

Abstract
The activation of the transcription factor NF-κB is critical for a number of physiological responses. Here, we provide evidence for a signaling pathway that mediates NF-κB activation in response to oxidative stress. We show that tyrosine phosphorylation of protein kinase D (PKD) at Y463 in the Pleckstrin Homology (PH) domain is mediated by the Src and Abl tyrosine kinase signaling pathway, and that this is both necessary and sufficient to activate NF-κB in response to oxidative stress. PKD activates NF-κB through the IKK complex and more specifically, IKKβ, leading to IκBα degradation. We also present evidence that this pathway is required for increased cellular survival in response to oxidative stress. We propose a model in which protection from oxidative stress-induced cell death requires the tyrosine phosphorylation of PKD leading to the activation of the transcription factor NF-κB.
Keywords: Abl/IKK/NF-κB/PKD/Src
Introduction
The continuous exposure of organisms to environmental threats such as viral infections, UV light, oxidative stress as well as chemotherapeutic drugs requires cellular responses to avoid the lethal damage of genes or cells, which typically occurs via necrosis or apoptosis. In response, cells are able to activate signaling pathways and recover from DNA damage, and several studies have shown that the inducible transcription factor NF-κB plays an important role in the survival of cells exposed to these insults (Li et al., 1999; Mercurio and Manning, 1999). However, it is also known that other cellular stimuli such as cytokines and growth factors can activate NF-κB by distinct pathways from those which participate in the stress response. The tumor necrosis factor-α (TNFα) and interleukin-1 (IL-1)-induced NF-κB activation pathways are well characterized. Regulation of NF-κB has been shown to occur through the activation of upstream protein kinases, including NIK (NF-κB-inducing kinase), MEKK1 (mitogen-activated protein kinase kinase kinase 1), NAK (NF-κB-activating kinase) and IKK-ε/i, all of which mediate NF-κB activation by converging on the IKK (IκB-kinase) complex (Peters and Maniatis, 2001). The IKK complex comprises three major components, the IκB-kinases IKKα and IKKβ as well as the adapter protein IKKγ/NEMO. Activation of this complex serves to mediate IκB phosphorylation, ubiquitination and degradation by the 28s proteasome. Similarly, an NF-κB activation pathway mediated by phosphoinositide 3-OH-kinase (PI 3-K) and the serine/threonine protein kinase Akt/PKB, has been linked to growth factor-induced NF-κB activation and anti-apoptotic responses (Romashkova and Makarov, 1999).
The regulation of NF-κB activation induced by reactive oxygen species (ROS) or oxidative stress has not been defined and appears to be cell type dependent. For example, tyrosine phosphorylation of IκBα occurs in some cells stimulated with both H2O2 and pervanadate (PV), leading to NF-κB activation. This can occur either in the presence or absence of IκB degradation and in an IKK-independent manner (Imbert et al., 1996). Activation of the PI 3-K pathway by oxidative stress also leads to dissociation of IκBα from NF-κB in the absence of IκB degradation (Beraud et al., 1999). These studies suggest that activation of NF-κB by oxidative stress may not occur through a common pathway in all cells.
ROS and oxidative stress are also known to induce the tyrosine phosphorylation of a number of signaling proteins, either indirectly through inhibition of tyrosine phosphatases, or by directly acting on tyrosine kinases. For example, the tyrosine kinases Src and Abl are known to be activated in response to ROS (Kumar et al., 2001). The serine/threonine kinase protein kinase D (PKD) can also be activated by treatment of cells with H2O2 (Waldron and Rozengurt, 2000). PKD was originally classified as a PKC family member and termed PKCµ (Johannes et al., 1994). However, more recent studies have shown that it is in fact a member of a distinct family of kinases, classified together with two other isoforms, PKD2 and PKD3/PKCν. Besides some similarity in the kinase and C1 domains, PKD has additional protein modules not found in PKCs, including a Pleckstrin Homology (PH) domain and a hydrophobic region whose functions are not well defined. However, the PH domain of PKD may play a regulatory role as deletion of this domain results in increased kinase activity (Iglesias and Rozengurt, 1998). PKD is activated by mitogens, G-protein coupled-receptor agonists or by triggering of the B- and T-cell receptors. In addition to its role in T- and B-cell signaling (Matthews et al., 2000), recent studies have highlighted an important role for PKD in the organization and function of the Golgi (reviewed in Van Lint et al., 2002). There is also some evidence that PKD may play a role in regulating apoptosis, as it can be activated by DNA-damaging agents.
In the present study, we sought to identify the mechanisms by which PKD mediates oxidative stress-induced cellular responses. We show that activation of PKD by ROS is mediated by the Src–Abl signaling pathway leading to tyrosine phosphorylation of PKD. This leads to activation of NF-κB and protection from oxidative-stress-induced cell death. These data provide evidence for a signaling pathway that mediates NF-κB activation and that may serve as a general mechanism by which cells respond to environmental stresses and increase their survival.
