


Abstract
Telomerase is a specialized reverse transcriptase that contains an integral RNA subunit including a short template sequence. It extends telomeric 3′ overhangs and chromosome breakpoints by catalyzing reiterative copying of this internal template into single-stranded telomeric DNA repeats. Here we report for the first time that in vitro the ciliate Tetrahymena telomerase can efficiently extend very short single-stranded DNA primers (<6 nt). These data indicate that interactions with nucleotides further upstream are not essential for elongation of longer primers. We also report that the minimal lengths required for primers to be extended by the telomerase depend on the positions along the template at which the primers are initially aligned. At a primer concentration of 2.5 µM, primers aligned in the beginning, middle and next to the end of the template, respectively, must consist of at least 4, 5 and 6 nt to be extended by the telomerase. At a primer concentration of 50 µM, the corresponding minimal lengths are 3, 4 and 5 nt. The systematic variation of the minimal required primer lengths supports the presence of a site within the telomerase ribonucleoprotein complex that mediates specific positioning of 3′ termini of telomeric and non-telomeric DNA in the beginning of the template during telomere synthesis.
INTRODUCTION
The telomeric DNA of most eukaryotes consists of short repeats, including clusters of G nucleotides in one strand and complementary clusters of C nucleotides in the other strand. The G-rich strand ends as a 3′ overhang (reviewed in 1). Telomerase is a specialized reverse transcriptase that catalyzes the extension of the telomeric single-stranded 3′ overhangs. It also catalyzes de novo addition of single-stranded G-rich telomeric repeats to breakpoints in chromosomes (reviewed in 2–5). The basic catalytic core of telomerase consists of an RNA subunit, designated telomerase RNA (TER) (6), and a protein subunit, designated telomerase reverse transcriptase (TERT) (7–9). The holoenzyme contains additional auxiliary subunits (reviewed in 2,4). The TER subunit includes a short sequence, which serves as a template for synthesis of the G-rich telomeric repeats. Other regions of TER include conserved secondary structure features that play essential roles in the telomerase ribonucleoprotein assembly and in the catalytic activity of the enzyme (10–14). The TERT subunit contains a cluster of short motifs (RT motifs), which are conserved among all reverse transcriptases (8,15). The region of TERT containing these motifs includes the active site of the enzyme. Another region of TERT, which is closer to the N-terminus of the protein, contains additional short motifs that are conserved among telomerases (16–18). This region is necessary and sufficient for binding TER. Auxiliary proteins, which have been reported so far in telomerases of ciliates, yeasts and mammals, do not appear to share common features (19–21).
The Tetrahymena telomerase, which is the subject of the present report, was the first telomerase to be discovered and the most thoroughly characterized (22; reviewed in 2). In vitro studies revealed that this enzyme catalyzes processive elongation of single-stranded oligodeoxyribonucleotide primers by the multiple repeats that constitute the Tetrahymena telomeres, d(GGGGTT)n (23). Based on these studies and on in vivo observations, a model was proposed for the elongation reaction (5,24). According to this model, the 3′ overhang of a Tetrahymena telomere, which serves as a primer, aligns and forms a double helix with the intrinsic TER template region (3′-AACCCCAAC-5′; see Figs 1 and 5). It is then extended by the enzyme up to the end (5′ terminus) of the template region. Subsequently, the Watson–Crick hydrogen bonds between the primer and the template are disrupted and the 3′ terminus of the primer is translocated to the beginning (3′ terminus) of the template region, along with the active site of the enzyme. The translocation is followed by synthesis of the next telomeric repeat, and so on. To account for the processivity of the telomerase, it was suggested that, in addition to the active site, the enzyme contains a second site (anchor site), to which an upstream region of the primer remains bound during the translocation (24,25).
Figure 1.

Three series of oligodeoxyribonucleotide primers used for telomerase assays. The Tetrahymena TER template region is shown at the top of each panel. The template region consists of a sequence of 9 nt extending between residues nos 43 and 51 (6,36). (A–C) Series of oligo primers that were designed to align in the middle (A), in the beginning (B) and next to the end (C) of the template region.
Figure 5.
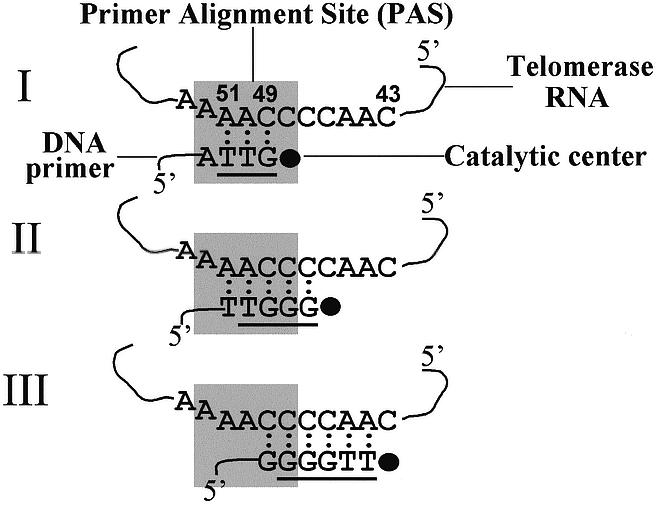
A scheme showing the minimal lengths required for primers aligned in the beginning, middle and next to the end of the template to be extended with the Tetrahymena telomerase. A segment of the Tetrahymena TER subunit is shown here (6). This segment includes the RNA template region, nucleotides 43–51. Nucleotides 43–49 serve as templating residues, i.e. they are copied by the telomerase into DNA telomeric repeats. Nucleotides 50–51 serve to align the primers along the template, but are not copied by the enzyme (36). The DNA nucleotide sequences indicated in schemes I, II and III designate the shortest primers that could be extended with the telomerase either by a single nucleotide, or by multiple repeats, at a primer concentration of 2.5 µM. The underlined sequences designate the shortest primers that could be extended with the telomerase by a single nucleotide at a primer concentration of 50 µM. The rectangle denotes the hypothetical PAS defined in the Discussion.
