

Abstract
Background
For effective exposition of biological information, especially with regard to analysis of large-scale data types, researchers need immediate access to multiple categorical knowledge bases and need summary information presented to them on collections of genes, as opposed to the typical one gene at a time.
Results
We present here a web-based tool (FunSpec) for statistical evaluation of groups of genes and proteins (e.g. co-regulated genes, protein complexes, genetic interactors) with respect to existing annotations (e.g. functional roles, biochemical properties, localization). FunSpec is available online at http://funspec.med.utoronto.ca
Conclusion
FunSpec is helpful for interpretation of any data type that generates groups of related genes and proteins, such as gene expression clustering and protein complexes, and is useful for predictive methods employing "guilt-by-association."
Background
A large proportion of the online resources compiling information about gene and protein functions present information one-gene-at-a-time [1-3]. Interpretation of large-scale data types, however, often requires a rapid means to access and present information on large numbers of genes simultaneously [4,5]. Recent tools that provide annotation of gene expression data involve text mining of biomedical literature [6] and the integration of a wide variety of data types including Pfam domains, SWISS-PROT keywords and known pathways (e.g. DRAGON View; see [7]). We have developed an online resource called FunSpec (for Functional Specification), which uses information from public databases to evaluate lists of yeast genes to determine if they are enriched for particular attributes, using a well-accepted statistical model. The data tables underlying FunSpec are updated regularly from major public-domain knowledge sources, and from our analyses of large-scale data sets. Hence, FunSpec is a convenient, current, and comprehensive resource for any application requiring interpretation of groups of genes, including clustering of microarray expression data, lists of protein IDs from mass spectrometry, and genetic interactions.
Results
Online database and published data repositories
Yeast, perhaps the most thoroughly-studied eukaryote, already has a number of comprehensive online databases containing functional annotation information for close to two-thirds of all known or predicted open reading frames (ORFs) in the genome, and numerous large-scale experimental results covering practically every gene/protein (eg., [8,9]). In many cases there are links between the data sources, though the majority of these are at the individual gene level. For example, the Comprehensive Yeast Genome Database at the Munich Information Center for Protein Sequences (MIPS) lists five catalogues encompassing a curation of several thousand publications and spanning functional classification, known protein complexes, protein classes, mutant phenotypes and subcellular localization [2]. Similarly, the Gene Ontology (GO) Consortium organizes the same information under the categorizations: Biological Process, Molecular Function and Cellular Component [10]. Other online knowledge bases encompass computationally-derived gene properties, such as domain and motif information (e.g. SMART [11], Pfam [12]). In addition, there are now mass stores of high-throughput biological data accumulating from experimental results such as synthetic lethal [13], protein interactions by yeast two hybrid [14,15] or mass spectrometry [16,17], immunolocalization [18], large-scale phenotypic analysis [19], and microarray analysis (eg., [8]).
A listing of the current databases that can be evaluated with FunSpec is given in Table 1. The published experimental information that can be queried is given in Table 2. Through our own analyses of recent literature on ribosomal RNA processing, additional categories are provided to elicit enrichment of pre-ribosome components. Also, a separate category has been added to encompass genes that are essential for viability [20].
Table 1.
FunSpec-searchable public databases
| Resource | Website | Publication |
|---|---|---|
| Munich Information Center for Protein Sequences (MIPS) | http://mips.gsf.de/proj/yeast/CYGD/db/index.html | [2] |
| Gene Ontology (GO) | http://www.geneontology.org/ | [10] |
| Simple Modular Architecture Research Tool | http://smart.embl-heidelberg.de/ | [11] |
| Protein families database of alignments and HMMs | http://www.sanger.ac.uk/Software/Pfam/ | [12] |
Table 2.