Results
Activation and tyrosine phosphorylation of PKD potentiates NF-κB in response to oxidative stress
Stimulation of HeLa cells with oxidative stress at concentrations that did not cause cell death (250 nM H2O2) resulted in increased NF-κB activity, which was significantly enhanced by overexpression of PKD (Figure 1A). We also investigated whether other stimuli known to activate PKD also lead to NF-κB activation in HeLa cells and NIH 3T3 fibroblasts. Stimulation of both cell types with either bradykinin or platelet-derived growth factor (PDGF) did not result in NF-κB activation, despite their ability to activate PKD (Figure 1B and C; Supplementary figures S1A and S1B). Because oxidative stress is known to increase protein tyrosine phosphorylation, we investigated the tyrosine phosphorylation of PKD. Both oxidative stress (H2O2) and treatment of HeLa cells or NIH 3T3 cells with the phosphatase inhibitor PV increased tyrosine phosphorylation of endogenous and transfected PKD (Figure 1D). Conversely, stimulation with bradykinin, PDGF or PMA (12-phorbol 13-myristate acetate) did not induce PKD tyrosine phosphorylation (Figure 1D; Supplementary figure S1C). These observations were extended to two other cell types, human embryonic kidney 293 cells (HEK293E) and human breast carcinoma cells MDA-MB-435. H2O2 induced tyrosine phosphorylation of PKD in HEK293E cells, and this correlated with increased NF-κB activity (Figure 1E). Conversely, MDA-MB-435 cells did not show PKD tyrosine phosphorylation in response to H2O2, and there was no activation of NF-κB (Figure 1E). Therefore, in two distinct cell types, H2O2-mediated tyrosine phosphorylation of PKD correlated with induction of NF-κB activity, whereas stimuli that did not promote PKD tyrosine phosphorylation also did not induce NF-κB.

Fig. 1. PKD potentiates NF-κB activation in response to oxidative stress. (A) HeLa cells transfected with HA-PKD were treated with 250 nM H2O2, and NF-κB reporter gene assays were performed. (B) HeLa cells were treated with bradykinin (50 ng/ml, left panel) over time or with H2O2 (right panel), and PKD in vitro kinase assays were carried out. Immunoblots of immunoprecipitated PKD revealed equivalent expression of PKD (bottom panels). (C) HeLa cells were transfected with the NF-κB reporter construct and treated with 50 ng/ml bradykinin or 250 nM H2O2, and luciferase reporter activity was measured. (D) HeLa cells transfected with HA-PKD were treated with bradykinin (50 ng/ml), PV (75 µM), H2O2 (10 µM) or PMA (100 nM) for 10 min and PKD immunoprecipitates were immunoblotted with anti-phosphotyrosine (α-pY, pY-PKD, top panel), stripped and re-probed with anti-PKD (PKD, bottom panel). HA-PKD refers to the transfected protein and PKD refers to the endogenous protein. (E) MDA-MB-435 and HEK 293E cells were incubated with H2O2 (10 µM) for 5 and 10 min and PKD tyrosine phosphorylation measured on anti-PKD immunoprecipitates (pY-PKD). Endogenous (PKD) and expressed PKD (HA-PKD) was detected on an anti-PKD immunoblot (left panels). NF-κB activity was measured in MDA-MB-435 and HEK 293E cells using the NF-κB–luciferase reporter construct in cells stimulated with the indicated doses of H2O2 (right panels). All results are typical of three independent experiments.
Src and Abl mediate stress-induced PKD tyrosine phosphorylation and activation
Elevation of protein tyrosine phosphorylation induced by oxidative stress is mediated in part by the indirect activation of the tyrosine kinases Src and Abl. We therefore analyzed the role of Src and Abl in H2O2-mediated tyrosine phosphorylation and activation of PKD. Overexpression of a kinase-inactive Src allele (Src*) blocked H2O2 and PV-stimulated PKD tyrosine phosphorylation (Figure 2A). Similarly, PKD tyrosine phosphorylation was partially blocked by the Abl inhibitor STI-571 after treatment with oxidative stress (Figure 2B). Transfection of constitutively active alleles of both Src (Src Y527F) and Abl (p120 v-Abl) induced tyrosine phosphorylation of endogenous PKD (Figure 2C). Because a hierarchical Src–Abl signaling pathway exists in some cells, we investigated the Src-dependent activation of Abl in H2O2-stimulated cells. First, H2O2-induced activation of endogenous Abl was potentiated by expression of wild-type Src (Figure 2D). Secondly, co-transfection of constitutively active Src with wild-type Abl led to an increase in Abl kinase activity (Figure 2E). Finally, tyrosine phosphorylation of PKD mediated by constitutively active Src was blunted by co-transfection with kinase-inactive Abl, but not wild-type Abl (Figure 2F). Therefore, Src is upstream of Abl in the H2O2-stimulated tyrosine phosphorylation of PKD.

