) | Mm.2314 | 461 | 297 | 353 | 263* | H5-K3 |
| Siat8e | sialyltransferase 8 (alpha 2, 8 sialytransferase) E | Mm.5173 | 128 | 207 | 49 | 20* | H5-K4 |
| Ttr | transthyretin | Mm.2108 | 304 | 490 | 157* | 687* | H1-K5 |
| (B) | |||||||
| Gene Symbol | Description | Unigene |
ADI (Average Difference Intensity) |
Cluster Coordinates | |||
|
Control |
FUNG |
FISH |
FUNG + FISH |
||||
| Nucleic Acid Binding | |||||||
| Atf4 | activating transcription factor 4 | Mm.641 | 1122 | 910 | 1045 | 647 | H1-K4 |
| Atf5 | activating transcription factor 5 | Mm.1566 | 870 | 505* | 563* | 281* | H3-K3 |
| Dbp | D site albumin promoter binding protein | Mm.3459 | 169 | 581* | 174 | 747* | H5-K5 |
| Eef1a1 | eukaryotic translation elongation factor 1 alpha 1 | Mm.196614 | 1517 | 845* | 1877 | 1642 | H1-K1 |
| Eif4a1 | eukaryotic translation initiation factor 4A1 | Mm.12858 | 125 | 180 | 68 | 218 | H5-K5 |
| Ercc2 | excision repair cross-complementing rodent repair deficiency, complementation group 2 | Mm.56990 | 148 | 142 | 319* | 298* | H1-K1 |
| Gtf2i | general transcription factor II I | Mm.22593 | 89 | 47 | 150 | 20 | H1-K1 |
| HNF-3g | forkhead box A3 (Foxa3) | Mm.42260 | 411 | 501 | 454 | 207* | H2-K4 |
| Hoxd12 | homeo box D12 | Mm.57124 | 127 | 177 | 85 | 44 | H2-K4 |
| Hzf-pending | hematopoietic zinc finger | Mm.14099 | 235 | 328 | 516* | 173 | H1-K4 |
| Klf13 | Kruppel-like factor 13 | Mm.41170 | 257 | 455 | 381 | 515* | H4-K2 |
| Miz1 | Msx-interacting-zinc finger | Mm.6370 | 283 | 453 | 429 | 146 | H2-K4 |
| Nfyb | nuclear transcription factor-Y beta (NF-Y) | Mm.3259 | 121 | 219 | 73 | 195 | H4-K2 |
| Pbx1 | pre B-cell leukemia transcription factor 1 | Mm.61526 | 1587 | 1039 | 1328 | 933* | H3-K3 |
| Rpo2tc1 | RNA polymerase II transcriptional coactivator | Mm.41746 | 461 | 560 | 368 | 292 | H2-K4 |
| Srebp-1 | ethanol induced 6 (sterol regulatory element binding protein-1); Etohi6 | Mm.30133 | 648 | 610 | 310* | 370* | H3-K3 |
| Tceb1l | transcription elongation factor B (SIII), polypeptide 1 (15 kDa),-like | Mm.42944 | 420 | 549 | 672* | 744* | H5-K5 |
| Tcf2 | transcription factor 2 | Mm.7226 | 151 | 121 | 244 | 20* | H1-K4 |
| Tcf7 | transcription factor 7, T-cell specific | Mm.31630 | 212 | 237 | 203 | 568* | H5-K5 |
| Tcfe3 | EST highly similar to transcription factor E3 | Mm.25762 | 114 | 135 | 203 | 20 | H1-K4 |
| Thrsp; Spot14 | thyroid hormone responsive SPOT14 homolog (Rattus) | Mm.28585 | 2481 | 2947 | 2353 | 1505* | H2-K4 |
| Usf2 | upstream transcription factor 2 | Mm.15781 | 180 | 209 | 32* | 191 | H5-K2 |
| Fatty Acid Synthesis | |||||||
| Acas1 | acetyl-Coenzyme A synthetase 1 (AMP forming) | Mm.22719 | 497 | 200* | 297* | 113* | H3-K3 |
| Acly | ATP citrate lyase (ACL) | N/A | 2513 | 939* | 1117* | 530* | H3-K3 |
| Acly | ATP citrate lyase (ACL) | N/A | 3197 | 1568* | 1754* | 878* | H3-K3 |
| Acly | ATP citrate lyase (ACL) | N/A | 456 | 115* | 64* | 51* | H3-K3 |
| Acly | ATP citrate lyase (ACL) | N/A | 603 | 278* | 252* | 163* | H3-K3 |
| Elovl2; Ssc2 | elongation of very long chain fatty acids (FEN1/Elo2, SUR4/Elo3, yeast)-like 2 | Mm.2567 | 3010 | 2291 | 1780* | 1690* | H3-K3 |
| Elovl3; Cig30 | elongation of very long chain fatty acids (FEN1/Elo2, SUR4/Elo3, yeast)-like 3 | Mm.21806 | 517 | 421 | 983* | 317 | H1-K1 |
| Facl2 | fatty acid Coenzyme A ligase, long chain 2 | Mm.28962 | 514 | 709 | 677 | 926* | H5-K5 |
| Fasn | fatty acid synthase (FAS) | Mm.3760 | 5214 | 2715* | 4104 | 966* | H1-K5 |
| Scd1 | stearoyl-Coenzyme A desaturase 1 | Mm.140785 | 9783 | 8749 | 9493 | 5541* | H1-K4 |
| Fatty Acid Oxidation | |||||||
| Acadm | acetyl-Coenzyme A dehydrogenase, medium chain (MCAD) | Mm.10530 | 1364 | 2014 | 1774 | 2261* | H4-K2 |
| Aldh1a1 | aldehyde dehydrogenase family 1, subfamily A1 | Mm.4514 | 2387 | 3917* | 4274* | 4091* | H4-K2 |
| Aldh1a7 | aldehyde dehydrogenase family 1, subfamily A7 | Mm.14609 | 1717 | 1977 | 3208* | 2264 | H1-K1 |
| Cpt1a | carnitine palmitoyltransferase 1, liver | Mm.18522 | 734 | 1285* | 1058 | 1272* | H4-K2 |
| Cpt2 | carnitine palmitoyltransferase 2 | Mm.29499 | 418 | 673 | 791* | 690* | H4-K1 |
| Crat | carnitine acetyltransferase | Mm.20396 | 58 | 82 | 190* | 71 | H1-K1 |
| Ech1 | enoyl coenzyme A hydratase 1 (delta 3,5-delta 2,4-dienoyl-CoA isomerase) | Mm.2112 | 1239 | 1388 | 2031* | 1731 | H1-K1 |
| Carbohydrate Metabolism | |||||||
| Camk2b | calcium/calmodulin-dependent protein kinase II, beta | Mm.4857 | 152 | 139 | 20* | 84 | H3-K3 |
| Cam-Pde 1c | calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1c | N/A | 98 | 61 | 75 | 584* | H5-K5 |
| G6pc | glucose-6-phosphatase, catalytic (G6Pase) | Mm.18064 | 1065 | 1224 | 1669* | 553* | H1-K4 |
| Got1 | glutamate oxaloacetate transaminase 1, soluble | Mm.19039 | 976 | 798 | 656 | 467* | H3-K3 |
| Gpam | glycerol-3-phosphate acyltransferase, mitochondrial (GPAT) | Mm.87773 | 1184 | 683* | 989 | 526* | H3-K4 |
| Hspa5 | heat shock 70kD protein 5 (glucose-regulated protein, 78kD) | Mm.918 | 2802 | 1996 | 3014 | 1820* | H1-K4 |
| Pck1 | phosphoenolpyruvate carboxykinase 1, cytosolic (PEPCK) | Mm.42246 | 1376 | 2913* | 2198* | 2369* | H4-K2 |
| Pck1 | phosphoenolpyruvate carboxykinase 1, cytosolic (PEPCK) | Mm.42246 | 964 | 1550 | 1352 | 1652* | H4-K2 |
| Pck1 | phosphoenolpyruvate carboxykinase 1, cytosolic (PEPCK) | Mm.42246 | 243 | 647* | 452* | 633* | H4-K2 |
| Pck1 | phosphoenolpyruvate carboxykinase 1, cytosolic (PEPCK) | Mm.42246 | 320 | 660* | 564* | 744* | H4-K2 |
| Cytochrome P450 Metabolism | |||||||
| Cyp1a2 | cytochrome P450, 1a2, aromatic compound inducible | Mm.15537 | 3952 | 5021 | 2839* | 4187 | H2-K2 |
| Cyp2a4 | cytochrome P450, 2a4 | Mm.14781 | 922 | 2682* | 1100 | 2362* | H4-K2 |
| Cyp2b9 | cytochrome P450, 2b9, phenobarbitol inducible type a | Mm.876 | 57 | 161 | 241* | 146 | H4-K1 |
| Cyp3a11 | cytochrome P450, steroid inducible 3a11 | Mm.21193 | 4777 | 7297 | 8429* | 6542 | H4-K1 |
| Cyp3a16 | cytochrome P450, 3a16 | Mm.30303 | 1438 | 2805* | 2835* | 2282* | H4-K2 |
| Cyp4a10 | cytochrome P450, 4a10 | Mm.10742 | 335 | 894* | 1566* | 1010* | H4-K1 |
| Cyp4a14 | cytochrome P450, 4a14 | Mm.7459 | 20 | 160* | 385* | 51 | H1-K1 |
| Cypf13 | cytochrome P450 CYP4F13 | Mm.22045 | 600 | 834 | 529 | 439 | H2-K2 |
| Por | P450 (cytochrome) oxidoreductase | Mm.3863 | 54 | 208* | 119 | 232* | H4-K2 |
| Transport | |||||||
| Abca1 | ATP-binding cassette, sub-family A (ABC1) member 1 | Mm.369 | 550 | 121* | 217* | 270* | H3-K3 |
| Abca7 | ATP-binding cassette, sub-family A (ABC1) member 7 | Mm.103351 | 20 | 20 | 20 | 144* | H5-K5 |
| Abce1 | ATP-binding cassette, sub-family E (OABP)member 1 | Mm.5831 | 118 | 116 | 310* | 124 | H1-K1 |
| Cd36 | CD36 antigen | Mm.18628 | 247 | 223 | 591* | 170 | H1-K1 |
| Fabp5 | fatty acid binding protein 5, epidermal (E-FABP) | Mm.741 | 3236 | 593* | 384* | 238* | H3-K3 |
| Slc10a1 | solute carrier family 10 (sodium/bile acid cotransporter family), member 1 (Ntcp) | Mm.104295 | 845 | 877 | 534* | 1005 | H5-K5 |
| Sterol Metabolism | |||||||
| Fpps | farnesyl pyrophosphate synthase | Mm.39472 | 794 | 327* | 510* | 421* | H3-K3 |
| Hmgcs2 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2, mitochondrial (HMG-CoA2) | Mm.10633 | 3592 | 4491 | 4590 | 5822* | H5-K5 |
| Lipoprotein Metabolism | |||||||
| Apoa4 | apolipoprotein A-IV | Mm.4533 | 2011 | 781* | 629* | 117* | H3-K3 |
| Apoa4 | apolipoprotein A-IV precursor | Mm.4533 | 194 | 60* | 46* | 20* | H3-K3 |
| ApoB-100 | EST highly similar to apolipoprotein B-100 precursor | Mm.29123 | 2675 | 5710* | 5005* | 5395* | H4-K2 |
| Apoc1 | apolipoprotein CI | Mm.182440 | 5719 | 3465* | 6337 | 5953 | H1-K1 |
| Apoc2 | apolipoprotein CII | Mm.28394 | 3199 | 2511 | 2143* | 2005* | H3-K3 |
| Apoe | apolipoprotein E | Mm.138866 | 6425 | 6438 | 9632* | 5253 | H1-K1 |
Hepatic peroxisome proliferator-activated receptors (PPARs)
PPARs are nuclear hormone receptors that are activated by micromolar concentrations of various FAs and FA analogues, such as fibrates and thiazolidinediones [29]. This subfamily can be divided into three isotypes, designated PPARα, PPARδ and PPARγ, each with tissue-specific expression. PPARs form heterodimers with retinoid X receptor (RXR), and interact with the peroxisome proliferator response element (PPREs) in gene promoters. PPREs are direct repeats (DR) of a hexanucleotide sequence AGGTCA separated by one nucleotide and are therefore referred to as a DR-1 response element [30]. PPARα and PPARγ play critical roles in the catabolism and storage of FA, whereas the function of PPARδ is less certain. PPARα is the predominant PPAR subtype expressed in liver, as confirmed presently. The other two subtypes were not likely expressed in the mouse liver, as negative values were obtained with the Affymetrix software. PPARα is reported to regulate genes involved in FA transport, synthesis and oxidation, glucose and lipid metabolism, ketogenesis and Δ5, Δ6, and Δ9-desaturation [17].