We have recently developed an interference footprinting technique for mapping the interactions that occur between the primers and the Tetrahymena telomerase during in vitro primer extension reactions (26). In this study, major functional interactions were detected between the telomerase and the six 3′ terminal residues of primers consisting of 17–35 nt residues. In addition, moderate interactions occurred with the seventh nucleotide residue. Only minor interactions occurred with additional nucleotides located elsewhere along these primers.
The observation that the telomerase interacted primarily with the six 3′-terminal residues of the primers suggested that sequences that map further upstream might not be essential for the elongation process. Other data indicated that the interactions between the primers and the telomerase may depend on the position that their 3′ termini initially occupy along the RNA template region (27). Therefore, we reasoned that the telomerase might be capable of extending oligonucleotide primers consisting of ≤6 nt and that the minimal length required for primer extension may depend on the initial alignment of the primer along the template. Here we report in vitro primer extension studies that supported these notions. At a moderate primer concentration of 2.5 µM, the minimal length required for a primer aligned at the beginning of the template to be extended either by a single nucleotide, or by multiple repeats, was 4 nt. The minimal lengths required for extension of primers aligned in the middle and next to the end of the template were 5 and 6 nt, respectively. The minimal primer lengths required for extension were found to be even smaller in assays performed at the very high primer concentration of 50 µM. The implications of these data for the mechanism of primer extension by telomerase are discussed.
MATERIALS AND METHODS
Oligonucleotides
DNA oligonucleotides were purchased from Sigma Genosis. Oligonucleotides longer than 8 nt residues were purified by electrophoresis in polyacrylamide gels, followed by ethanol precipitation and Sephadex G-50 spun column chromatography (26). Smaller oligonucleotides were not purified by this procedure, because their recovery was very low. We found that the purification only slightly improved the efficiency of extension of the oligonucleotides by telomerase.
Plasmids
The following plasmids were kindly sent to us by Dr K. Collins, University of California, Berkeley: pT7159, encoding the wild-type Tetrahymena TER (28); p133CITE, encoding the Tetrahymena TERT (29); pT7159C48G, encoding the mutant TER C48G (30). A plasmid encoding TERT that contained a C-terminal FLAG tag was used to express the tagged TERT.
Preparation of native telomerase
Tetrahymena thermophila vegetative cells were grown to logarithmic phase and starved as described (31,32). An S100 extract was prepared and telomerase was partially purified by chromatography in DEAE–Sepharose and Octyl–Sepharose columns (26,33).
Preparation of reconstituted core telomerase
Telomerase was reconstituted by co-expression of TER and TERT, using the coupled transcription/translation system in rabbit reticulocyte lysates (TNT T7 Quick System; Promega). Reconstitution reactions contained 200 µl of lysate, 20 µM methionine and equimolar amounts of TER encoding plasmid that had been digested with FokI, and a TERT encoding plasmid (7.5 and 15 µg/ml, respectively, of the plasmids). The mixtures were incubated at 30°C for 2 h. Then, 750 µl of TMG buffer [10 mM Tris–HCL (pH 8.0), 1 mM MgCl2, 10% glycerol, 5 mM β-mercaptoehanol] were added and aliquots were stored at –70°C, as described (30).
Immunopurification of the reconstituted telomerase
Immunopurified FLAG-tagged telomerase was prepared by modification of published procedures (12,34). ANTI-FLAG M2 affinity gel beads (Sigma) were washed in buffer A [20 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 0.1 M NaCl, 0.1 mM EDTA, 10% glycerol] and blocked in buffer A containing 100 µg/ml tRNA and 100µg/ml BSA. The reconstituted telomerase was mixed with the beads at a volume ratio of 3:1 and binding was accomplished by agitation at 4°C for 2 h. The beads were washed thoroughly first in buffer A containing 0.3 M NaCl and then in TMG buffer. Finally, the beads containing the bound telomerase were resuspended in an equal volume of TMG to make a 1:1 slurry.
Telomerase assays
Primer extension by a single nucleotide. Reactions were carried out in 20 µl mixtures containing 50 mM Tris–HCl (pH 8.0), 2 mM MgCl2, 0.1 M sodium acetate, 1 mM spermidine, 6 mM β-mercaptoethanol, 0.2 U/ml RNasin, 0.1 µM [α-32P]dGTP (specific radioactivity, 3000 Ci/mmol; Amersham Biosciences) or 0.3 µM [α-32P]dCTP (specific radioactivity, 3000 Ci/mmol), 2.5 or 50 µM of heat-denatured DNA primer and 5–10 µl of telomerase. In reactions containing immunopurified telomerase, 10 µl of 1:1 slurry were added. The reaction mixtures were incubated for 45 min at 10°C. Reactions were terminated by addition of 20 µl of TES2 solution [10 mM Tris–HCl (pH 8.0), 20 mM EDTA, 0.2% SDS]. Four microliter samples of each assay were mixed with sequencing loading buffer and analyzed without any further purification by electrophoresis in 16% Long Ranger (FMC) sequencing gels containing 7 M urea, as described before (26). The gels were dried and exposed to phosphorimaging screens that were subsequently scanned with a Molecular Dynamics PhosphorImager. Based on the specific radioactivity of the bands observed in the phosphorimaging screens, we calculated that in all the assays presented in this study, <2.5 × 10–4 of the input primer molecules were extended.
Primer extension by multiple repeats. Primer extension reactions were carried out as described in the section above, with the following changes: all reactions contained 1 µM [α-32P]dGTP (specific radioactivity, 300 Ci/mmol), 100 µM dTTP, 2.5 µM DNA primer and 5 µl of native telomerase. Reactions were terminated by addition of 110 µl of TES1 solution [10 mM Tris–HCl (pH 8.0), 20 mM EDTA, 0.1% SDS] and proteinase K to a final concentration of 0.05 mg/ml, and the mixtures were incubated at 45°C for 45 min. 32P-labeled 35mer oligonucleotide was added to each assay as a recovery marker and reaction products were precipitated with ethanol in the presence of 2.5 M ammonium acetate and 100 µg/ml yeast tRNA, as described (23,26,28).
Radioactive labeling of oligonucleotides
Oligonucleotides were labeled with 32PO4 at their 5′ end, using T4 polynucleotide kinase (New England Biolabs). Alternatively, the oligonucleotides were labeled at their 3′ end by extension with 32P-labeled dGTP, using terminal deoxynucleotidyl transferase (Promega) (35). The concentration of the [32P]dGTP in these reactions was limiting, such that the major product was a primer extended by a single [32P]dGMP residue. The radioactive oligonucleotides were not precipitated with ethanol before being electrophoresed in denaturing gels.