FunSpec-searchable published datasets
| Data Type | Publication |
|---|---|
| Protein-Protein Interactions via Yeast Two Hybrid Method | [14] |
| Protein-Protein Interactions via Yeast Two Hybrid Method (core + full datasets) | [15] |
| Synthetic Lethal Interactions via Synthetic Genetic Array analysis | [13] |
| Protein Complex Identification using TAP tagging and mass spectrometry | [16] |
| Protein Complex Identification using Flag tagging and mass spectrometry | [17] |
| Proteome Localization | [18] |
| Essentiality | [20] |
Interpretation of clusters
Upon collection of data resulting in clusters of genes/proteins having similar properties, responses, or other associations, often a researcher may wish to summarize and interpret the current state of knowledge of genes in such a cluster. A cluster of genes can be said to be "functionally enriched" for an attribute if the proportion of genes within the cluster known to have that attribute exceeds the number that could reasonably be expected from random chance. For example, 215 of the 6267 genes in yeast [1] are known to function in ribosome biogenesis. If, in a clustering analysis of gene expression profiles, one finds a cluster of 100 genes to be co-regulated and of those, 60 are known to function in ribosome biogenesis, then intuitively the cluster is enriched for ribosome biogenesis (60% in the cluster versus 3% in the genome). The degree of functional enrichment for a given cluster and category can be quantitatively assessed by the hypergeometric distribution [21]. For each category, the probability (p-value) of observing such an overlap by chance is calculated as: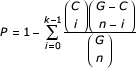
where G is the size of the genome, C is the number of genes in the genome having that attribute, n is the size of the query cluster, of which k are known to possess the attribute.
If this probability is sufficiently low for a given category, then a list of genes (e.g., a cluster) is said to be enriched for that attribute (see examples below). FunSpec inputs a list of genes and computes the hypergeometric P-value in many knowledge sources (MIPS, GO, SMART and Pfam domains, published protein complexes, 2-hybrid interactions, and subcellular localizations). A Bonferroni correction may be applied to compensate for multiple testing over many categories of a knowledge base.
Examples
In this section, we describe instances where FunSpec may be useful. In Figure 1, a 2-dimensional clustering of gene expression data from [8], covering 237 genes and 271 experiments is presented. The 76 genes indicated by the red bar were submitted to MIPS Functional Classification database on the FunSpec website. The categories "amino acid biosynthesis" and "amino acid metabolism" were deemed most unlikely to occur by chance (p < 10-14 in both cases). To show the specificity of evaluating clusters in this manner, the 6 genes indicated in blue (isolated by visual inspection) have the same overall expression profile as genes generally involved in amino acid metabolism, but with a few distinct differences. FunSpec identifies these as involved in "methionine metabolism." (p = 1.0 × 10-7).
Figure 1.

A 2-D clustering analysis of yeast gene expression data from [8].
Components of purified protein complexes can also be interpreted using FunSpec. It is noteworthy that functional annotations, most of which are based on phenotypic rather than biochemical data, generally correlate with large protein complexes in the literature (rand index = 0.94, adjusted rand index = 0.15; rand index measures the similarity of two partitions where 0 represents no similarity and 1 is complete correspondence; the adjusted rand index is a much more conservative measure [22]) (data not shown). This underscores the utility of interpreting one data type with information from another [23,24]. Subcellular localization correlates positively with membership in protein complexes, as would be expected to enable physical association (among all protein complexes in the MIPS database, 17% are enriched (P < 0.01) for a specific subcellular compartment in the Kumar experimental data [18], in comparison to 1% for random lists of proteins with the same size distribution) (data not shown). For newly-identified complexes, FunSpec provides not only information about localization and potential function, but also a measure of confidence regarding the biological validity of the complex.
Discussion
The current FunSpec implementation utilizes the hypergeometric distribution (following [21]), which assumes independent tests (i.e. each gene has a single classification). In reality, the MIPS and GO categories are hierarchical. We are currently considering other statistical models to account for the hierarchical nature of the categorization, as well as alternatives to the Bonferroni correction (eg. False Discovery Rate [25]).
There are other methods of determining significance (e.g. binomial distribution [26]; representation factor [27]) and other means of relating clusters to functional annotation (e.g. associated terms and phrases in Medline abstracts [6]) but the hypergeometric distribution has proven to be the simplest to use. The advantages of FunSpec over current tools is the integration of many relevant and recent data sources and a convenient website enabling the user to explore special features of gene groupings quickly and exhaustively.
In the near future, as more functional annotation information becomes available for other organisms such as mouse and human, the FunSpec resource will be updated to accommodate the new knowledge bases on these organisms, presumably including a great deal of new experimental data. The underlying assembly of categorical annotations is useful on its own and can be downloaded from the FunSpec web site as a text file.