In spite of dramatic differences in the expression of a variety of PPAR-responsive genes, PPAR gene expression itself was quite constant in all mice analyzed, as previously reported [31]. This constancy of PPAR expression indicates that differences in the transcription of genes containing a PPRE are likely due to the direct activation of PPARs. Feeding our diets affected various genes containing a PPRE, with roles in triacylglycerol synthesis (mitochondrial glycerol-3-phosphate acyltransferase, Gpam), FA oxidation (carnitine palmitoyl transferase, Cpt1; dienoyl CoA isomerase, Ech1), FA desaturation (stearoyl-coenzyme A desaturase, Scd1 or Δ9 desaturase), gluconeogenesis (cytosolic phosphoenolpyruvate carboxykinase 1, Pck1 or PEPCK; glucose-6-phosphatase, G6pc), cytochrome p450 metabolism (Cyp4a-10 and -14), FA transport (Cd36; FA binding protein, Fabp5), cholesterol metabolism and transport (apolipoprotein A4, Apoa4; apolipoprotein B-100, ApoB-100), and ketone body formation (mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2, Hmgcs2).
Various genes involved in cholesterol metabolism (microsomal transfer protein, MTP; apolipoprotein C3, Apoc3; cholesterol 7α hydroxylase, Cyp7a1), carbohydrate metabolism (pyruvate kinase; facilitative glucose transporter gene, GLUT2), and coagulation (vitamin K dependent coagulation factors VII, IX, X) have been previously found to be induced by PPARs [32], but were not transcriptionally regulated in the present experiment.
A recent study examined effects of PPARα-specific induction in murine livers using cDNA microarrays [30]. Despite the different strain of mice examined (C57BL/6J) and the different genomic technology utilized for the examination of differentially expressed genes (cDNA microarrays), several genes were found differentially regulated in both experiments, indicating that the PPAR agonist and LC-PUFA affect gene expression through similar pathways. Genes such as Cd36, Cyp2b9, Hmgcs2, and Ech1 were induced more than 4-fold when Wy-14,643 (a PPARα-specific agonist) was administered to mice for 2 weeks. These transcripts were similarly regulated in the current study, providing further indication that many of the differentially expressed genes were likely PPARα-activated.
Preliminary evidence suggests preferred ligands for PPAR activation are 18 C and 20 C FAs, as FAs shorter than 14 C or longer than 20 C will not fit correctly into the PPAR binding pocket and stabilize the AF-2 helix [17]. The net effect would be that PPAR co-activators are not recruited to relax nucleosomal DNA and allow transcription machinery to interact with various promoter elements. Therefore DHA (22:6:n3) may require retroconversion via β-oxidation to an 18 C or 20 C LC-PUFA prior to activating PPARs [17,33]. Without the use of purified FA it is difficult to distinguish whether the DHA in our fish oil preparation was retroconverted since the fish oil also provided the retroconversion products, 20:5n3 and 22:5n3 (Table 3). Likewise, AA was not provided as a pure FA component making comparisons to the control to detect retroconversion difficult. However, earlier work clearly shows that both DHA and AA can be retroconverted to 2 carbon shortened intermediates.
Table 3.
Ratios of FA with n+2 carbons/FA with n carbons in the diet and various hepatic PL pools. There was some evidence to indicate elongase activity may have been impaired in some cases, as explained in the text. n, refers to the number of carbons in the FA chain length; Eth, Ether; refer to previous Tables for other abbreviations. Groups sharing a superscript in common are not statistically significant, ANOVA, P < 0.05.
| Dietary Group |
16:0/ 14:0 |
18:0/ 16:0 |
20:0/ 18:0 |
22:0/ 20:0 |
24:0/ 22:0 |
18:1n7/ 16:1n7 |
20:1n9/ 18:1n9 |
22:1n9/ 20:1n9 |
24:1n9/ 22:1n9 |
20:2n6/ 18:2n6 |
22:2n6/ 20:2n6 |
20:3n6/ 18:3n6 |
20:3n3/ 18:3n3 |
20:4n3/ 18:4n3 |
22:4n6/ 20:4n6 |
22:5n3/ 20:5n3 |
Eth18:0/ 16:0 |
| Diet ratio | |||||||||||||||||
| Control | 12.60 | 0.29 | 0.06 | 1.57 | 0.32 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | - | 0.00 | - | - | 0.00 | 0.00 | 0.29 |
| FUNG | 13.57 | 0.33 | 0.08 | 0.96 | 4.10 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | - | 1.63 | - | - | 0.01 | 0.00 | 0.33 |
| FISH | 8.95 | 0.29 | 0.09 | 0.91 | 1.14 | 0.00 | 0.01 | 0.44 | 1.75 | 0.01 | - | 0.43 | - | - | 0.05 | 0.37 | 0.29 |
| FUNG+FISH | 9.61 | 0.32 | 0.10 | 0.71 | 4.00 | 0.00 | 0.01 | 0.36 | 2.38 | 0.02 | - | 1.22 | - | - | 0.01 | 0.29 | 0.32 |
| SPN | |||||||||||||||||
| Control | - | 0.26a | 0.43b | 5.99a | 0.47b | 1.02c | - | - | - | - | - | 0.23a | - | - | 0.00a | 0.40a | - |
| FUNG | - | 0.33b | 0.40b | 7.89b | 0.49b | 0.54ab | - | - | - | - | - | 0.15a | - | - | 0.18b | 0.39a | - |
| FISH | - | 0.37b | 0.23a | 7.49ab | 0.37a | 0.91bc | - | - | - | - | - | 0.45b | - | - | 0.00a | 0.99b | - |
| FUNG+FISH | - | 0.34b | 0.38b | 9.72c | 0.43ab | 0.36a | - | - | - | - | - | 0.25a | - | - | 0.00a | 0.32a | - |
| PC | |||||||||||||||||
| Control | 141.61a | 0.34b | 0.02a | 0.17b | - | 2.29d | 0.02b | 0.08a | - | 0.02b | - | 10.79b | - | 0.51c | 0.02b | 0.61b | - |
| FUNG | 162.36b | 0.35b | 0.01a | 0.22b | - | 1.82c | 0.02b | 0.11ab | - | 0.01a | - | 4.81a | - | 0.04a | 0.02b | 1.98c | - |
| FISH | 191.31c | 0.26a | 0.02a | 0.14a | - | 1.24b | 0.02b | 0.14b | - | 0.02b | - | 19.56c | - | 0.73d | 0.01a | 0.12a | - |
| FUNG+FISH | 216.95d | 0.26a | 0.02a | 0.10a | - | 0.98ab | 0.01a | 0.23c | - | 0.01a | - | 7.66ab | - | 0.17ab | 0.01a | 0.69b | - |
| PS/PI | |||||||||||||||||
| Control | 10.65a | 5.57c | 0.01a | 0.29b | 0.58b | - | 0.07b | 0.35a | 0.99ab | 0.08c | 0.86c | 5.68a | - | - | 0.02b | 0.40a | - |
| FUNG | 32.30b | 4.96bc | 0.01a | 0.31b | 0.41ab | - | 0.06a | 0.50ab | 1.14b | 0.05ab | 0.39b | 4.73a | - | - | 0.02b | 2.41b | - |
| FISH | 17.66a | 4.41ab | 0.02b | 0.15a | 0.31a | - | 0.06a | 0.36a | 0.56a | 0.05ab | 0.37ab | 14.63b | - | - | 0.02b | 0.58a | - |
| FUNG+FISH | 42.49b | 3.83a | 0.02b | 0.16a | 0.35a | - | 0.07b | 0.84b | 0.57a | 0.03a | 0.23ab | 6.18a | - | - | 0.01a | 1.61ab | - |
| PE | |||||||||||||||||
| Control | 82.93a | 0.80a | 0.01a | 0.18c | - | 3.02c | 0.05b | 0.16a | - | 0.02a | 0.00a | 4.70b | 0.12a | 0.047a | 0.02b | 43.94c | 0.38d |
| FUNG | 120.36b | 0.92c | 0.01a | 0.13bc | - | 2.19b | 0.04a | 0.15a | - | 0.03b | 0.00a | 3.10a | 0.00a | 0.174a | 0.04c | 121.04d | 0.59c |
| FISH | 141.69b | 0.71b | 0.02b | 0.10ab | - | 1.49a | 0.05a | 0.18a | - | 0.02a | 0.29b | 6.78c | 0.11a | 0.793b | 0.01a | 3.98a | 0.33b |
| FUNG+FISH | 137.18b | 0.78ab | 0.02b | 0.08a | - | 1.23a | 0.05a | 0.32b | - | 0.03b | 0.10a | 3.06a | 0.35b | 0.134a | 0.02b | 22.62b | 0.53ab |
| CL | |||||||||||||||||
| Control | 4.14a | 0.84ab | 0.08b | 0.32a | - | 1.44b | 0.05b | 0.14a | 1.87c | 0.01a | 0.00a | 5.80a | 0.00a | - | 0.09b | - | - |
| FUNG | 4.58b | 0.66b | 0.06a | 0.72b | - | 1.06a | 0.04a | 0.51b | 0.59a | 0.01a | 0.03b | 15.91b | 0.22b | - | 0.09b | - | - |
| FISH | 5.69bc | 1.09a | 0.08b | 0.21a | - | 1.45b | 0.05b | 0.11a | 1.15ab | 0.01a | 0.00a | 7.20ab | 0.26b | - | 0.05a | - | - |
| FUNG+FISH | 6.70c | 1.02a | 0.09c | 0.33a | - | 1.39b | 0.05b | 0.50b | 0.66a | 0.01a | 0.01ab | 15.38b | 0.42c | - | 0.05a | - | - |
Hepatic sterol regulatory element-binding proteins (SREBPs)
SREBPs are critically important basic helix-loop helix (bHLH) transcription factors activated in response to variations in intracellular cholesterol levels that induce transcription of genes with sterol regulatory element (SRE) and E-box containing promoters. Within the SREBP promoter element, the liver X receptor (LXR)-responsive elements (LXRE) may have an important role [34]. LC-PUFA may affect SREBP-1c mediated transcription via several mechanisms. One mechanism is to antagonize activation of the LXR receptor leading to fewer LXR/RXR heterodimer complexes to bind the LXRE in the SREBP-1c promoter and less SREBP-1c-mediated transcription of SRE-containing genes [34]. The PPARα signaling cascade intertwines with SREBP pathways since LC-PUFA also activate PPARα, which can induce LXRα expression [17].
In the present experiment, saturated FA replaced AA and DHA to equilibrate total fat content. Yoshikawa et al.[34] found AA to be more potent than DHA (saturated and monounsaturated being ineffective) at inhibiting activation of SREBP-1c promoter and LXRE enhancer constructs, induced by co-expression of LXRα or -β. An increase in transcription of SREBP-1 may be associated with increased levels of the active SREBP-1 protein.
Numerous hepatic genes containing SRE promoters were regulated by LC-PUFA in the present experiments, including SREBP-1 itself (consistent with [35,36]). Also regulated were genes involved in triacylglycerol synthesis (Gpam), FA synthesis and elongation (Acas1, Acly, FA synthase, Spot14, Elovl2; Elovl2 may speculatively contain an SRE as described by Moon and colleagues [35]), FA desaturation (Scd1), gluconeogenesis (Pck1), cytochrome p450 metabolism (Cyp4a-10 and -14), FA transport (Cd36), cholesterol metabolism and transport (Fpps), and ketone body formation (mitochondrial Hmgcs2). It is evident that genes containing a PPRE have roles in FA oxidation but not FA synthesis, whereas the converse is true for genes containing SRE promoters.
Several genes that may contain SRE promoters such as SREBP-1 and Acly, and Elovl2 and Cd36, were found in the same cluster. However, similar to PPRE-containing genes, not all SRE-containing genes co-clustered, indicating that not all SRE-containing genes were regulated identically by the three LC-PUFA diets. Overall, the number of regulated genes with SREs and PPREs indicates the breadth of molecular responses to dietary LC-PUFA, many of which appear to be co-orchestrated not only by these two transcription factors, but by additional elements as well.
Hepatic SREBP-1 was down regulated with FISH and FUNG+FISH, but importantly, not affected with FUNG (-1.1, -2.1*, -1.8*). Thus, chronic AA feeding alone does not appear, in our experiments, to interact with the SREBP-1 signaling cascade. It is not clear how AA is handled differently in the liver to prevent its interaction with SREBP. The probe set in the Affymetrix Murine 11 k chip could not differentiate between the -1a and -1c splicing variants of SREBP-1. There was however high sequence identity (93%) between the probe set sequence and rat ADD1 mRNA, the murine homologue of human SREBP-1c. The SREBP-1c/ADD1 isoform regulates transcription of genes involved in FA biosynthesis and desaturation [37].