RESULTS
Characteristics of the oligodeoxynucleotides used as telomerase primers
Figure 1 shows three series of oligodeoxyribonucleotides ranging in length between 2 and 24 nt, which were tested in the present study for their ability to serve as primers for telomerase. Each series is shown in a separate panel along with the TER template region, which is illustrated at the top. The template region consists of a sequence of 9 nt extending between residues 43 and 51 (6,36). Nucleotides 43–49 serve as templating residues, that is, they are copied by the telomerase into DNA telomeric repeats. Nucleotides 50–51 serve to align the primers along the template, but are not copied by the enzyme (36). Figure 1A presents oligos of one series, which were designed to align in the middle of the RNA template region. The longest oligo in this series consisted of 21 nt, and included a non-telomeric sequence flanked by telomeric repeats at the 3′ and the 5′ termini. This oligo has been previously utilized for the interference footprinting studies mentioned in the Introduction [oligo tel1 (26)]. The other oligos shown in Figure 1A are shorter derivatives of the 21 nt oligo, in which 5′-terminal sequences of various lengths have been deleted. Figure 1B and C show the other two series of oligos, which were designed to align in the beginning and next to the end of the template, respectively.
Before using the oligos for telomerase primer extension assays, a sample of each oligo was electrophoresed in a denaturing gel and the bands were inspected by exposure to ultraviolet (UV) light at 260 nm. This analysis revealed that the mobilities of the short oligos were not correlated with their lengths (data not shown). To further check the authenticity of the oligos, the molecular masses of the 4mer, the 5mer and the 6mer oligos of the series shown in Figure 1A were determined by mass spectrometry and found to correspond to the expected values. We reasoned that the cause for the apparently abnormal mobilities of these oligos might be that they were unphosphorylated at both their 5′ and 3′ ends. To examine this hypothesis, samples of the oligos shown in Figure 1A were phosphorylated with DNA kinase at the 5′ termini, using γ-32P-labeled ATP. Other samples of the same oligos were extended with terminal transferase by a 32P-labeled dGMP residue at the 3′ termini. These samples were electrophoresed in a denaturing gel that was subsequently exposed to a phosphorimager screen and analyzed in a phosphorimager apparatus. Figure 2 shows these data. It can be seen that the samples that were extended with terminal transferase (TT oligos in Fig. 2) displayed a single major band, which contained the indicated oligos that were extended by a single 32P-labeled dGMP. The minor bands observed in these lanes contained the same oligos that were further extended by additional dGMP residues. These oligos remained unphosphorylated at both ends. The samples that were labeled with kinase displayed single major bands containing the indicated oligos that were phosphorylated at their 5′ termini (K oligos in Fig. 2). Clearly, the mobilities of the TT oligos were considerably slower than the mobilities of corresponding K oligos. Moreover, the mobilities of the short (≤10 nt) TT oligos were apparently not correlated with their lengths. For example, the mobility of the oligo containing 10 nt (9TT, a 9mer to which a single dGMP residue was added by terminal transferase) was faster than that of the oligo containing 5 nt (4TT, a 4mer to which a single dGMP residue was added by terminal transferase). On the other hand, the mobilities of the phosphorylated oligos were correlated with their lengths, as expected. Gel electrophoresis assays of oligos of the other two series showed that these oligos too had similar anomalous mobilities (data not shown). We expected that oligos extended with the telomerase by a single nucleotide would have the same mobilities as those of the corresponding terminal transferase extension products.
Figure 2.
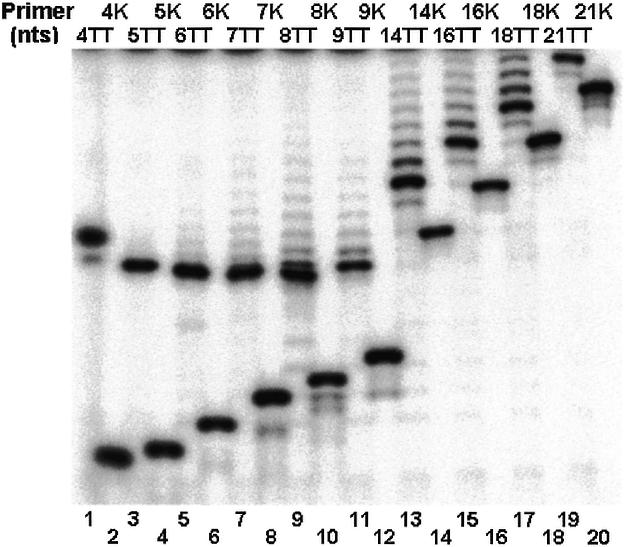
Anomalous electrophoretic mobilities of short unphosphorylated oligonucleotides. The oligos shown in Figure 1A were labeled with 32P either by addition of a single labeled dGMP residue to the 3′ terminus with terminal transferase (TT oligos; odd lanes), or by addition of 32PO4 to the 5′ terminus with polynucleotide kinase (K oligos; even lanes). The labeled oligos were electrophoresed in a denaturing gel and the bands were detected by exposure to a phosphorimager screen and subsequent analysis in a phosphorimager apparatus.