Conclusions
FunSpec takes clusters (or any collection of genes or proteins) and identifies attributes (based on prior knowledge) for which the cluster is enriched. In our own research we use FunSpec routinely for rapid interpretation of all clusters in full-genome clustering analyses, as well as for "guilt-by-association" predictive methodologies (e.g. [5,8,28]). We and others have also found it useful as a filter for prioritizing screening hits, interpreting genetic interactions, and selecting thresholds/boundaries for membership in protein complexes (data not shown).
Methods
The FunSpec backend is implemented in C++. Upon receiving the information from the web page, the relevant databases are queried, the statistics are calculated and the results are sent back to the user. At present, the databases are stored in flat text files.
Authors' contributions
MR downloaded and organized the biological knowledge bases, coded the statistical routines, and drafted the manuscript. JG created a FunSpec prototype and participated in locating and evaluating online databases. NM formed the website. TH coordinated execution of the project.
All authors read and approved the final manuscript.
Contributor Information
Mark D Robinson, Email: m.robinson@utoronto.ca.
Jörg Grigull, Email: jorg.grigull@utoronto.ca.
Naveed Mohammad, Email: naveed.mohammad@utoronto.ca.
Timothy R Hughes, Email: t.hughes@utoronto.ca.
Acknowledgements
The authors thank members of the Best Institute (University of Toronto) for testing the web implementation, and Gary Bader, Mike Tyers, and Andrew Emili for critical evaluation of this manuscript. This work was supported by Genome Canada, the CIHR, and the University of Toronto Connaught Foundation.
References
- Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Gruber C, Geier B, Haase D, Kaps A, Lemcke K, Mannhaupt G, Pfeiffer F, Schuller C. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2000;28:37–40. doi: 10.1093/nar/28.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo MC, Crawford ME, Hirschman JE, Kranz JE, Olsen P, Robertson LS, Skrzypek MS, Braun BR, Hopkins KL, Kondu P. YPD, PombePD and WormPD: model organism volumes of the BioKnowledge library, an integrated resource for protein information. Nucleic Acids Res. 2001;29:75–79. doi: 10.1093/nar/29.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum D, Luscombe NM, Jansen R, Qian J, Gerstein M. Interrelating different types of genomic data, from proteome to secretome: 'oming in on function. Genome Res. 2001;11:1463–1468. doi: 10.1101/gr.207401. [DOI] [PubMed] [Google Scholar]
- Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, Altschuler SJ. Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet. 2002;31:255–265. doi: 10.1038/ng906. [DOI] [PubMed] [Google Scholar]
- Blaschke C, Oliveros JC, Valencia A. Mining functional information associated with expression arrays. Funct Integr Genomics. 2001;1:256–268. doi: 10.1007/s101420000036. [DOI] [PubMed] [Google Scholar]
- Bouton CM, Pevsner J. DRAGON View: information visualization for annotated microarray data. Bioinformatics. 2002;18:323–324. doi: 10.1093/bioinformatics/18.2.323. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Kumar A, Cheung KH, Ross-Macdonald P, Coelho PS, Miller P, Snyder M. TRIPLES: a database of gene function in Saccharomyces cerevisiae. Nucleic Acids Res. 2000;28:81–84. doi: 10.1093/nar/28.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. Creating the gene ontology resource: design and implementation. Genome Res. 2001;11:1425–1433. doi: 10.1101/gr.180801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, Jansen R, Cheung KH, Sheehan A, Symoniatis D, Umansky L. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;402:413–418. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Hubert L, Arabie P. Comparing Partitions. Journal of Classification. 1985;2:193–218. [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Kemmeren P, van Berkum NL, Vilo J, Bijma T, Donders R, Brazma A, Holstege FC. Protein interaction verification and functional annotation by integrated analysis of genome-scale data. Mol Cell. 2002;9:1133–1143. doi: 10.1016/s1097-2765(02)00531-2. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B. 1995;57:289–300. [Google Scholar]
- Cho RJ, Huang M, Campbell MJ, Dong H, Steinmetz L, Sapinoso L, Hampton G, Elledge SJ, Davis RW, Lockhart DJ. Transcriptional regulation and function during the human cell cycle. Nat Genet. 2001;27:48–54. doi: 10.1038/83751. [DOI] [PubMed] [Google Scholar]
- Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS. A gene expression map for Caenorhabditis elegans. Science. 2001;293:2087–2092. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome-wide prediction of protein function. Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]