SREBP-1c, in addition to interacting with SREs, binds E-box containing promoters [38]. E-box containing promoters were not examined so the current data analysis could have underestimated SREBP-1-linked effects. Some E-box or bHLH binding factors activate the same genes as SREBP-1. For example, both upstream stimulating factor (USF; an E-box binding factor) and SREBP bind the FA synthase (Fasn) E-box [38]. Hepatic Usf2 transcription was significantly down regulated with FISH, but not affected by FUNG or the combination diet using microarrays, but this result could not fully be confirmed using RT-PCR (1.2, -5.6*, 1.1 with microarray; 2.5, -1.1, 2.0 with RT-PCR).
The NF-Y transcription factor adds further complexity to transcriptional regulation since it can interact and modulate the expression of SREBP-1c target genes containing Y-box motifs, such as those found in Fasn [37]. Mutating the NF-Y binding site in Fasn relieved LC-PUFA-induced Fasn down regulation [39]. Nfyb was differentially expressed after dietary LC-PUFA addition (1.8, -1.6, 1.6). Although no significant changes were observed between dietary treatments and control diet, a significant difference was observed between the FUNG and FISH diets. Thus, LC-PUFA could have induced a joint SREBP-1c/NFY effect on common promoter elements.
Hepatic nuclear factors (HNFs)
Hepatic nuclear factors have been recognized to be transcription factors mediating the transcription of a variety of genes putatively involved in energy metabolism. HNFs may compete with PPARs for binding to DR-1 elements. Thus, LC-PUFA can activate PPARα/RXRα, leading to displacement of HNF4α from PPRE; or be converted to CoA derivatives, which in turn can suppress HNF4α transcriptional activity. Alternatively, these transcription factors may respond differently to chronic versus acute administration of LC-PUFA.
Hnf4α was not differentially regulated in the current experiment. The related transcription factor HNF3γ (winged helix protein, fork head, Foxa3) was however down regulated -2.0* fold with FUNG+FISH, but not with the individual LC-PUFA. HNF3γ protein may be involved in etiology of obesity, hyperlipidemia, and diabetes, by regulating glucagon transcription, insulin resistance, and pancreatic γ cell function [40,41]. To our knowledge, the regulation of hepatic HNF3γ expression by LC-PUFA has not been previously reported.
HNF3γ-signaling may regulate transcription of Pck1 (up regulated ca. 2* fold with each diet, presently), and G6pc (differentially regulated in the current experiment) [42]. HNF3γ is also reported to increase transcription of several genes that were not differentially regulated in the present experiment including, transferrin, tyrosine amino transferase [43], and IPF-1/PDX-1 [40].
Hepatic fatty acid synthesis
In the cytoplasm, FA synthesis is controlled via Acly, acetyl CoA carboxylase (ACC), and Fasn. The Acly reaction enhances FA synthesis by providing more acetyl CoA and NADPH substrates and enhancing ACC activity. The malonyl CoA generated in the ACC reaction then inhibits FA oxidation by reversibly inhibiting Cpt1.
It has previously been shown that the Acly gene in human cultured hepatic cells has a regulatory region from -61 to -49 in the 5' flanking region similar to that of Fasn, which is responsive to insulin and LC-PUFA [44]. In the present experiment, there was coordinated down regulation of both Acly (between -1.8* and -8.9* fold regulation for four different probe sets) and Fasn (-1.9*, -1.3, -5.4*; FUNG and FISH were not statistically significant from one another, but each was significantly different with the combination diet). It is intriguing that, in general, both Acly and Fasn were more strongly down regulated with the combination diet than the individual LC-PUFA. This synergy represents just one of the many unexpected findings revealed with a transcriptomic approach. Although ACC was not regulated at the transcriptional level, cytoplasmic citrate (generated from the Acly reaction) is an allosteric activator of ACC, so less cytoplasmic citrate would lead to less activation of ACC [45]. Transcript levels of hepatic acetyl CoA synthetase 1 AMP forming (acetate:CoA ligase AMP forming; Acas1) were consistently down regulated by LC-PUFA (-2.5*, -1.7*, -4.4*). This enzyme typically has specificity for 2–3 C FA chain lengths. In the liver, intestine, adipose, and mammary gland, the cytoplasmic form synthesizes acetyl CoA for lipogenesis [46]. It is regulated by SREBP-1a, -1c- and -2, as well as SP1 and SP3 binding to GC-boxes; the promoter also contains an E-box [47]. It is down regulated with fasting, and induced with re-feeding; such dietary regulation does not exist in SREBP-1 knockout mice [48]. More generally, acyl CoA synthase is reported to be PPARα-activated [49]. The observed down regulation is consistent with less acetyl CoA available for the ACC reaction, as well as a plethora of other reactions. Overall, it is plausible that both FUNG and FISH feeding decreased FA synthesis in the current experiment.
Hepatic FA oxidaton
As described above, Acly expression (and possibly ACC activity) was significantly down regulated by LC-PUFAs, likely leading to less FA synthesis. Malonyl CoA inhibits FA oxidation by reversibly inhibiting Cpt1. Cpt1 transfers acyl residues from CoA to carnitine in the intermembrane space of the inner mitochondrial membrane, and has a regulatory role in controlling hepatic β-oxidation [50]. Cpt2 transfers carnitinoyl residues to CoA in the mitochondrial matrix for β-oxidation. Carnitine acetyltransferase (Crat) may have the same role as CPT2, but with specificity for 2–10 C units. Mitochondrial Cpt1 (1.8*, 1.4, 1.7*), Cpt2 (1.6, 1.9*, 1.7*), and Crat (1.4, 3.3*, 1.2) were all up regulated by LC-PUFA, but the three LC-PUFA were not identical in this regard. For example, Crat expression was significantly up regulated 3.3* fold with FISH treatment, but was less effected with FUNG and the combination diet.
Acyl-Coenzyme A dehydrogenase catalyzes the first step in mitochondrial β-oxidation, converting acyl-CoA to 2-trans-enoyl CoA. Isoforms with specificity for short, medium, and long chain FA exist. Medium-chain acyl-Coenzyme A dehydrogenase (Acadm) was up regulated, particularly with the combination diet (1.5, 1.3, 1.7*).
Dienoyl CoA isomerase (also known as Δ3,5 Δ2,4 dienoyl CoA isomerase; Ech1 transcripts) converts double bonds to trans bonds during β-oxidation of LC-PUFA. Ech1 was significantly up regulated with FISH alone (1.1, 1.6*, 1.4), consistent with the need to isomerize numerous double bonds during β-oxidation. It is not clear why FUNG did not also up regulate transcripts, as LC-PUFA are generally reported to up regulate this isomerase [51]. The present data generally suggest LC-PUFA increased FA oxidation by down regulating Acly; and up regulating Cpt-1 and -2, Ech1 and Acadm. However, FUNG, FISH, and FUNG+FISH did not affect regulation of these transcripts identically.
Hepatic carbohydrate metabolism
Phosphoenolpyruvate carboxykinase (Pck1) is the rate-limiting enzyme of gluconeogenesis, and most of its activity is regulated transcriptionally. Feeding of all three LC-PUFA diets led to an increase in hepatic Pck1 transcription, indicating gluconeogenesis was favored over glycolysis. Specifically, four probe sets with high sequence similarity to rat Pck1 (accession numbers AA106463, AA110781, AA080172, AA063800) demonstrated differential gene expression for the mouse Pck1 homologue. The signaling pathways affecting Pck1 transcription are discussed in the section "cAMP signaling/links to carbohydrate metabolism".
G6pc converts glucose-6 phosphate to free glucose during gluconeogenesis. G6pc was unaffected with FUNG, significantly up regulated 1.6* fold with FISH (consistent with up regulated gluconeogenesis), but down regulated -1.9* fold with the combination diet (1.1, 1.6*, -1.9*). Rat G6pc has HNF4α consensus binding sites [52]. In HeLa cells and HepG2 hepatoma cells, PUFA down regulated G6pc, possibly via inhibition of HNF 4α promoter activity [52]. In our current mouse experiment, the transcriptional effects of LC-PUFA on G6pc may thus have been HNF4α- or PPARα mediated.
Hepatic cAMP signaling/links to carbohydrate metabolism
cAMP is an important signaling molecule that can be found in specific cellular domains [53]. Levels are controlled through synthesis via adenylate cyclase, and degradation via phosphodiesterases (PDEs). Calmodulin PDEs (CaM-PDEs) are sensitive to calmodulin and calcium up regulation [54]. cAMP can then affect cell signaling via interactions with the regulatory subunits of cAMP activated kinases, including protein kinase A, leading to their subsequent activation, and via binding of cAMP response element binding proteins (CREBs) to cAMP response elements (CREs) or cAMP response units (CRU) consisting of CREs.
In the liver, cAMP regulates Pck1 transcription via the binding of CREB, as well as CCAAT/enhancer binding protein (C/EBP), to the CRU. The Pck1 promoter can thus exist in altered states of cAMP responsiveness, depending on the transcription factors bound to CRU elements [55]. In hepatocytes, SREBP-1c can block transcriptional induction of cAMP on PEPCK-C metabolic action [56].
In the present study, liver Cam-Pde 1c was not regulated by FUNG and FISH feeding alone, but was significantly up regulated 6.0* fold with FUNG+FISH together. A calcium/calmodulin-dependent protein kinase 2 (CKII; Camk2b) was not affected with FUNG feeding or the combination diet, but was significantly down regulated -7.6* fold with FISH alone. CKII is a calmodulin target in the brain, involved in synaptic plasticity. In the liver, CKII may be associated with SAG protein (sensitive to apoptosis gene) and phosphorylate it. SAG in turn has a role in preventing oxidation by inhibiting cytochrome c release and caspase activation. Further, CKII may activate CPT-1 via cytoskeletal phosphorylation of cytokeratin. Thus, an activation of CKII may lead to increased β-oxidation and antioxidant protection. Feeding of all three LC-PUFA diets led to an approximately 2* fold increase in hepatic Pck1 for each probe set identified.
Overall, LC-PUFA may up regulate Pck1, the rate-limiting enzyme in gluconeogenesis, via cAMP-, SREBP-1- and/or PPAR-signaling [57]. This has the overall effect of limiting fat accumulation and shunting metabolic flux to gluconeogenesis. In a molecular sense, FUNG and FISH feeding mimics (or accentuates) the fasted metabolic state.
Hepatic Cytochrome p450 metabolism of LC-PUFA
There are at least 17 cytochrome p450 gene families (Cyps), with four hepatic types (Cyps 1–4) that metabolize foreign compounds and lipophilic substrates, including hormones, FAs, drugs and xenobiotics. Cyp 1 transcription is activated when cytosolic hydrocarbons activate the Ah receptor which then translocates to the nucleus and heterodimerizes with Arnt, leading to binding to an upstream xenobiotic response element on Cyp 1. Cyps 2–4 are induced through nuclear hormone receptors. Phenobarbitol and phenobarbitol-like substances activate CAR-RXR heterodimers (CAR, constitutive androstane receptor), that bind DR-4 response elements on the Cyp 2 promoter. Cyps 3 and 4 are activated by PXR-RXR (PXR, pregnane X receptor) and PPARα-RXR, respectively, also binding DR-4 response elements in both cases.
Nine hepatic cytochrome p450 family members were differentially regulated by LC-PUFA (Cyp1a2, Cyp2a4, Cyp2b9, Cyp3a11, Cyp3a16, Cyp4a10, Cyp4a14, Cypf13, and Por). Two broad clusters (hierarchical clustering alone) and 3 specific clusters (hierarchical- and k-means clustering) of co-expression exist within this subset, indicating transcriptional co-regulation (Table 2). Furthermore, additional specific clusters were close to each other, indicating only a fine split in expression behavior across diets.
Cyps 4a-10, -12, and -14 are specifically involved in ω/ω-1 FA hydroxylation [37]. Cyp4a10 (2.7*, 4.7*, 3.0* with microarray; 15.0*, 18.8*, 6.8* with RT-PCR) and Cyp4a14 (8.0*, 19.3*, 2.6) were both up regulated by LC-PUFA. The murine P450 4a14 is highly homologous to rat 4A2 and 4A3. Rat 4A2 and 4A3, and murine 4a10 are reported to ω/ω-1 hydroxylate C12:0 and 20:4n6, whereas, murine 4a14 hydroxylates only C12:0 [58]. Levels of hydroxylated FA were not examined. Changes in other hepatic cytochrome p450 genes are included in Table 2.