Minimal lengths required for primers to be extended with telomerase by a single nucleotide
We first performed primer extension assays with native telomerase, which was partially purified from Tetrahymena cells (26,33), using oligos of the series shown in Figure 1A. In these experiments, each of the oligos was incubated with the telomerase in the presence of 32P-labeled dGTP alone. Under these conditions, it was expected that oligos that could be utilized by the telomerase as primers, would be extended by a single 32P-labeled dGMP residue (26). It was also expected that the enzyme would not extend oligos that were shorter than a certain minimal size. The reactions were performed at 10°C, the temperature that has been previously employed for footprinting telomerase elongation complexes (26). Two different concentrations of primers, 2.5 and 50 µM, were used. The extension products were subsequently resolved by electrophoresis in a denaturing gel and analyzed as described in the previous section. It should be noted that these samples were not purified and ethanol-precipitated before being loaded on the gel, as is usually done in this type of analysis; for unphosphorylated oligos consisting of ≤9 nt could not be quantitatively recovered after ethanol precipitation (N. Baran, B. Paul, Y. Haviv and H. Manor, unpublished results). Figure 3A shows the extension products obtained in these reactions. Each product was manifested as a single prominent band that had a mobility of an oligo consisting of the original primer, to which a single dGMP residue was added (lanes 2–12; see the terminal transferase markers in lanes 14 and 15 and Fig. 2). In addition, several other diffuse bands were detected in lanes 1–12 and also in lane 13, which contained the products of a control reaction that was carried out in the absence of any exogeneous primer. Most of these non-specific products were not detected in samples that were further purified and precipitated with ethanol. Control assays performed with mixtures of the 6mer and the 21mer oligos, or the 7mer and the 21mer oligos, showed that the synthesis of the molecules found in the specific bands was sensitive to pretreatment of the telomerase with RNase (lanes 16–19). Hence, they were authentic telomerase extension products. It should be noted that similar data were obtained in assays performed at 30°C, instead of 10°C (data not shown).
Figure 3.

Determination of the minimal length required for a primer aligned in the middle of the TER template region, to be extended by a single nucleotide. Primers of the series shown in Figure 1A were extended with native Tetrahymena telomerase (A), or with a telomerase core that was reconstituted by co-expression of the Tetrahymena TERT and TER in a rabbit reticulocyte lysate (B), or with an immunopurified reconstituted core telomerase (C). In the assays shown in (A) and (B) (lanes 1–12), the primers at the two indicated concentrations were incubated with the telomerase in the presence of 32P-labeled dGTP alone. In (C) (lanes 1–10), each reaction was similarly performed with equimolar concentrations (2.5 µM) of the 21mer oligo and the indicated primer. Lane 13 in (A) and (B) and 11 in (C) show control assays performed with no primer. Lanes 14 and 15, respectively, in (A) show markers produced by extension of the 4mer and the 5mer oligos with terminal transferase. The marker bands are designated with open arrows. The strong bands at the top of these lanes are non-specific bands that were often observed in the terminal transferase reaction mixtures. Samples of the products were directly loaded and electrophoresed in a denaturing gel without being purified and ethanol precipitated. The arrows point at single bands that contained oligos, which were extended by a single 32P-labeled dGMP residue. Note that the 6mer, 7mer and 8mer products were not resolved in the gel. In the control assays shown in (A), the reactions were performed with 2.5 µM of primers and with telomerase that was preincubated with (lanes 17 and 19), or without RNase (lanes 16 and 18). Similar control assays are also shown in (B) (lanes 14–17). In other control assays shown in (B), the reactions were performed with reticulocyte lysates that produced only TERT (lanes 18 and 20), or only TER (lanes 19 and 21).
Further inspection of the data presented in Figure 3A revealed that at the lower concentration of DNA of 2.5 µM an extension product of the 4mer oligo was not detected (lane 1). However, the 4mer oligo was clearly extended by the telomerase in the assay performed at the higher DNA concentration of 50 µM (lane 2). The 5mer oligo was efficiently extended even at the lower DNA concentration of 2.5 µM (lane 3). The 6mer and 7mer oligos were extended at a somewhat higher efficiency than the 5mer oligo (lanes 5–8) and at a much higher efficiency than the 21mer oligo (lanes 11 and 12). It should be noted that in all these assays only a very small fraction of primer molecules (<2.5 × 10–4) were extended by the telomerase and thereby became radioactively labeled (see Materials and Methods). To assess the reason for the high efficiency of labeling of the short oligos relative to that of the 21mer oligo, we have carried out single-nucleotide extension assays, in which the 6mer oligo and the 21mer oligo were mixed at different concentration ratios. The rates of extension observed in these assays for each oligo were compared to the corresponding rates obtained in separate assays carried out under the same conditions with each of these oligos. These data revealed that the kcat of the telomerase for the 6mer oligo was larger than the kcat for the 21mer oligo, and that the KM of the enzyme for the shorter oligo was also larger than the KM for the 21mer oligo (N. Baran and H. Manor, unpublished results). This result indicated that the relatively high efficiency of labeling of the 6mer oligo (and of the other short oligos) versus the 21mer oligo, was due to a faster turnover that the telomerase underwent in the reactions performed with the shorter oligos and not to a higher affinity of the enzyme to these oligos. In other words, enzyme molecules more rapidly dissociated from the shorter extended primers after each round of synthesis and became available for extension of additional molecules of the same type.
Figure 3B shows similar assays performed with the same primers and a core Tetrahymena telomerase that was reconstituted in vitro by co-expression of plasmids encoding the TERT and the TER subunits in a rabbit reticulocyte lysate (29). The reconstituted telomerase differs from the native enzyme in that it is much less processive (29,34; N. Baran, unpublished data). This difference could be due to the presence of auxiliary subunits in the native enzyme, which the reconstituted enzyme lacks. As lanes 1–13 in Figure 3B show, the data obtained in these assays were similar to those obtained with the native enzyme. Lanes 14–21 show control assays carried out with the 6mer and the 21mer oligos, or the 7mer and the 21mer oligos, which confirmed the authenticity of the reconstituted telomerase. First, RNase treatment abolished the activity of the enzyme (lanes 15 and 17). Secondly, the extension products were only synthesized in lysates incubated in the presence of both plasmids encoding TERT and TER (lanes 14 and 16), but not in lysates incubated in the presence of only one of these two plasmids (lanes 18–21).
For the next experiment we used an immunopurified Tetrahymena telomerase. This telomerase has been reconstituted from TER and a TERT subunit containing a FLAG tag at the C-terminus. We used this tag to purify the enzyme by binding it to anti-FLAG antibodies adsorbed to Sepharose beads. Figure 3C shows primer extension assays performed with the immobilized enzyme and with the oligos shown in Figure 1A at a concentration of 2.5 µM. In this experiment, each reaction was carried out in the presence of both the 21mer and one of the shorter oligos, at a 1:1 molar ratio. It can be seen that the background was much lower in these assays. However, the results were similar to the results of the assays shown in Figure 3A and B, in regard to the minimal length required for a primer to be extended, and the more efficient radioactive labeling of the shorter oligos. Thus, if the native enzyme contained auxiliary subunits, their presence did not substantially affect the initial steps of the primer extension reactions, that is the binding of the primers to the enzyme and the addition of the first nucleotide to the chains.