Hepatic desaturation of LC-PUFA
As previously noted, SCD-1 is an important enzyme whose transcription is mediated by both PPAR- and SREBP-mediated signaling [59]. SCD-1 introduces cis double bonds at position C9 in FA chains. It has preferred substrate specificity for C18:0, but will also desaturate other saturated chain lengths. FUNG+FISH showed a significant down regulation of transcripts (-1.1, -1.0, -1.8* with microarray; 2.5*, -1.4, -2.7* with RT-PCR). In this instance, the microarray indicated no change with FUNG; however, RT-PCR indicated an up regulation. This may be expected as RT-PCR is more sensitive than microarrays. It is important to note however, that changes deemed significant by the microarray were also confirmed to be significant by RT-PCR, as is the case for FUNG+FISH induced changes in Scd1. The transcriptional changes in Scd1 expression were in some cases consistent with a marker for apparent decreased enzymatic activity, namely the ratios of FA in hepatic PL pools with a Δ9 double bond/same FA without a Δ9 double bond (Table 4). For example, in PS/PI and phosphatidylethanolamine (PE), the ratio of 16:1n7/16:0 and 18:1n9/18:0 was lowest for the FUNG+FISH diet.
Table 4.
Ratios of FA in hepatic PL pools with a Δ9 double bond/equivalent FA without a Δ9 double bond. There was some evidence to indicate Δ9 desaturase activity may have been impaired, as explained in the text. Abbreviations: FA, fatty acid; PL, phospholipid; see Table 1 for additional abbreviations. Δ9 desaturase is also known as stearoyl-Coenzyme A desaturase (Scd1). Groups sharing a superscript in common are not statistically significant, ANOVA, P < 0.05.
| Group | SPN | PC | PS/PI | PE | Ether PE | ||||
|
16:1n7/ 16:0 |
18:1n9/ 18:0 |
16:1n7/ 16:0 |
18:1n9/ 18:0 |
16:1n7/ 16:0 |
18:1n9/ 18:0 |
16:1n7/ 16:0 |
18:1n9/ 18:0 |
18:1n9/ 18:0 |
|
| Control | 0.03b | 0.18b | 0.05c | 1.27b | 0.04c | 0.09b | 0.04c | 0.49c | 0.50b |
| FUNG | 0.01a | 0.08a | 0.03b | 0.81a | 0.03b | 0.09b | 0.04c | 0.37b | 0.30a |
| FISH | 0.01a | 0.25b | 0.03b | 1.28b | 0.02b | 0.10b | 0.03b | 0.37b | 0.49b |
| FUNG+ FISH | 0.01a | 0.09a | 0.02a | 0.80a | 0.02b | 0.06a | 0.02a | 0.24a | 0.23a |
Hepatic elongation of LC-PUFA
There are suggestions in the literature that FA elongation may also be controlled by transcription factor signaling. The elongase Elovl2 may contain an SRE, and therefore be under the transcriptional control of SREBP [35]). Due to observations in the metabolite data, which suggests a repression of elongase activity, the dietary regulation of Elovl2 and Elovl3 was focused on.
Changes in genes with known elongase activity were examined. The yeast gene Elo1 is associated with elongating FAs with 14 to 16 C; yet, this activity is often masked by cytosolic FA synthase activity [60]. Evidence shows Elovl2 elongates FA to 24C; and Elovl3 elongates FA to 26C, with preference for converting FA from 24C to 26C, in yeast [61].
The probe set for Elovl2 was down regulated, particularly with FISH and the combined diet (-1.3, -1.7*, -1.8*), consistent with some of the lipid data described below; whereas the probe set for Elovl3 was not affected by FUNG, up regulated with FISH, and down regulated, although not significantly, with the combination diet (-1.2, 1.9*, -1.6). An NCBI BLAST search identified the murine homologue of yeast ELO2 (Elovl2) to be Ssc2 (Sequence similarity to Cig30), predominating in liver and testes [62]; complementation studies in yeast mutants indicated the functional equivalent of ELO3 (Elovl3) is Ssc1 [35]. Substrates of mouse Ssc2 were AA and EPA (DHA was not tested) [35]. Thus, FUNG and FISH feeding may transcriptionally regulate their subsequent conversion to elongated products.
Our metabolic data showed evidence that elongase activities may have been altered by LC-PUFA feeding. There was reduced conversion of 18 to 16C FA following LC-PUFA feeding. Specifically, the ratio of C18:0/16:0 in phosphatidylcholine (PC), PS/PI, PE and ether PE was decreased by FISH (Table 3). The ratio of 18:1n7/16:1n7 in sphingomyelin was decreased by FUNG and FUNG+FISH; in PC, PE, and CL only FUNG feeding decreased the 18:1n7/16:1n7 ratio. There was also possible reduced conversion of 20C to 22C FA, and 22C to 24C FA. The ratios of 24:0/22:0 in sphingomyelin and PS/PI, 24:1n9/22:1n9 in PS/PI and CL, 22:4n6/20:4n6 in PC, PE, and CL, and 22:5n3/20:5n3 in PC and PE, were all reduced by FISH, whereas FUNG had either a neutral effect or partly reversed effects seen with FISH (with exception of CL 24:1n9/22:1n9 ratio).
Hepatic Lipid Transport of FAs
Transcription factors such as PPARs and SREBPs are also known to have an effect on various lipid transport genes. Hepatic Abca1, Abca7 and Abce1 were differentially regulated; however, their roles in the liver are not precisely known. Several members of the ATP-binding cassette (ABC) superfamily, such as Abca1, are induced via PPARγ and LXRα activation [63]. Abca1 has been shown to play a role in reverse cholesterol pathways and may be linked to HDL deficiency in metabolic syndromes [64]. In macrophages, 18:2n6 and AA increased ABCA1 protein degradation without affecting transcriptional levels [65]. In the current study, Abca1 was significantly decreased by FUNG and FISH.
FA translocase (Cd36) was significantly up regulated with FISH alone (-1.1, 2.4*, -1.5). Cd36 co-clustered with E-FABP, Apoe, Apoc1, and Abce1 transcript, all involved in FA and sterol transport. CD36 protein mediates long chain free FA uptake in adipocytes and other tissues, including liver. In murine pre-adipocytes, it is transcriptionally up regulated by long chain free FAs via a PPARα-mediated process [66]. Approaches using antisense technology, null mice, transgenic mice, and microarrays revealed important additional roles of CD36 protein as a receptor for thrombospondin, collagen type I, and oxidized LDL on macrophages [67]. The up regulation of Cd36 with FISH, and antagonism in presence of FUNG, merits further study.
FA binding proteins (FABPs) are small cytosolic proteins with virtually identical backbone structures that facilitate the solubility and intracellular transport of FAs. E-FABP is found in liver and other organs [68], but it has been most studied in psoriatic skin lesions [69] where it has putative roles in FA cytoplasmic transport, skin barrier function and keratinocyte differentiation [70]. Although links between PPARs and E-FABP have not currently been identified, this gene clustered with other known PPAR-affected genes suggesting a possibly important regulatory link. Purified E-FABP has five fold higher affinity for 18:0, than for 18:1n9 and 20:4n6 (22:6n3 not studied) [69]. Herein, hepatic E-FABP expression (Fabp5 transcript) was down regulated by all three diets (-5.5*, -8.4*, -13.6* with microarray; -2.9*, -8.5*, -16.4* with RT-PCR;). The significance of this large decrease in hepatic expression by both FUNG and FISH, and particularly the combination of the two, is a potentially important finding whose physiological consequence must be further investigated. Whether LC-PUFA induces the same decrease in Fabp5 in psoriatic skin is not known to our knowledge. N3 LC-PUFA, particularly 20:5n3, are known to be of clinical benefit in treating psoriasis [71].
Hepatic cholesterol metabolism, ketogenesis, and apolipoproteins
Mitochondrial Hmgcs2, which is involved in ketogenesis, was up regulated, particularly with the combination diet (1.2, 1.3, 1.6*). Mitochondrial Hmgcs2 is known to be induced by PPARα activation in experiments with hepatocytes [72,73]. The cytosolic form involved in cholesterol biosynthesis was not affected. Fpps was consistently down regulated (-2.4*, -1.6*, -1.9*) across diets, which could lead to reduced cholesterol synthesis; cholesterol synthesis and plasma cholesterol levels were not determined. Oils rich in DHA, such as fish oil, have cholesterol and triacylglycerol lowering properties [74].
LC-PUFA altered expression of various apolipoproteins. Apolipoprotein C1 (Apoc1) was only down regulated with FUNG alone (-1.7*, 1.1, 1.0). Apolipoprotein E (Apoe) was only up regulated with FISH alone (1.0, 1.5*, -1.2). There was consistent up regulation with all three LC-PUFA diets of an EST (expressed sequence tag) highly similar to human apolipoprotein B-100 (ApoB-100) precursor (2.1*, 1.9*, 2.0*). Both apolipoprotein C2 (Apoc2; -1.3, -1.5*, -1.6*) and apolipoprotein A4 (Apoa4; described in detail below) were consistently down regulated across diets.
Apoe and Apoc1, and Apoa4 and Apoc2, were similarly co-expressed/co-clustered across diets. When clustered with genes in the more general "transport" category, categories of co-expressed genes with underlying roles in lipid and sterol transport were evident. For example, Apoe and Apoc1 co-clustered with Abce1 and Cd36; and Apoa4 and Apoc2 co-clustered with Abca1 and Fabp5.
FUNG and FISH alone moderately down regulated Apoa4, whereas the combination diet resulted in a large down regulation (-2.6*, -3.2*, -17.2* with microarray; -1.3, -3.0, -4.9* with RT-PCR). A similar trend was seen for Apoa4 precursor transcript (-3.2*, -4.2*, -9.7*). ApoAIV protein is an anti-atherogenic component of high-density lipoprotein (HDL) particles and is also found in very low-density lipoproteins and chylomicrons [75]. It is involved in cholesterol absorption; low-density lipoprotein (LDL) clearance as a cofactor for hepatic lipase; increasing dolichol acylation; and increasing PE, but not PC, hydrolysis. FUNG and FISH may decrease Apoa4 expression through PPARα [76,77], and in the current experiment, LC-PUFA may have affected PPARα transactivation and signaling. ApoAIV activity could also be affected by diet induced structural changes to membrane PL in lipoprotein particles, as Apoa4 intercalates with PC in model membranes [78]. The biological consequences of reduced Apoa4 transcription, particularly with the combination diet, merits further investigation. Rodents fed low cholesterol diets, as in the present study, carry most of their cholesterol in the HDL fraction, and under these conditions, fish oil feeding reduces cholesterol from the HDL fraction, so the above mentioned changes in apolipoproteins, although fascinating, must be interpreted within the context of the chosen models.
FISH and FUNG-mediated effects in the Hippocampus
In the present study, 356 differentially expressed genes were identified in the hippocampus using a 5% LFC model. The trend in the data was similar to that seen with liver data, as shown in Figure 1 (see Materials and Methods for details). These data revealed that the consumption of dietary oils led to differences in the expression of a broad range of hippocampal genes that have been previously indicated to be involved in cell signaling and signal transduction, including cation transport, calcium binding, protein phosphorylation, protein kinases, and ATP- and GTP-binding as revealed by examining GO identifiers across all of the diet-regulated genes (Figure 2). In striking contrast to the liver, a number of hippocampal LC-PUFA-induced differentially expressed genes had annotations suggesting immune related functions, as well as general defense responses specific to the hippocampus, including: "MHC-interacting proteins, lymphocyte antigens, immunoglobulins, and defense response." Additionally, there were numerous ontological assignments for genes which suggested dietary LC-PUFA feeding affected cell cycle control including the following categories: "cytoskeleton, cell cycle control, and cell cycle regulator".
In the hippocampus, dietary LC-PUFA activated a different set of transcription factors than observed in the liver, including: nuclear factor of κ light chain gene enhancer in B cells 1 p105 (Nfkb1); nescient helix loop helix 1 (Nhlh1); E2F transcription factor 3 (E2f3); basic transcription factor 3 (Btf3); inhibitor of DNA binding 1 (Idb1); D site albumin promoter binding protein (Dbp); paired box gene 9 (Pax9); and the ELK1 member of the ETS oncogene family. Although dietary LC-PUFA activated numerous transcriptional factors in both liver and hippocampus, it is a noteworthy difference that the transcriptional signatures for PPAR, SREBP, and HNF could not be detected in the hippocampal dataset.
The interpretation of all 356 hippocampal transcripts is beyond the scope of the current manuscript, but all raw data is deposited at the NCBI gene expression Omnibus [79]. The present data provides evidence that LC-PUFA were incorporated into brain and hippocampal tissues (as previously discussed) and thereby influenced novel signaling pathways via receptor interactions. Some of these signaling pathways are highlighted below.
Hippocampal signaling pathways affected by dietary LC-PUFA
Transthyretin signaling
Thyroid hormones including transthyretin are generally involved in neuronal proliferation and differentiation, and are required for normal cytoskeletal assembly. Transthyretin is specifically involved in transporting thyroxine (T4) and retinol-binding protein in cerebrospinal fluid and brain serum. AA has been found to bind transthyretin and inhibit thyroxin associations [80]. Transthyretin may also sequester β amyloid, thereby having neuroprotective properties. Dietary FISH had the effect of decreasing Ttr; whereas, FUNG and particularly the combination diet, increased Ttr (1.6, -1.9*, 2.2*). Similar to our findings in mice, feeding rats fish oil significantly decreased Ttr expression in whole brain 2.9 fold [18]. Interestingly, feeding of Ginkgo biloba extract (publicized to affect mental function) in mice increased hippocampal Ttr [81]. Levels of transthyretin, and associations of transthyretin with thyroxine and retinol-binding protein should be assessed in future LC-PUFA feeding experiments.
5-hydroxytryptamine signaling
Dietary LC-PUFA may affect 5-hydroxytryptamine (serotonin, 5-HT) signaling. Specifically, 20:4 N-acylethanolamine can displace binding of ligands to HT receptors [82], and displace 5-HT3 receptor currents [83], and we have previously reported that dietary AA feeding increased whole brain 20:4 N-acylethanolamine, as a separate component of the present work [84]. In the present work, dietary FUNG and FISH, alone or together, increased Htr4 receptor levels (6.5*, 3.8, 4.4). 5-HT4 receptor (Htr4 transcript) increases in expression have been shown to augment hippocampal acetylcholine outflow, thereby affecting cognitive processes [85].
Alpha-2,8-sialyltransferase
Hippocampal α-2,8-sialyltransferase (Siat8e) was down regulated with FISH and the combination diet, but not with FUNG; difference relative to control was only significant for the combination diet (1.6, -2.6, -6.4*). Hippocampal protein kinase Cδ (Prkcd) was slightly down regulated with the three LC-PUFA diets (-1.6, -1.3, -1.8*). PUFA feeding can influence PKC activity [86] and one mechanism of action may be via generation of unique diacylglycerol molecular species [87].
Alpha-2,8-linked disialic acid residues are found in glycoproteins [88]. During learning, there is a transient increase in neuronal polysialyation in the dentate gyrus of the hippocampus. This has been associated with selective retention and/or elimination of synapses that are transiently overproduced during memory consolidation [89]. In rat hippocampus, during development, PKCδ can negatively regulate polysialyltransferase activity and neural cell adhesion molecule (NCAM) polysialylation state [89]. NCAM is implicated in neural differentiation and cellular plasticity [90]. Our observed decrease in Prkcd expression may have affected hippocampal polysialyltransferase activity. It is not known if Prkcd regulates expression of Siat8e.
Proopiomelanocortin (POMC)
POMC is a hypothalamic neuropeptide, and a target for leptin. POMC is cleaved post-translationally producing bioactive peptides (adrenocorticotrophin, β-endorphin and α-, β- and γ-melanocyte stimulating hormones) that interact with neurons expressing melanocortin receptors, leading to appetite suppression [91]. Feeding high fat diets is known to increase Pomc expression [92]. Herein, FUNG feeding resulted in a 6.0* fold increase in hippocampal Pomc, whereas FISH and the combination diet had smaller, non-statistically significant effects (6.0*, 1.6, 1.4). The physiological effect of potentially increasing POMC protein with FUNG feeding is not known. As a next step, levels of POMC protein and downstream POMC peptides should be quantified
Conclusions and key findings
The molecular effects of dietary fish oil fed in reasonable quantities over prolonged periods has been found in humans and animals to be related to alterations in health outcome. In the present study, mice were fed relevant quantities of fish oil and AA rich fungal oils and the responses were studied in murine liver and hippocampus using a combined gene expression- and lipid-profiling strategy. The simple addition of these oils to the habitual diet of mice influenced hundreds of gene products [79]. Furthermore, in a striking number of examples, inter-diet (FUNG vs. FISH vs. FUNG+FISH) and inter-organ differences in the transcriptional profile were observed, indicating that the predominant bioactive FAs in fish oil (20:5n3, 22:5n3 and 22:6n3) and AA enriched fungal oil (20:4n6) do not affect gene transcription identically in the two organs studied.
Hepatic gene transcriptional profiles and ontology classifications indicated that fish oil LC-PUFA likely increased FA β-oxidation and gluconeogenesis, and decreased FA synthesis. Furthermore, the transcriptional profile observed indicated that these LC-PUFA regulated hepatic energy processes through the tight coordination of several critical transcriptional factors. Feeding both LC-PUFA containing oils mimicked (or accentuated) the fasted metabolic state, confirming this aspect of the beneficial health effects of dietary LC-PUFA for both normal and clinical populations, and even household pets.
These same conclusions regarding energy expenditure were reached in experiments assessing enzymatic activities, and transcriptional changes in rat liver using Northern blots, where fish oil feeding: increased palmitoyl CoA mitochondrial and peroxisomal oxidation rate; increased Cpt1, Cpt2, Δ3, Δ2-enoyl CoA isomerase, and Ech1 expression and activity; and decreased expression and activity of FA synthase, malic enzyme, glucose 6-phosphate dehydrogenase, and pyruvate kinase, compared to palm and/or safflower oil feeding [93]. Likewise, in rat white retroperitoneal adipose tissue, fish oil feeding also decreased Fasn expression [94]. Intriguingly, the hormone leptin, which also acts through SREBP-1 and PPARα, affected many transcripts similarly to LC-PUFA (e.g., leptin decreased Gpam and Apoa4 transcripts; and up regulated Ech1, mitochondrial Hmgcs2, and Cyp4a14 transcripts [95].
Chronic feeding of fish and AA-enriched fungal oils broadly affected genes regulated by PPARα, HNFα, and SREBP-1 in the liver; transcription factors implicated in lipid metabolism as well as other processes. Only with future studies in which PPAR-specific agonists and antagonists are included, along with receptor antagonists, will the precise role of PPAR-signaling in response to LC-PUFA feeding be elucidated. The overlapping actions/effects of these various transcription factors present a challenge that will require tools, such as genomics and metabolomics, to provide a stepping stone towards the complete understanding of LC-PUFA metabolism.
There were striking differences in the genes and transcriptional factors activated by LC-PUFA in the liver and hippocampus, despite these organs being metabolically linked, as AA and DHA cross the blood brain barrier. There are at least three reasons for these inter-organ differences. Firstly, the dietary FAs were not identically incorporated into both tissues, as indicated by tissue lipid profiling. Secondly, different transcription factors exist and were activated in the two organs, which would affect downstream signaling. Thirdly, there is the possibility, that even when the same transcription factors were activated in both organs, factors including dimerization partners, multiple binding of various transcription factors, post-translational modifications, and signaling kinetics could affect the transcriptional profile.
Currently, nutritional FA recommendations are made largely by examining levels of key LC-PUFA in plasma; and less often, red blood cells, platelets, and biopsied tissue. Transcriptional gene profiles have not been considered in making such recommendations. The present work convincingly shows the potential importance of examining transcriptional profiles in addition to FA profiles; and the importance of examining more than one tissue, in making such LC-PUFA nutritional and clinical recommendations.
Materials and Materials
Experimental diets
All diets contained 90% fat-free AIN93G rodent diet in powder form (Lot 9350–5, Dyets, Inc., Bethlehem, PA), 0.4% milk fat, 1.2% palm olein, 1.9% Trisun sunflower oil, 1.5% soybean oil and 2.1–5.1% medium chain triacylglycerol oil (all of the above ingredients except the AIN93G were from Nestlé affiliated companies) (Tables 5 and 6). Part of the medium chain triacylglycerol oil in control diet was replaced with: 1.1% fungal oil (providing 0.5 dietary wt% AA and 1.0en%) in the FUNG diet; 1.9% fish oil (providing 0.5 dietary wt% DHA and 1.0en%) in the FISH diet; and 1.1% fungal oil and 1.9% fish oil in the combined FUNG+FISH diet. The levels of AA and DHA chosen were similar to that previously shown in rats to affect neurotransmitter levels and behavior [96]; and were not excessively high, being 2–3 fold higher than that recommended for human babies who have a slower Δ6 desaturase activity than rodents. Food was maintained at -80°C in daily aliquots under liquid N2, thawed each afternoon before administration to mice, and uneaten food was discarded daily.
Table 5.
Dietary composition of the experimental diets. Values represent the weight percent of each component in the diet, wet weight basis. Fungal oil was the source of AA; fish oil was the source of DHA. Suppliers are confidential. Fungal and fish oils were replaced with MCT (medium chain triacylgylcerols) to provide the same total fat content of each diet.
| Diet ingredient | Control | FUNG | FISH | FUNG+FISH |
| Powdered Diet AIN 93, fat free | 90.0 | 90.0 | 90.0 | 90.0 |
| Milk fat | 0.4 | 0.4 | 0.4 | 0.4 |
| MCT oil | 5.1 | 4.0 | 3.1 | 2.1 |
| Palm olein | 1.2 | 1.2 | 1.2 | 1.2 |
| Trisun oil | 1.9 | 1.9 | 1.9 | 1.9 |
| Soy oil | 1.5 | 1.5 | 1.5 | 1.5 |
| Fungal oil rich in AA | 0.0 | 1.1 | 0.0 | 1.1 |
| Fish oil rich in DHA | 0.0 | 0.0 | 1.9 | 1.9 |
Table 6.
Fatty acid composition of the experimental diets. The sources of all oils are given in Table 5. Values represent g FA/100 g total FA, following 3 determinations. The FUNG diet contained ca. 5% AA; the FISH diet contained ca. 5% DHA; and the FUNG+FISH diet contained ca. 5% of AA and 5% of DHA.
| Fatty Acid | Control | FUNG | FISH | FUNG+FISH |
| 4:0 + 6:0 | 0.2 | 0.4 | 0.1 | 0.3 |
| 8:0 | 27.2 | 21.3 | 16.9 | 11.1 |
| 10:0 | 22.9 | 18.0 | 14.3 | 9.4 |
| 12:0+13:0+14:0+ 14:1n5+15:0+17:0 | 1.6 | 1.6 | 2.7 | 2.7 |
| 16:0 | 7.9 | 9.2 | 11.9 | 13.2 |
| 18:0 | 2.3 | 3.0 | 3.5 | 4.2 |
| 18:1n9 | 24.8 | 25.4 | 27.0 | 27.6 |
| 18:1n9 trans | 0.3 | 0.3 | 0.8 | 0.8 |
| 18:2n6 | 11.4 | 12.3 | 11.9 | 12.8 |
| 18:3n6 | - | 0.3 | 0.1 | 0.4 |
| 18:3n3 | 1.0 | 1.0 | 1.1 | 1.1 |
| 18:4n3 | - | - | 0.1 | 0.1 |
| 20:0 | 0.1 | 0.2 | 0.3 | 0.4 |
| 20:1n9 | 0.1 | 0.1 | 0.2 | 0.2 |
| 20:2n6 | - | 0.1 | 0.1 | 0.2 |
| 20:3n6 | - | 0.4 | 0.1 | 0.5 |
| 20:4n6 | - | 4.7 | 0.4 | 5.1 |
| 20:5n3 | - | 0.3 | 1.1 | 1.4 |
| 22:0+22:1n9+24:0+24:1n9+26:0+unknown | 0.3 | 1.3 | 1.9 | 2.9 |
| 22:4n6 | - | 0.1 | 0.0 | 0.1 |
| 22:5n3 | - | - | 0.4 | 0.4 |
| 22:6n3 | - | - | 5.2 | 5.2 |
Mice, feeding, and dissection conditions
Prior to actual experimentation, test dissections were made on identical mice to perfect the dissection techniques at Porsolt & Partners Pharmacology (Paris, France).
Male Rj:NMRI mice from Elevage Janvier, Le Genest-Saint-Isle France, weighing 10–11 g at delivery and 33–51 grams on experimental d 42, were housed 10 per cage in wire cages with bedding and normal light cycle. Mice received ad libitum quantities of distilled water and purified powdered diets (7.5 g/mouse) in ceramic cups (10/group) from d 1–57 or 58, and were thereafter sacrificed in the morning by cervical dislocation over these last 2 experimental days. Food was not removed the night before, and since nocturnal mice are known to eat during the normal period dark cycle employed, the mice could have been fasted 0–6 h at sacrifice. Mice were thus not completely fasted at sacrifice, a complete fast being 12–16 h.
After feeding diets to 10 mice per group, 5 mice were randomly selected for inclusion in hepatic and hippocampal gene expression analysis. From these 5 mice, livers were dissected according to standard protocols (Porsolt & Partners Pharmacology, Boulogne-Billancourt, France), which consisted of carefully dissecting out the liver, rinsing with 70% EtOH to remove adhering blood, trimming of adhering connective tissue with scissors, cutting into 100–150 mg subsections, freezing in liquid N2, and storing at -80°C until gene expression and lipid analysis. From these same 5 mice, the hippocampus was dissected from the mice according to standard protocols (Porsolt & Partners Pharmacology, Boulogne-Billancourt, France), rinsed as described for livers, cut into 17–50 mg subsections, and frozen as described for livers. Hepatic and hippocampal lipid analysis was performed according to standard methods at Lipomics, Inc. (U.S.A) on individual mice (n = 5).