Figure 4A shows primer extension assays performed with oligos of the series aligned in the beginning of the TER template region (Fig. 1B). The enzyme used for these assays was reconstituted from TER having a mutation in the template region. It was designated C48G, because the C residue 48 in the template region had been replaced with G (compare the illustrations of the template shown in Fig. 1A and B). In these experiments, the primers were incubated with the telomerase in the presence of 32P-labeled dCTP alone and were, therefore, extended by a single 32P-labeled dCMP residue (a single-nucleotide extension reaction could not have been performed with the wild-type enzyme at this position). It can be seen that among the primers used for these assays, even a 3mer was extended at the higher concentration of 50 µM (lane 4) and a 4mer was extended at the lower concentration of 2.5 µM (lane 5). Clearly, the efficiencies of extension of the 4mer, 5mer, 6mer and 7mer oligos were considerably higher at the concentration of 50 µM. Further inspection of the data revealed that at the concentration of 50 µM, the bands containing the extension products of the 4mer, the 5mer and the 6mer (lanes 6, 8 and 10) were stronger than the corresponding bands of the 7mer and the 8mer (lanes 12 and 14). Thus, as in the previous series of primers, the enzyme apparently underwent a faster turnover during extension of the shorter primers.
Figure 4.

Determination of the minimal lengths required for primers aligned in the beginning or next to the end of the TER template region to be extended by a single nucleotide. Primers aligned in the beginning (A), or next to the end (B) of the template, were extended by a single 32P-labeled dCMP (A), or by a single 32P-labeled dGMP (B) and analyzed as described in the legend to Figure 2. A mutant core telomerase, C48G, was used for both experiments. This mutant was reconstituted from TERT and a mutant TER, in which the C48 in the template region was replaced with G.
Figure 4B presents similar assays performed with oligos of the series shown in Figure 1C, which aligned next to the end of the TER template region. The enzyme used for these assays was the reconstituted C48G telomerase and the primers were extended by a single 32P-labeled dGMP residue. It can be seen that, among this series of oligos, a 5mer was the shortest primer that the telomerase extended at the higher concentration of 50 µM (lane 4) and only a 6mer was extended at the lower concentration of 2.5 µM (lane 5). It is also evident that the 6mer and the 7mer oligos were more efficiently extended than the longer oligos (compare lanes 5–8 with lanes 9–14). Thus, like the other two series of oligos, in this series too the enzyme underwent a faster turnover during extension of the shorter primers. It should be noted that similar results were obtained in assays that were carried out with the wild-type reconstituted telomerase and the appropriate series of primers (data not shown).
Figure 5 shows a schematic illustration of the minimal primers that were extendable with telomerase. At an oligo concentration of 2.5 µM, the shortest oligo that could be extended with telomerase by a single nucleotide consisted of 4 nt (Fig. 5, I). The 3′ end of this primer aligned at the beginning (3′ terminus) of the template region. Among the oligos that aligned in the middle of the template region, the shortest that could be extended with the telomerase by a single nucleotide consisted of 5 nt (Fig. 5, II). The shortest extendable oligo in the series that aligned next to the end (5′ terminus) of the template consisted of 6 nt (Fig. 5, III). The corresponding minimal primer lengths observed in assays performed at the exceedingly high concentration of primers of 50 µM, were 3, 4 and 5 nt, respectively, for primers aligned in the beginning, middle and next to the end of the template. Thus, to be extended by a single nucleotide, the minimal primer must be longer, the closer its 3′ terminus is to the end of the template.
Minimal lengths required for primers to be extended with the telomerase by multiple repeats
Next, we determined the minimal lengths required for the oligos of the three series described in the previous sections to be extended with the telomerase by multiple repeats. These reactions (designated complete reactions) were all carried out with the native telomerase, since, as already mentioned above, the reconstituted enzyme is much less processive, i.e. it does not extend primers beyond completion of a few repeats (29,34; N. Baran, unpublished results). Oligos of each of the three series (at a concentration of 2.5 µM) were extended with the telomerase at 10°C in the presence of both 32P-labeled dGTP and unlabeled dTTP, and the products were resolved by electrophoresis in denaturing polyacrylamide gels, as described above. Unlike the single nucleotide extension assays shown in Figures 2–4, the products of these reactions were purified and precipitated with ethanol prior to electrophoresis, as described in Materials and Methods. This was essential because, otherwise, the background of radioactivity along the lanes was rather high and masked the bands containing the products of the reactions.
Figure 6A shows the extension products that were obtained in assays of the primers aligned in the middle of the RNA template region. It can be seen that long extension products containing multiple telomeric repeats were detectable in the assay performed with the 5mer oligo (lane 2), but not in the assay performed with the 4mer oligo (lane 1). The yield of these products increased as the length of the primers increased from 5 to 7 nt (lanes 2–4). The yields obtained in the assays performed with the longer oligos were only slightly higher than the yield obtained with the 7mer oligo (lanes 5–7 and data not shown). In addition to these high molecular weight products, radioactively labeled bands containing shorter products were also detected in this gel. Some prominent bands had mobilities that were faster than the mobilities of the input primers. These bands presumably contained oligos that were phosphorylated at either their 5′ or 3′ termini; for their mobilities were the same as those of marker oligonucleotides, which were phosphorylated with kinase and were electrophoresed in other lanes of the same gel (6K–9K; see also Fig. 2). These molecules could not be intact telomerase extension products, because such products would have slower mobilities, as indicated by terminal transferase markers that were run in the same gel. Instead, they could have been generated by enzymic cleavage of the telomerase extension products that left a phosphate at a terminus. Such phosphorylated cleavage products could be produced by the previously reported nucleolytic activity of the telomerase itself (37,38), or by a nuclease contaminant of our telomerase preparations, which were only partially purified. However, such phosphorylated degradation products were not generated in the reaction performed with the 21mer oligo (lane 7) and with 14mer, 16mer and 18mer oligos (data not shown). Based on the mobilities of the terminal transferase markers, we identified some of the extension products. As expected, extension products of lengths ≤10 nt were not resolved in the gel (see Fig. 2). The arrowheads indicate the bands containing the longest products of the second round of elongation of each primer, which have been also identified by use of independent markers. For example, the indicated products of the 5mer and the 6mer primers were oligos consisting of 15 and 16 nt, respectively (lanes 2 and 3). Interestingly, these products were less abundant than the corresponding 14mer and 15mer products (compare the bands indicated with arrowheads to the bands below them in the same lanes). Such relative abundances of these products were only observed in the reactions performed with the short primers, while in the reactions performed with the longer primers, the relative abundances were the opposite, as expected (compare lane 7 with lanes 2 and 3).