Nucleic acid preparation
Liver tissue was extracted from 5 individual mice per group and extracted separately using Qiagen RNeasy mini-kits (Basel, Switzerland) according to manufacturer instructions, except that RNeasy columns were impregnated with DNase I (Roche, Basel, Switzerland) to remove any genomic DNA contamination.
For the group fed a control diet, two independent hepatic pools (n = 2) of 10 μg each were created, consisting of equal amounts (2 μg) of total RNA from each of the 5 aforementioned mice. This was done to assess measurement variability. Secondly, a separate piece of liver was excised from three of the individual control mice (n = 3) to additionally and independently extract a second set of RNA; these three mice were chosen at random from the group of five. This second round of extraction was used to independently assess the variability of normal control mice housed under the prevailing conditions of the study.
For each of the other dietary groups, a single pool (n = 1) of 10 μg total hepatic RNA was created for comparison against the control. Hippocampal RNA was prepared as described above for hepatic RNA, except only 1 pooled control RNA sample from 5 mice was prepared; hence there was a total of 4 pooled hippocampal samples, i.e. one from each of the 4 diets (Control, FUNG, FISH, and FUNG+FISH). In sum, eight liver samples and four hippocampal RNA samples were hybridized on Affymetrix GeneChips.
All RNA samples were quantified with the RiboGreen RNA Quantification Kit (Molecular Probes, Eugene Oregon), then analyzed via agarose gel electrophoresis for intact 18S and 28S rRNA. All study samples were judged to contain sufficient amounts of high-quality RNA for hybridization to GeneChips. As another quality control measure, prior to hybridization with Affymetrix GeneChips (Affymetrix, Inc., Santa Clara, CA), we confirmed that pooled samples gave strong signals using Affymetrix test chips.
Gene expression analysis using the murine 11k GeneChip
cRNA preparation
For the control diet group, each of three individual mouse liver RNA samples (n = 3) were hybridized to Affymetrix GeneChips. For both the control and LC-PUFA diets, 15 μg pooled total RNA was the starting material for hybridization to the Mu11kA & B GeneChips. Pooled liver controls were evaluated in duplicate on the Affymetrix probe array cartridge to estimate experimental variation in gene chip hybridization, as previously described in detail [27].
In all cases, total RNA was converted to biotinylated cRNA, hybridized in the Affymetrix probe array cartridge, stained, and then quantified. First and second strand cDNA synthesis was performed using the SuperScript Choice System (Invitrogen AG, Basel, Switzerland), according to manufacturer instructions, but using an oligo-dT primer containing a T7 RNA polymerase binding site. Labeled cRNA was prepared with the MEGAscript In Vitro Transcription kit (Ambion, Inc., Austin, TX). Biotinylated CTP and UTP (Enzo Biochem Inc., NY) were used together with unlabeled dNTPs in the reaction, and unincorporated nucleotides were removed with RNeasy columns.
Array hybridization and scanning
cRNA (10 μg) was fragmented at 94°C for 35 min in buffer containing 40 mM Tris-acetate pH 8.1, 100mM KOAc, 30 mM MgOAc. Prior to hybridization, fragmented cRNA in a 6X SSPE-T hybridization buffer (6X: 1M NaCl, 10mM Tris pH 7.6, 0.005% Triton), was heated to 95°C for 5 min, cooled to 40°C and loaded onto an Affymetrix probe array cartridge. The probe array was incubated for 16 h at 40°C at constant rotation (60 rpm), then exposed to 10 washes in 6X SSPE-T at 25°C followed by 4 washes in 0.5X SSPE-T at 50°C. Biotinylated cRNA was stained with 10 g/mL streptavidin-phycoerythrin conjugate (Molecular Probes) in 6X SSPE-T for 30 min at 25°C followed by 10 washes in 6xSSPE-T at 25°C. Probe arrays were scanned at 560 nm using a confocal laser scanning microscope (made for Affymetrix by Hewlett-Packard). Readings from the quantitative scanning were analyzed with Affymetrix Gene Expression Analysis Software.
Selection of differentially regulated genes and data analysis
A mathematical method was developed and applied to the raw GeneChip data for the selection of differentially regulated genes. This method moves beyond setting a single fold change cut-off by considering fold change in the context of absolute expression [27]. In brief, this method includes: (A) the determination of the upper 5% of fold changes within narrow bins of absolute expression levels to determine a limit fold change (LFC) function; and (B) subsequent ranking of genes by a combined fold change/absolute difference calculation. Herein, absolute expression is referred to as average difference intensity (ADI) in all mathematical equations. The function describing the upper 5% of hippocampal and liver data was described with the following equations: LFChippocampus = 1.36 + (90/min ADI); and LFCliver = 1.52+(100/min ADI), respectively.
The selection of differentially expressed genes was corroborated in three ways for liver samples: (1) assessing the measurement variability and (2) assessing individual mouse variability in order to establish confidence intervals with which to calculate the significance of differences with pooled diet response groups and (3) confirmation by RT-PCR for a sub-selection of genes. Replicate analysis established that all genes selected by this method also laid above a more conventional 99.9% confidence interval, when compared to the variation of pooled replicates (n = 2). Subsequent analysis of the variation between individual mice (n = 3) indicated that this variation is comparable to the pooled variation and that all genes selected as differentially expressed lied above the 99.0% confidence interval for individual mouse variability. The gene expression means of the three individual mice were calculated and compared to pooled data in a scatter plot. A correlation co-efficient of 0.93 was obtained indicating that the pools are accurate representations of the individual mice used to create them. Lastly, validation studies showed up to 85.7% concordance of microarray data with real time polymerase chain reaction (RT-PCR) across the range of absolute expression for specific genes selected as differentially regulated [27]. All RT-PCR data, including information about the low false positive rate, for all of the liver samples mentioned in the present work, can be found in the aforementioned reference.
The 5% LFC method indicated that 513 genes in the hippocampus and 489 genes in the liver were differentially regulated by LC-PUFA from the original 13,179 genes represented on the Mu11K Affymetrix GeneChip. Absence/presence calls were not considered a priori in selecting significantly regulated genes, but were a post-selection criterion. After removing those genes that were identified as absent across all dietary conditions, 356 hippocampal and 329 hepatic differentially expressed probe sets remained in the list for further analysis and interpretation. Of these, 221 and 214 genes represented annotated sequences in the hippocampus and liver, respectively.
Fold changes are always listed in the respective order: FUNG, FISH, and FUNG+FISH without repeating these labels; and negative signs indicate down regulation. To determine if the ADI for a treatment group was significantly different from the control diet, the 5% LFC best-fit equations were utilized: LFChippocampus = 1.36 + (90/min ADI); LFCliver = 1.52+(100/min ADI). Groups significantly different from the control, as determined by a pair-wise comparison, are labeled with an asterisk in the text and Table 2. The mathematical model utilized also selected some genes due to significant differences between experimental treatments, even when no significant differences versus the control were observed. For reasons of simplicity, significant differences between experimental treatments are not indicated in Table 2, but are discussed in the text.
Data analysis
Selected genes were grouped by expression patterns through a combined k-means and hierarchical clustering approach (Spotfire, Inc., Sommerville, MA) by setting the maximum number of clusters in either method to 5 (a compromise between not having too many or too few clusters) and then graphing on a 2-D gene plot (Figures not shown; hierarchical cluster numbers (h1-h5), and k-means cluster number (k1-5) included in Table 2). Genes were then annotated using gene ontology (GO) classifications; a dynamic, structured, yet precisely defined system which characterizes genes based on defined categories of molecular function, biological process, and cellular component [28]. Finally, genes were assigned names through NCBI Locuslink (Table 2).
Gene Confirmation
A subset of the genes selected by the 5% LFC model were examined by RT-PCR using SYBR® green dye (Molecular Probes) which interacts with double-stranded nucleotide sequences. Data represented means of triplicates for each experimental treatment using pooled RNA samples (n = 5). Amplification was performed using an ABI 5700 machine (Applied Biosystems, Foster City, CA, USA) with a hot start at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min for denaturation, annealing and elongation. Data was normalized to β-actin and GAPDH, which were not statistically, differentially expressed. A students t-test (α = 0.05) was used to confirm significantly differentially expressed genes following RT-PCR.
Lipid analysis
Following sacrifice of the animals, the livers (250 mg piece), hippocampus (from half of the mice), and whole brains (from the other half of mice) from each animal were rapidly frozen in liquid N2 and stored at -80°C. Each whole brain weighed between 395–454 mg. A slurry of each whole brain was made by thawing the tissue samples at room temperature for the minimum time necessary, adding 12.6 mL of CHCL3:MeOH (2:1, v/v), briefly homogenizing on ice, and re-freezing the samples under N2 at -80°C (concentration was ca. 30 mg brain/mL solvent). Livers, hippocampus, and whole brain (4 individual samples/tissue) were quantitatively analyzed for FA distribution (μg FA/g tissue) in each PL class, by Lipomics Technologies, Inc. (West Sacramento, CA). Brain samples were also separately extracted, and N-acylethanolamines (NAEs) and monoacylglycerols (MAGs) purified and characterized as previously described in detail [84].
Abbreviations
AA arachidonic acid
ACC acetyl CoA carboxylase
ADI average difference intensity
DHA docosahexaenoic acid
EPA eicosapentaenoic acid
FA(s) fatty acid(s)
FISH fish oil
FUNG fungal oil
FUNG+FISH refers to the combination of fungal and fish oils fed together
GO gene ontology
HNF hepatic nuclear factor
LC-PUFA long chain polyunsaturated fatty acid
LFC limit fold change
LXR liver X receptor
LXRE LXR responsive elements
PL phospholipid
PPAR peroxisome proliferator activated receptor
RT-PCR real time-polymerase chain reaction
SRE(BP) sterol regulatory element (binding protein)
Authors' contributions
AB designed and processed the in vivo mice experiments that led to the basic structure of this work. AB also was responsible for identifying many of the specific lipid-gene correlations in the two datasets and putting them in the context of nutritional biochemistry. DMM analyzed, integrated and organized all of the microarray data. DMM performed and analyzed all of the RT-PCR confirmation studies. Both DMM and MR were responsible for developing the mathematical algorithms used for the identification of differential gene expression. MR led the overall integration of the lipid-gene datasets and organized the writing of this manuscript. JBG put the data and conclusions into context with previous literature and emerging trends in the field of nutrition.
All authors have read and approved the final manuscript.
Acknowledgments
Acknowledgments
The authors thank Andreas Rytz and Robert Mansourian for aid in statistical selection of genes; Thomas Thykjaer and Toben Orntoft from Aarhus University for GeneChip hybridizations and technical advice on data analysis; Joshua Levy from Incellomics for gene ontology classifications; and David Vilanova for submission of raw data to NCBI's Gene Expression Omnibus. We would also like to thank Muriel Fiaux, Irina Monnard & Patricia Leone for contributing to the many technical tasks that were part of these experiments. We thank Steve Watkins from Lipomics for helpful discussion in interpreting quantitative fatty acid profiling data. We thank Porsolt & Partners Pharmacology (Paris, France) for performing the feeding experiments and organ dissections. Lastly, we thank Olivier Ballèvre and Andrea Pfeifer for their commitment to the project.
Contributor Information
Alvin Berger, Email: alvin@cytochroma.com.
David M Mutch, Email: david.mutch@rdls.nestle.com.
J Bruce German, Email: jbgerman@ucdavis.edu.
Matthew A Roberts, Email: matthew-alan.roberts@rdls.nestle.com.
References
- Albert CM, Campos H, Stampfer MJ, Ridker PM, Manson JE, Willett WC, Ma J. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N Engl J Med. 2002;346:1113–1118. doi: 10.1056/NEJMoa012918. [DOI] [PubMed] [Google Scholar]
- Connor WE. Importance of n-3 fatty acids in health and disease. Am J Clin Nutr. 2000;71:171S–175S. doi: 10.1093/ajcn/71.1.171S. [DOI] [PubMed] [Google Scholar]
- Hu FB, Bronner L, Willett WC, Stampfer MJ, Rexrode KM, Albert CM, Hunter D, Manson JE. Fish and omega-3 fatty acid intake and risk of coronary heart disease in women. Jama. 2002;287:1815–1821. doi: 10.1001/jama.287.14.1815. [DOI] [PubMed] [Google Scholar]
- Kang JX, Leaf A. Prevention of fatal cardiac arrhythmias by polyunsaturated fatty acids. Am J Clin Nutr. 2000;71:202S–207S. doi: 10.1093/ajcn/71.1.202S. [DOI] [PubMed] [Google Scholar]
- Kromhout D, Bosschieter EB, de Lezenne Coulander C. The inverse relation between fish consumption and 20-year mortality from coronary heart disease. N Engl J Med. 1985;312:1205–1209. doi: 10.1056/NEJM198505093121901. [DOI] [PubMed] [Google Scholar]
- Calder PC. n-3 polyunsaturated fatty acids and cytokine production in health and disease. Ann Nutr Metab. 1997;41:203–234. doi: 10.1159/000177997. [DOI] [PubMed] [Google Scholar]
- Volker D, Fitzgerald P, Major G, Garg M. Efficacy of fish oil concentrate in the treatment of rheumatoid arthritis. J Rheumatol. 2000;27:2343–2346. [PubMed] [Google Scholar]
- Ziboh VA, Miller CC, Cho Y. Metabolism of polyunsaturated fatty acids by skin epidermal enzymes: generation of antiinflammatory and antiproliferative metabolites. Am J Clin Nutr. 2000;71:361S–366S. doi: 10.1093/ajcn/71.1.361s. [DOI] [PubMed] [Google Scholar]
- Martinez M, Vazquez E, Garcia-Silva MT, Manzanares J, Bertran JM, Castello F, Mougan I. Therapeutic effects of docosahexaenoic acid ethyl ester in patients with generalized peroxisomal disorders. Am J Clin Nutr. 2000;71:376S–385S. doi: 10.1093/ajcn/71.1.376s. [DOI] [PubMed] [Google Scholar]
- Bougnoux P. n-3 polyunsaturated fatty acids and cancer. Curr Opin Clin Nutr Metab Care. 1999;2:121–126. doi: 10.1097/00075197-199903000-00005. [DOI] [PubMed] [Google Scholar]
- Hu FB, van Dam RM, Liu S. Diet and risk of Type II diabetes: the role of types of fat and carbohydrate. Diabetologia. 2001;44:805–817. doi: 10.1007/s001250100547. [DOI] [PubMed] [Google Scholar]
- Innis SM, Gilley J, Werker J. Are human milk long-chain polyunsaturated fatty acids related to visual and neural development in breast-fed term infants? J Pediatr. 2001;139:532–537. doi: 10.1067/mpd.2001.118429. [DOI] [PubMed] [Google Scholar]
- Kelley DS. Modulation of human immune and inflammatory responses by dietary fatty acids. Nutrition. 2001;17:669–673. doi: 10.1016/S0899-9007(01)00576-7. [DOI] [PubMed] [Google Scholar]
- Roche HM, Gibney MJ. Effect of long-chain n-3 polyunsaturated fatty acids on fasting and postprandial triacylglycerol metabolism. Am J Clin Nutr. 2000;71:232S–237S. doi: 10.1093/ajcn/71.1.232s. [DOI] [PubMed] [Google Scholar]
- Abeywardena MY, Head RJ. Longchain n-3 polyunsaturated fatty acids and blood vessel function. Cardiovasc Res. 2001;52:361–371. doi: 10.1016/S0008-6363(01)00406-0. [DOI] [PubMed] [Google Scholar]
- Escudero A, Montilla JC, Garcia JM, Sanchez-Quevedo MC, Periago JL, Hortelano P, Suarez MD. Effect of dietary (n-9), (n-6) and (n-3) fatty acids on membrane lipid composition and morphology of rat erythrocytes. Biochim Biophys Acta. 1998;1394:65–73. doi: 10.1016/S0005-2760(98)00095-2. [DOI] [PubMed] [Google Scholar]
- Jump DB. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr Opin Lipidol. 2002;13:155–164. doi: 10.1097/00041433-200204000-00007. [DOI] [PubMed] [Google Scholar]
- Kitajka K, Puskas LG, Zvara A, Hackler L, Jr, Barcelo-Coblijn G, Yeo YK, Farkas T. The role of n-3 polyunsaturated fatty acids in brain: Modulation of rat brain gene expression by dietary n-3 fatty acids. Proc Natl Acad Sci U S A. 2002;99:2619–2624. doi: 10.1073/pnas.042698699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura MT, Cho HP, Xu J, Tang Z, Clarke SD. Metabolism and functions of highly unsaturated fatty acids: an update. Lipids. 2001;36:961–964. doi: 10.1007/s11745-001-0806-5. [DOI] [PubMed] [Google Scholar]
- Lauritzen L, Hansen HS, Jorgensen MH, Michaelsen KF. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog Lipid Res. 2001;40:1–94. doi: 10.1016/S0163-7827(00)00017-5. [DOI] [PubMed] [Google Scholar]
- Schiefermeier M, Yavin E. n-3 Deficient and docosahexaenoic acid-enriched diets during critical periods of the developing prenatal rat brain. J Lipid Res. 2002;43:124–131. [PubMed] [Google Scholar]
- Ferdinandusse S, Denis S, Mooijer P, Zhang Z, Reddy J, Spector A, Wanders R. Identification of the peroxisomal β-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J Lipid Res. 2001;42:1987–1995. [PubMed] [Google Scholar]
- Duplus E, Forest C. Is there a single mechanism for fatty acid regulation of gene transcription? Biochem Pharmacol. 2002;64:893. doi: 10.1016/S0006-2952(02)01157-7. [DOI] [PubMed] [Google Scholar]
- Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol. 2001;11:180–187. doi: 10.1016/S0959-4388(00)00194-X. [DOI] [PubMed] [Google Scholar]
- Murthy M, Hamilton J, Greiner RS, Moriguchi T, Salem N, Jr, Kim HY. Differential effects of n-3 fatty acid deficiency on phospholipid molecular species composition in the rat hippocampus. J Lipid Res. 2002;43:611–617. [PubMed] [Google Scholar]
- Qi K, Hall M, Deckelbaum RJ. Long-chain polyunsaturated fatty acid accretion in brain. Curr Opin Clin Nutr Metab Care. 2002;5:133–138. doi: 10.1097/00075197-200203000-00003. [DOI] [PubMed] [Google Scholar]
- Mutch DM, Berger A, Mansourian R, Rytz A, Roberts MA. The limit fold change model: A practical approach for selecting differentially expressed genes from microarray data. BMC Bioinformatics. 2002;3:17. doi: 10.1186/1471-2105-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. http://www.geneontology.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkaoui-Malki M, Meyer K, Cao WQ, Latruffe N, Yeldandi AV, Rao MS, Bradfield CA, Reddy JK. Identification of novel peroxisome proliferator-activated receptor alpha (PPARα) target genes in mouse liver using cDNA microarray analysis. Gene Expr. 2001;9:291–304. doi: 10.3727/000000001783992533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson L, Linden D, Jalouli M, Oscarsson J. Effects of fatty acids and growth hormone on liver fatty acid binding protein and PPARα in rat liver. Am J Physiol Endocrinol Metab. 2001;281:E772–E781. doi: 10.1152/ajpendo.2001.281.4.E772. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Fox-Robichaud A, Kikkawa R, Kashiwagi A, Kojima H, Fujimiya M, Wong NC. Transcription factors and age-related decline in apolipoprotein A-I expression. J Lipid Res. 1999;40:1709–1718. [PubMed] [Google Scholar]
- Sprecher H. Metabolism of highly unsaturated n-3 and n-6 fatty acids. Biochim Biophys Acta. 2000;1486:219–231. doi: 10.1016/S1388-1981(00)00077-9. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem. 2002;277:1705–1711. doi: 10.1074/jbc.M105711200. [DOI] [PubMed] [Google Scholar]
- Moon YA, Shah NA, Mohapatra S, Warrington JA, Horton JD. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J Biol Chem. 2001;276:45358–45366. doi: 10.1074/jbc.M108413200. [DOI] [PubMed] [Google Scholar]
- Shimano H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res. 2001;40:439–452. doi: 10.1016/S0163-7827(01)00010-8. [DOI] [PubMed] [Google Scholar]
- Jump D, Thelen A, Mater M. Functional interaction between sterol regulatory element-binding. J Biol Chem. 2001;276:34419–34427. doi: 10.1074/jbc.M105471200. [DOI] [PubMed] [Google Scholar]
- Moon YS, Latasa M-J, Griffin MJ, Sul HS. Suppression of fatty acid synthase promoter by polyunsaturated fatty acids. J Lipid Res. 2002;43:691–698. [PubMed] [Google Scholar]
- Teran-Garcia M, Rufo C, Nakamura MT, Osborne TF, Clarke SD. NF-Y involvement in the polyunsaturated fat inhibition of fatty acid synthase gene transcription. Biochem Biophys Res Commun. 2002;290:1295–1299. doi: 10.1006/bbrc.2002.6341. [DOI] [PubMed] [Google Scholar]
- Navas MA, Vaisse C, Boger S, Heimesaat M, Kollee LA, Stoffel M. The human HNF-3 genes: cloning, partial sequence and mutation screening in patients with impaired glucose homeostasis. Hum Hered. 2000;50:370–381. doi: 10.1159/000022943. [DOI] [PubMed] [Google Scholar]
- Cederberg A, Gronning LM, Ahren B, Tasken K, Carlsson P, Enerback S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell. 2001;106:563–573. doi: 10.1016/s0092-8674(01)00474-3. [DOI] [PubMed] [Google Scholar]
- Lin B, Morris DW, Chou JY. The role of HNF1α, HNF3γ, and cyclic AMP in glucose-6-phosphatase gene activation. Biochemistry. 1997;36:14096–14106. doi: 10.1021/bi9703249. [DOI] [PubMed] [Google Scholar]
- Kaestner K, Hiemisch H, Luckow B, Schutz G. The HNF-3 gene family of transcription factors in mice: gene structure. Genomics. 1994;20:377–385. doi: 10.1006/geno.1994.1191. [DOI] [PubMed] [Google Scholar]
- Fukuda H, Iritani N, Katsurada A, Noguchi T. Insulin- and polyunsaturated fatty acid-responsive region(s) of rat ATP citrate lyase gene promoter. FEBS Lett. 1996;380:204–207. doi: 10.1016/0014-5793(96)00031-2. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Vavvas D, Kurowski T, Laybutt DR, Schmitz-Peiffer C, Biden T, Kraegen EW. Malonyl CoA as a metabolic switch and a regulator of insulin sensitivity. Adv Exp Med Biol. 1998;441:263–270. doi: 10.1007/978-1-4899-1928-1_24. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Kaneyuki T, Tagawa K. Production of acetate in the liver and its utilization in peripheral tissues. Biochim Biophys Acta. 2001;1532:79–87. doi: 10.1016/S1388-1981(01)00117-2. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Yamamoto J, Okamura M, Fujino T, Takahashi S, Takeuchi K, Osborne TF, Yamamoto TT, Ito S, Sakai J. Transcriptional regulation of the murine acetyl-CoA synthetase 1 gene through multiple clustered binding sites for sterol regulatory element-binding proteins and a single neighboring site for Sp1. J Biol Chem. 2001;276:34259–34269. doi: 10.1074/jbc.M103848200. [DOI] [PubMed] [Google Scholar]
- Sone H, et al. Acetyl-coenzyme A synthetase is a lipogenic enzyme controlled by SREBP-1 and energy status. Am J Physiol Endocrinol Metab. 2002;282:E222–230. doi: 10.1152/ajpendo.00189.2001. [DOI] [PubMed] [Google Scholar]
- Martin G, Poirier H, Hennuyer N, Crombie D, Fruchart JC, Heyman RA, Besnard P, Auwerx J. Induction of the fatty acid transport protein 1 and acyl-CoA synthase genes by dimer-selective rexinoids suggests that the peroxisome proliferator-activated receptor-retinoid X receptor heterodimer is their molecular target. J Biol Chem. 2000;275:12612–12618. doi: 10.1074/jbc.275.17.12612. [DOI] [PubMed] [Google Scholar]
- Eaton S. Control of mitochondrial β-oxidation flux. Prog Lipid Res. 2002;41:197–239. doi: 10.1016/S0163-7827(01)00024-8. [DOI] [PubMed] [Google Scholar]
- Chen Q, Luthria DL, Sprecher H. Analysis of the acyl-CoAs that accumulate during the peroxisomal beta-oxidation of arachidonic acid and 6,9,12-octadecatrienoic acid. Arch Biochem Biophys. 1998;349:371–375. doi: 10.1006/abbi.1997.0461. [DOI] [PubMed] [Google Scholar]