Figure 6.

Determination of the minimal lengths required for primers aligned in the beginning, middle and next to the end of the template, to be extended with telomerase by multiple repeats. Primers of the series shown in Figure 1 were extended with the native telomerase in a reaction mixture containing 32P-labeled dGTP, unlabeled dTTP and 2.5 µM of primer. (A–C) Assays of primers aligned in the middle (Fig. 1A), the beginning (Fig. 1B) and next to the end of the template (Fig. 1C), respectively. Lanes 8 (A) and 7 (B and C) show control assays performed with no primers. The arrows in (A) indicate 7mer, 8mer, 9mer, 10mer unphosphorylated extension products of these primers, which were not resolved in the gel, and phosphorylated degradation products (6K–9K; described in the Results). The arrowheads indicate the completed products of the second round of primer extension. These various species were identified by running, in other lanes of the same gel, oligo markers that were labeled either with kinase, or with terminal transferase, as described in the legend to Figure 2. The marker designates a 32P-labeled 35mer oligo that was used as a recovery marker.
Figure 6B shows similar assays carried out with oligos illustrated in Figure 1B that aligned at the beginning of the RNA template region. It can be seen that the minimal length required for an oligo of this series to be extended with the telomerase by multiple repeats was 4 nt (lane 2). Furthermore, although the abundance of the long high molecular weight products increased in the reaction performed with the 5mer primer (lane 3), no further increase was observed in the abundance of these products in the reactions performed with the longer primers (lanes 4–6).
Figure 6C shows primer extension assays of a similar type carried out with oligos corresponding to those shown in Figure 1C, except that the C-facing residue 48 in the RNA was replaced with a G, such that these oligos could pair with the wild-type RNA of the native enzyme. These oligos aligned next to the end of the RNA template region. It can be seen that the minimal length required for a primer of this type to be extended with the telomerase by multiple repeats was 6 nt (lane 2). In this experiment, the abundance of the high molecular weight products increased, as the length of the primer was increased (lanes 3–6). In Figure 6B and C, molecules having faster mobilities than those of the input primers could be seen. As in Figure 6A, these molecules were probably phosphorylated degradation products of the extended molecules.
Thus, as in the single-nucleotide extension reactions, the minimal lengths required for a primer to be extended with the telomerase by multiple repeats at the primer concentration of 2.5 µM, were 4, 5 and 6 nt, respectively, for primers aligned in the beginning, middle and next to the end of the RNA template region. It should be noted that in similar complete reactions performed at the much higher primer concentration of 50 µM, the minimal lengths were the same as those obtained at the lower concentration (data not shown).
DISCUSSION
The studies reported here revealed, first, that both the native Tetrahymena telomerase and the reconstituted core enzyme can efficiently bind and extend single-stranded DNA primers that are shorter than 6 nt, the length of the shortest primers that have been utilized so far for telomerase assays in vitro (37,39). In assays performed at the very high primer concentration of 50 µM, a primer consisting of 3 nt was the shortest that could be extended with the telomerase by a single nucleotide, while at the moderate concentration of 2.5 µM, the shortest length of an extendable primer was 4 nt. Secondly, as illustrated in Figure 5, the minimal lengths required for primers to be extended with telomerase depended on the positions that these primers initially occupied along the RNA template region. Moreover, at the primer concentration of 2.5 µM, the required minimal lengths for extension of primers by a single nucleotide and by multiple repeats were found to be the same at the various positions along the template. This result indicated that the minimal length required for primers to be extended by multiple repeats was determined at an early step in the elongation process, possibly the initial binding of the primers to the enzyme.
A plausible explanation for the position dependence of the minimal primer length is that the telomerase ribonucleoprotein complex (RNP) contains a site, which is schematically depicted in Figure 5 as a rectangle and is designated primer alignment site (PAS). PAS may interact with the RNA–DNA duplex near the beginning of the template region and with the adjacent unpaired primer nucleotides. As primers are elongated and the active site of the enzyme moves downstream along the template, PAS remains at a fixed position relative to the template. The results obtained at a primer concentration of 2.5 µM, as illustrated in Figure 5, can be explained by changes in the interactions of the primers with PAS, as follows: the shortest extendable primer in the series of oligos designed to align in the beginning of the template, that is the 4mer oligo, could only form a short, thermodynamically unstable, 3mer duplex with the template (Fig. 5, I). Hence, the ability of the telomerase to bind and extend this primer depended on its strong association with PAS. However, the telomerase failed to bind and extend a 4mer oligo of the series of oligos that were designed to align in the middle of the template, even though this oligo could form a more stable 4mer duplex (Fig. 5, II). We suggest that this result was due to reduced interactions of this 4mer oligo with PAS, as indicated by comparison of schemes II and I in Figure 5. The telomerase could bind and extend the minimal 5mer primer among the series of oligos that aligned at the position illustrated in scheme II, because it generated a more stable 5mer duplex with the template, and because the 5′ end of this longer duplex could better interact with PAS. Similar arguments can be provided as to the reason for the telomerase being capable of positioning and extending the minimal 6mer primer (but not a 5mer oligo) of the series of oligos designed to align next to the end of the template region (Fig. 5, III). The existence of PAS in the telomerase could also be inferred from the results of the assays performed at the very high primer concentration of 50 µM. It should be noted that one component of PAS might be the recently discovered ‘template recognition’ element sequence that maps within the template loop in the TER (13).
Some of the arguments presented above are also compatible with previous data obtained by Wang et al. (27) on mismatch tolerance of primer extension by the Tetrahymena telomerase. These authors examined the ability of the telomerase to extend longer primers (>20 nt) containing a single mismatch at various positions along their 3′-terminal sequences (which were otherwise complementary to RNA template sequences). Based on their data, they concluded that as the active site moves downstream along the template, the length of the RNA–DNA duplexes required for proper alignment and extension of the primers increases from 0 to 5 bp.
The existence of PAS in the telomerase RNP is also supported by the observations that the telomerase can extend entirely non-telomeric DNA primers and that the extension of such primers most frequently begins with the sequence d(GGGGT) (38,40,41). The latter observation indicated that the non-telomeric primers, which cannot form a duplex with any sequences in the template, align next to the beginning of the template. We propose that this ‘default’ positioning of the non-telomeric primers is at least partly due to interactions of such primers with PAS. Furthermore, in the case of these primers, the association with PAS completely substitutes for Watson–Crick base-pairing with the RNA template. In vivo, this ‘default’ positioning could account for the phenomenon of chromosome healing by telomerases, which is also frequently initiated with d(GGGGT) (42,43).
What are the functions that PAS may have in vivo? One function could be to mediate proper alignment of the single-stranded 3′ extensions of telomeres along the template region of the telomerase during initiation of telomere extension. PAS could also function during ongoing synthesis of multiple telomeric repeats. In this situation, it may mediate proper repositioning of the 3′ ends of primers in the beginning of the template region, following their dissociation and translocation from the end of the template.
Our finding that the telomerase efficiently extends primers consisting of three to six residues by a single nucleotide implies that in longer primers sequences upstream of the sixth nucleotide are not required for this reaction. Hence, interactions of the telomerase with such upstream sequences do not appear to be essential for the initial binding of the primers to the enzyme and their proper positioning along the template, or for the subsequent chemical steps of the reaction. These inferences are in line with the results obtained in our previous footprinting studies on single-nucleotide extension of longer primers (26). Therefore, we suggest that, in vivo, such interactions with upstream sequences in the telomeric DNA are not required for initiation of telomere extension by telomerase.
A more complex issue is the requirement for upstream sequences in DNA primers for processive elongation of the primers by multiple repeats. Interactions of such sequences with a putative anchor site of the telomerase were postulated to be required, in the course of this reiterative process, for translocation of the 3′ termini of the primers to the beginning of the template, as discussed above (24,25). In the context of the present study of the extension of short primers, the crucial step was the first translocation event, since in subsequent translocation events the DNA products became as long as the primers used for previous studies, i.e. ≥13 nt. The results presented in Figure 6 are pertinent to this issue. Consider, for example, the data on the series of oligos aligned next to the end of the template (Fig. 6C). The shortest primer in this series that was found to be elongated by multiple repeats was the 6mer oligo. This primer was extended by a single nucleotide when it reached the end of the template and became a 7mer oligo; it was this oligo that underwent the first translocation event. It appears, therefore, that sequences upstream of the seventh residue were not essential for translocation. However, as Figure 6C also shows, the yields of the long products in the assays performed with other primers of the same series were substantially increased, as the length of these primers increased from 6 to 9 nt. This increase could reflect an enhancement of the efficiency of translocation due to the presence of the three nucleotides upstream of the seventh residue (nucleotides 8–10). A similar consideration of the data presented in Figure 6A and B indicates that interactions of the enzyme with nucleotides 8–11 of the primers could play a role in the translocation.
In conclusion, our data indicated that precise repositioning of the primers at the beginning of the template does not require interactions other than those that occur between the six 3′-terminal primer residues, or a subset of these residues, with a specific site in the telomerase RNP that we have designated PAS. However, interactions of the telomerase with primer nucleotides that reside further upstream (up to the 11th residue) may augment the above interactions and stimulate the translocation process. It is not known whether these upstream nucleotide residues and the six 3′-terminal nucleotides interact with the same region of the telomerase. Identification of the protein and RNA residues in the telomerase RNP, which interact with the nucleotide residues in primers of various lengths, will help to resolve these issues.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Kathleen Collins for sending us the plasmids and protocols used for reconstitution of the Tetrahymena telomerase. We also thank Drs Elizabeth H. Blackburn and Yechiel Shalitin for helpful discussions. This study was supported by the Israel Science Foundation (grant no. 373/99).
REFERENCES
- 1.McEachern M.J., Krauskopf,A. and Blackburn,E.H. (2000) Telomeres and their control. Annu. Rev. Genet., 34, 331–358. [DOI] [PubMed] [Google Scholar]
- 2.Manor H., Haviv,I. and Baran,N. (2002) DNA primer extension by telomerase. In Krupp,G. and Parwaresch,R. (eds), Telomeres and Telomerases: Cancer and Biology. Landes Bioscience, Georgetown, TX.