- Rajas F, Gautier A, Bady I, Montano S, Mithieux G. Polyunsaturated fatty acyl-coenzyme A suppress the glucose-6 phosphatase promoter activity by modulating the DNA binding of hepatocyte nuclear factor 4 α. J Biol Chem. 2002;10.1074/jbc.M200971200:25. doi: 10.1074/jbc.M200971200. [DOI] [PubMed] [Google Scholar]
- Schwartz JH. The many dimensions of cAMP signaling. Proc Natl Acad Sci U S A. 2001;98:13482–13484. doi: 10.1073/pnas.251533998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Zhao AZ, Sonnenburg WK, Beavo JA. Stage and cell-specific expression of calmodulin-dependent phosphodiesterases in mouse testis. Biol Reprod. 2001;64:1746–1754. doi: 10.1095/biolreprod64.6.1746. [DOI] [PubMed] [Google Scholar]
- Roesler WJ. What is a cAMP response unit? Mol Cell Endocrinol. 2000;162:1–7. doi: 10.1016/S0303-7207(00)00198-2. [DOI] [PubMed] [Google Scholar]
- Chakravarty K, Leahy P, Becard D, Hakimi P, Foretz M, Ferre P, Foufelle F, Hanson RW. Sterol regulatory element-binding protein-1c mimics the negative effect of insulin on phosphoenolpyruvate carboxykinase (GTP) gene transcription. J Biol Chem. 2001;276:34816–34823. doi: 10.1074/jbc.M103310200. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli W. PPAR: Nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Capdevila JH, Falck JR. The CYP P450 arachidonic acid monooxygenases: from cell signaling to blood pressure regulation. Biochem Biophys Res Commun. 2001;285:571–576. doi: 10.1006/bbrc.2001.5167. [DOI] [PubMed] [Google Scholar]
- Ntambi JM, Bene H. Polyunsaturated fatty acid regulation of gene expression. J Mol Neurosci. 2001;16:273–278. doi: 10.1385/JMN:16:2-3:273. [DOI] [PubMed] [Google Scholar]
- Toke DA, Martin CE. Isolation and characterization of a gene affecting fatty acid elongation in Saccharomyces cerevisiae. J Biol Chem. 1996;271:18413–18422. doi: 10.1074/jbc.271.31.18413. [DOI] [PubMed] [Google Scholar]
- Oh CS, Toke DA, Mandala S, Martin CE. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J Biol Chem. 1997;272:17376–17384. doi: 10.1074/jbc.272.28.17376. [DOI] [PubMed] [Google Scholar]
- Tvrdik P, Westerberg R, Silve S, Asadi A, Jakobsson A, Cannon B, Loison G, Jacobsson A. Role of a new mammalian gene family in the biosynthesis of very long chain fatty acids and sphingolipids. J Cell Biol. 2000;149:707–718. doi: 10.1083/jcb.149.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinetti G, et al. PPARα and PPARγ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Langmann T. Structure, function and regulation of the ABC1 gene product. Curr Opin Lipidol. 2001;12:129–140. doi: 10.1097/00041433-200104000-00006. [DOI] [PubMed] [Google Scholar]
- Wang Y, Oram JF. Unsaturated fatty acids inhibit cholesterol efflux from macrophages by increasing degradation of ATP-binding cassette transporter A1. J Biol Chem. 2002;277:5692–5697. doi: 10.1074/jbc.M109977200. [DOI] [PubMed] [Google Scholar]
- Teboul L, Febbraio M, Gaillard D, Amri EZ, Silverstein R, Grimaldi PA. Structural and functional characterization of the mouse fatty acid translocase promoter: activation during adipose differentiation. Biochem J. 2001;360:305–312. doi: 10.1042/0264-6021:3600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohnert BI, Bernlohr DA. Regulation of fatty acid transporters in mammalian cells. Prog Lipid Res. 2000;39:83–107. doi: 10.1016/S0163-7827(99)00018-1. [DOI] [PubMed] [Google Scholar]
- Yu LR, Zeng R, Shao XX, Wang N, Xu YH, Xia QC. Identification of differentially expressed proteins between human hepatoma and normal liver cell lines by two-dimensional electrophoresis and liquid chromatography-ion trap mass spectrometry. Electrophoresis. 2000;21:3058–3068. doi: 10.1002/1522-2683(20000801)21:14<3058::AID-ELPS3058>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Hohoff C, Borchers T, Rustow B, Spener F, van-Tilbeurgh H. Expression, purification, and crystal structure determination of recombinant human epidermal-type fatty acid binding protein. Biochemistry. 1999;38:12229–12239. doi: 10.1021/bi990305u. [DOI] [PubMed] [Google Scholar]
- Hagens G, Masouye I, Augsburger E, Hotz R, Saurat JH, Siegenthaler G. Calcium-binding protein S100A7 and epidermal-type fatty acid-binding protein are associated in the cytosol of human keratinocytes. Biochem J. 1999;339:419–427. doi: 10.1042/0264-6021:3390419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayser P, Grimm H, Grimminger F. n-3 fatty acids in psoriasis. Br J Nutr. 2002;87:S77–S82. doi: 10.1079/BJN2001459. [DOI] [PubMed] [Google Scholar]
- Hsu MH, Savas U, Griffin KJ, Johnson EF. Identification of peroxisome proliferator-responsive human genes by elevated expression of the peroxisome proliferator-activated receptor α in HepG2 cells. J Biol Chem. 2001;276:27950–27958. doi: 10.1074/jbc.M100258200. [DOI] [PubMed] [Google Scholar]
- Lawrence JW, Li Y, Chen S, DeLuca JG, Berger JP, Umbenhauer DR, Moller DE, Zhou G. Differential gene regulation in human versus rodent hepatocytes by peroxisome proliferator-activated receptor (PPAR) α. PPARα fails to induce peroxisome proliferation-associated genes in human cells independently of the level of receptor expresson. J Biol Chem. 2001;276:31521–31527. doi: 10.1074/jbc.M103306200. [DOI] [PubMed] [Google Scholar]
- Nelson GJ, Schmidt PC, Bartolini GL, Kelley DS, Kyle D. The effect of dietary docosahexaenoic acid on plasma lipoproteins and tissue fatty acid composition in humans. Lipids. 1997;32:1137–1146. doi: 10.1007/s11745-997-0146-5. [DOI] [PubMed] [Google Scholar]
- Cohen RD, Castellani LW, Qiao JH, Van Lenten B, Lusis AJ, Reue K. Reduced aortic lesions and elevated high density lipoprotein levels in transgenic mice overexpressing mouse apolipoprotein A-IV. J Clin Invest. 1997;99:1906–1916. doi: 10.1172/JCI119358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raspe E, Madsen L, Lefebvre AM, Leitersdorf I, Gelman L, Peinado-Onsurbe J, Dallongeville J, Fruchart JC, Berge R, Staels B. Modulation of rat liver apolipoprotein gene expression and serum lipid levels by tetradecylthioacetic acid (TTA) via PPARα activation. J Lipid Res. 1999;40:2099–2110. [PubMed] [Google Scholar]
- Ricote M, Glass CK. New roles for PPARs in cholesterol homeostasis. Trends Pharmacol Sci. 2001;22:441–443. doi: 10.1016/S0165-6147(00)01802-2. [DOI] [PubMed] [Google Scholar]
- Weinberg RB, Patton CS. Binding of human apolipoprotein A-IV to human hepatocellular plasma membranes. Biochim Biophys Acta. 1990;1044:255–261. doi: 10.1016/0005-2760(90)90311-K. [DOI] [PubMed] [Google Scholar]
- The complete dataset is posted at the NCBI gene expression Omnibus http://www.ncbi.nlm.nih.gov/geo/ Accession Number: GSE91.
- Lim CF, Munro SL, Wynne KN, Topliss DJ, Stockigt JR. Influence of nonesterified fatty acids and lysolecithins on thyroxine binding to thyroxine-binding globulin and transthyretin. Thyroid. 1995;5:319–324. doi: 10.1089/thy.1995.5.319. [DOI] [PubMed] [Google Scholar]
- Watanabe CM, Wolffram S, Ader P, Rimbach G, Packer L, Maguire JJ, Schultz PG, Gohil K. The in vivo neuromodulatory effects of the herbal medicine Ginkgo biloba. Proc Natl Acad Sci U S A. 2001;98:6577–6580. doi: 10.1073/pnas.111126298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Ohta T, Watanabe K, Yoshimura H, Yamamoto I. Anandamide, an endogenous cannabinoid receptor ligand, also interacts with 5-hydroxytryptamine (5-HT) receptor. Biol Pharm Bull. 1998;21:224–226. doi: 10.1248/bpb.21.224. [DOI] [PubMed] [Google Scholar]
- Fan P. Cannabinoid agonists inhibit the activation of 5-HT3 receptors in rat nodose ganglion neurons. J Neurophysiol. 1995;73:907–910. doi: 10.1152/jn.1995.73.2.907. [DOI] [PubMed] [Google Scholar]
- Berger A, Crozier G, Bisogno T, Cavaliere P, Innis S, Di-Marzo V. Anandamide and diet: inclusion of dietary arachidonate and docosahexaenoate leads to increased brain levels of the corresponding N-acylethanolamines in piglets. Proc Natl Acad Sci U S A. 2001;98:6402–6406. doi: 10.1073/pnas.101119098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Togashi H, Mori K, Ueno- K, Ohashi S, Kojima T, Yoshioka M. Evidence for involvement of central 5-HT4 receptors in cholinergic function associated with cognitive processes: behavioral, electrophysiological, and neurochemical studies. J Pharmacol Exp Ther. 2001;296:676–682. [PubMed] [Google Scholar]
- Raygada M, Cho E, Hilakivi-Clarke L. High maternal intake of polyunsaturated fatty acids during pregnancy in mice alters offsprings' aggressive behavior, immobility in the swim test, locomotor activity and brain protein kinase C activity. J Nutr. 1998;128:2505–2511. doi: 10.1093/jn/128.12.2505. [DOI] [PubMed] [Google Scholar]
- Madani S, Hichami A, Legrand A, Belleville J, Khan NA. Implication of acyl chain of diacylglycerols in activation of different isoforms of protein kinase C. FASEB J. 2001;15:2595–2601. doi: 10.1096/fj.01-0753int. [DOI] [PubMed] [Google Scholar]
- Sato C, Fukuoka H, Ohta K, Matsuda T, Koshino R, Kobayashi K, Troy FA, Kitajima K. Frequent occurrence of pre-existing α2→8-linked disialic and oligosialic acids with chain lengths up to 7 Sia residues in mammalian brain glycoproteins. Prevalence revealed by highly sensitive chemical methods and anti-, di-, oligo-, and poly-Sia antibodies specific for defined chain lengths. J Biol Chem. 2000;275:15422–15431. doi: 10.1074/jbc.275.20.15422. [DOI] [PubMed] [Google Scholar]
- Gallagher HC, Murphy KJ, Foley AG, Regan CM. Protein kinase C delta regulates neural cell adhesion molecule polysialylation state in the rat brain. J Neurochem. 2001;77:425–434. doi: 10.1046/j.1471-4159.2001.00235.x. [DOI] [PubMed] [Google Scholar]
- Seidenfaden R, Hildebrandt H. Retinoic acid-induced changes in polysialyltransferase mRNA expression and NCAM polysialylation in human neuroblastoma cells. J Neurobiol. 2001;46:11–28. doi: 10.1002/1097-4695(200101)46:1<11::AID-NEU2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Pritchard LE, Turnbull AV, White A. Pro-opiomelanocortin processing in the hypothalamus: impact on melanocortin signalling and obesity. J Endocrinol. 2002;172:411–421. doi: 10.1677/joe.0.1720411. [DOI] [PubMed] [Google Scholar]
- Ziotopoulou M, Mantzoros CS, Hileman SM, Flier JS. Differential expression of hypothalamic neuropeptides in the early phase of diet-induced obesity in mice. Am J Physiol Endocrinol Metab. 2000;279:E838–845. doi: 10.1152/ajpendo.2000.279.4.E838. [DOI] [PubMed] [Google Scholar]
- Ide T, Kobayashi H, Ashakumary L, Rouyer IA, Takahashi Y, Aoyama T, Hashimoto T, Mizugaki M. Comparative effects of perilla and fish oils on the activity and gene expression of fatty acid oxidation enzymes in rat liver. Biochim Biophys Acta. 2000;1485:23–35. doi: 10.1016/S1388-1981(00)00026-3. [DOI] [PubMed] [Google Scholar]
- Raclot T, Groscolas R, Langin D, Ferre P. Site-specific regulation of gene expression by n-3 polyunsaturated fatty acids in rat white adipose tissues. J Lipid Res. 1997;38:1963–1972. [PubMed] [Google Scholar]
- Liang CP, Tall AR. Transcriptional profiling reveals global defects in energy metabolism, lipoprotein, and bile acid synthesis and transport with reversal by leptin treatment in Ob/ob mouse liver. J Biol Chem. 2001;276:49066–49076. doi: 10.1074/jbc.M107250200. [DOI] [PubMed] [Google Scholar]
- Chalon S, Delion-Vancassel S, Belzung C, Guilloteau D, Leguisquet AM, Besnard JC, Durand G. Dietary fish oil affects monoaminergic neurotransmission and behavior in rats. J Nutr. 1998;128:2512–2519. doi: 10.1093/jn/128.12.2512. [DOI] [PubMed] [Google Scholar]