- 3.Blackburn E.H. (2000) The end of the (DNA) line. Nature Struct. Biol., 7, 847–850. [DOI] [PubMed] [Google Scholar]
- 4.Collins K. and Mitchell,J.R. (2002) Telomerase in the human organism. Oncogene, 21, 564–579. [DOI] [PubMed] [Google Scholar]
- 5.Collins K. (1999) Ciliate telomerase biochemistry. Annu. Rev. Biochem., 68, 187–218. [DOI] [PubMed] [Google Scholar]
- 6.Greider C.W. and Blackburn,E.H. (1989) A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature, 337, 331–337. [DOI] [PubMed] [Google Scholar]
- 7.Lingner J. and Cech,T.R. (1996) Purification of telomerase from Euplotes aediculatus: requirement of a primer 3′ overhang. Proc. Natl Acad. Sci. USA, 93, 10712–10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lingner J., Hughes,T.R., Shevchenko,A., Mann,M., Lundblad,V. and Cech,T.R. (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science, 276, 561–567. [DOI] [PubMed] [Google Scholar]
- 9.Meyerson M., Counter,C.M., Eaton,E.N., Ellisen,L.W., Steiner,P., Caddle,S.D., Ziaugra,L., Beijersbergen,R.L., Davidoff,M.J., Liu,Q. et al. (1997) hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell, 90, 785–795. [DOI] [PubMed] [Google Scholar]
- 10.Romero D.P. and Blackburn,E.H. (1991) A conserved secondary structure for telomerase RNA. Cell, 67, 343–353. [DOI] [PubMed] [Google Scholar]
- 11.Lingner J., Hendrick,L.L. and Cech,T.R. (1994) Telomerase RNAs of different ciliates have a common secondary structure and a permuted template. Genes Dev., 8, 1984–1998. [DOI] [PubMed] [Google Scholar]
- 12.Licht J.D. and Collins,K. (1999) Telomerase RNA function in recombinant Tetrahymena telomerase. Genes Dev., 13, 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller M.C. and Collins,K. (2002) Telomerase recognizes its template by using an adjacent RNA motif. Proc. Natl Acad. Sci. USA, 99, 6585–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J.L., Blasco,M.A. and Greider,C.W. (2000) Secondary structure of vertebrate telomerase RNA. Cell, 100, 503–514. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura T.M. and Cech,T.R. (1998) Reversing time: origin of telomerase. Cell, 92, 587–590. [DOI] [PubMed] [Google Scholar]
- 16.Lai C.K., Mitchell,J.R. and Collins,K. (2001) RNA binding domain of telomerase reverse transcriptase. Mol. Cell. Biol., 21, 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman K.L. and Cech,T.R. (1999) Essential functions of amino-terminal domains in the yeast telomerase catalytic subunit revealed by selection for viable mutants. Genes Dev., 13, 2863–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bachand F. and Autexier,C. (2001) Functional regions of human telomerase reverse transcriptase and human telomerase RNA required for telomerase activity and RNA-protein interactions. Mol. Cell. Biol., 21, 1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller M.C. and Collins,K. (2000) The Tetrahymena p80/p95 complex is required for proper telomere length maintenance and micronuclear genome stability. Mol. Cell, 6, 827–837. [DOI] [PubMed] [Google Scholar]
- 20.Evans S.K. and Lundblad,V. (2000) Positive and negative regulation of telomerase access to the telomere. J. Cell Sci., 113, 3357–3364. [DOI] [PubMed] [Google Scholar]
- 21.Holt S.E., Aisner,D.L., Baur,J., Tesmer,V.M., Dy,M., Ouellette,M., Trager,J.B., Morin,G.B., Toft,D.O., Shay,J.W. et al. (1999) Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev., 13, 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greider C.W. and Blackburn,E.H. (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell, 43, 405–413. [DOI] [PubMed] [Google Scholar]
- 23.Greider C.W. (1991) Telomerase is processive. Mol. Cell. Biol., 11, 4572–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greider C.W. (1995) Telomerase biochemistry and regulation. In Blackburn,E.H. and Greider,C.W. (eds), Telomeres. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp. 35–68.
- 25.Hammond P.W., Lively,T.N. and Cech,T.R. (1997) The anchor site of telomerase from Euplotes aediculatus revealed by photo-cross-linking to single- and double- stranded DNA primers. Mol. Cell. Biol., 17, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benjamin S., Baran,N. and Manor,H. (2000) Interference footprinting analysis of telomerase elongation complexes. Mol. Cell. Biol., 20, 4224–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H., Gilley,D. and Blackburn,E.H. (1998) A novel specificity for the primer-template pairing requirement in Tetrahymena telomerase. EMBO J., 17, 1152–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Autexier C. and Greider,C.W. (1994) Functional reconstitution of wild-type and mutant Tetrahymena telomerase. Genes Dev., 8, 563–575. [DOI] [PubMed] [Google Scholar]
- 29.Collins K. and Gandhi,L. (1998) The reverse transcriptase component of the Tetrahymena telomerase ribonucleoprotein complex. Proc. Natl Acad. Sci. USA, 95, 8485–8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy C.D., Schultz,C.S. and Collins,K. (2001) Requirements for the dGTP-dependent repeat addition processivity of recombinant Tetrahymena telomerase. J. Biol. Chem., 276, 4863–4871. [DOI] [PubMed] [Google Scholar]
- 31.Greider C.W. and Blackburn,E.H. (1987) The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell, 51, 887–898. [DOI] [PubMed] [Google Scholar]
- 32.Harrington L., Hull,C., Crittenden,J. and Greider,C. (1995) Gel shift and UV cross-linking analysis of Tetrahymena telomerase. J. Biol. Chem., 270, 8893–8901. [DOI] [PubMed] [Google Scholar]
- 33.Gilley D., Lee,M.S. and Blackburn,E.H. (1995) Altering specific telomerase RNA template residues affects active site function. Genes Dev., 9, 2214–2226. [DOI] [PubMed] [Google Scholar]
- 34.Bryan T.M., Goodrich,K.J. and Cech,T.R. (2000) A mutant of Tetrahymena telomerase reverse transcriptase with increased processivity. J. Biol. Chem., 275, 24199–24207. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 36.Gilley D. and Blackburn,E.H. (1996) Specific RNA residue interactions required for enzymatic functions of Tetrahymena telomerase. Mol. Cell. Biol., 16, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collins K. and Greider,C.W. (1993) Tetrahymena telomerase catalyzes nucleolytic cleavage and nonprocessive elongation. Genes Dev., 7, 1364–1376. [DOI] [PubMed] [Google Scholar]
- 38.Melek M., Greene,E.C. and Shippen,D.E. (1996) Processing of nontelomeric 3′ ends by telomerase: default template alignment and endonucleolytic cleavage. Mol. Cell. Biol., 16, 3437–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee M.S. and Blackburn,E.H. (1993) Sequence-specific DNA primer effects on telomerase polymerization activity. Mol. Cell. Biol., 13, 6586–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu G.L. and Blackburn,E.H. (1991) Developmentally programmed healing of chromosomes by telomerase in Tetrahymena. Cell, 67, 823–832. [DOI] [PubMed] [Google Scholar]
- 41.Wang H. and Blackburn,E.H. (1997) De novo telomere addition by Tetrahymena telomerase in vitro. EMBO J., 16, 866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forney J.D. and Blackburn,E.H. (1988) Developmentally controlled telomere addition in wild-type and mutant paramecia. Mol. Cell. Biol., 8, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morin G.B. (1991) Recognition of a chromosome truncation site associated with alpha-thalassaemia by human telomerase. Nature, 353, 454–456. [DOI] [PubMed] [Google Scholar]