Fig. 2. Src and Abl mediate stress-induced tyrosine phosphorylation and activation of PKD. (A) HeLa cells transiently co-transfected with wild-type (WT) or kinase-inactive Src (Src*), and PKD were treated with H2O2 or PV. PKD immunoprecipitates were immunoblotted with anti-pY (pY-PKD), stripped and re-probed with anti-PKD (PKD). Src expression was evaluated by immunoblotting with anti-Src. (B) HeLa cells pre-treated with STI-571 (5 µM, 1 h) and stimulated with H2O2 were immunoprecipitated with anti-PKD, followed by anti-pY immunoblotting. (C) Endogenous PKD from HeLa cells transiently transfected with active Src (Src.Y527F) or Abl (v-Abl p120) was immunoprecipitated, immunoblotted with anti-pY (pY-PKD), stripped and re-probed with anti-PKD (PKD). Src and Abl expression was evaluated by immunoblotting with anti-Src and anti-Abl. (D) Cells were transfected with wild-type Src or dominant-negative Src (Src*) and stimulated with H2O2 as indicated. Anti-Abl immunoprecipitates were assayed for Abl kinase activity using GST–Crk as substrate. Total lysates were immunoblotted with anti-Src. (E) Cells were transiently co-transfected with active Src (Src.Y527F) or vector alone, and with wild-type Abl (WT-Abl) or vector control, as indicated. Abl immunoprecipitates were assayed for Abl protein kinase activity. Total lysates were immunoblotted with anti-Src (Src Y527F) or ant-Abl (Abl) (bottom panels). (F) Cells were co-transfected with either vector alone, activated Src (Src.Y527F), wild-type Abl (WT-Abl) or kinase-dead Abl (Abl KD), and PKD as indicated. PKD was immunoprecipitated and immunoblotted with anti-pY (pY-PKD), stripped and re-probed with anti-PKD. Total lysates were immunoblotted with anti-Src and anti-Abl. (G) Recombinant, purified PKD was incubated either alone or together with purified Abl in vitro with cold ATP, resolved by SDS–PAGE and immunoblotted with anti-pY (pY-PKD and pY-Abl, bottom panel). The nitrocellulose membrane was stripped and reprobed with anti-PKD (top panel). (H) Cells were transiently co-transfected with PKD and either empty vector, active Src (Src Y527F) or active Abl (v-Abl p120). Cells were either left untreated or stimulated with PV (PV, 75 µM) or PMA (100 nM) for 10 min, as indicated. PKD activity was measured by immune-complex kinase assay. Total lysates were immunoblotted with anti-PKD (PKD, bottom panel). All results are typical of three independent experiments.
We then investigated the direct phosphorylation of PKD by Abl. A purified kinase fragment of Abl directly tyrosine phosphorylated recombinant PKD in vitro (Figure 2G). We also investigated the activation of PKD by Src–Abl in HeLa cells, and co-expressed a wild-type PKD allele with constitutively active Src or Abl. Both tyrosine kinases induced maximal PKD kinase activity when compared with the control stimuli PV or PMA (Figure 2H). Thus, tyrosine phosphorylation of PKD mediated by the Src–Abl pathway is sufficient to fully activate the enzyme.
Activation of PKD in response to oxidative stress is mediated by tyrosine phosphorylation of Y463
We next sought to identify the tyrosine residue(s) in PKD phosphorylated in response to H2O2. We first compared the tyrosine phosphorylation of a PKD mutant deleted in the PH domain. This mutant showed no appreciable tyrosine phosphorylation compared with wild-type PKD in cells subjected to oxidative stress (our unpublished data). We next mapped candidate tyrosine residues within the PH domain of PKD by co-expression of PKD mutants (Tyr to Phe) together with active Src or Abl. Of these, we found that a PKD Y463F mutant was almost completely devoid of tyrosine phosphorylation after co-expression with the tyrosine kinases (Figure 3A), and also in response to oxidative stress (Figure 3B).

Fig. 3. Tyrosine phosphorylation of Y463 is necessary for the activation of PKD mediated by Src, Abl and oxidative stress. (A) HeLa cells were transiently co-transfected with wild-type PKD (WT) or a PKD.Y463F mutant and active Abl (v-Abl p120, left panel) or Src (Src.Y527F, right panel). PKD was immunoprecipitated and immunoblotted with anti-pY (pY-PKD). The nitrocellulose membrane was stripped and re-probed with anti-PKD (PKD). Src and Abl expression was evaluated by immunoblotting with anti-Src and anti-Abl. (B) Cells were transiently transfected with empty vector, wild-type PKD (WT) or PKD.Y463F mutant and stimulated with H2O2. PKD (anti-HA) was immunoprecipitated and immunoblotted with anti-pY (pY-PKD), stripped and re-probed with anti-PKD. (C) Cells were transfected with active Src (Src.Y527F), active Abl (v-Abl p120) or empty vector, either alone or in combination with wild-type PKD (WT) or PKD.Y463F, as indicated. PKD immunoprecipitates were immunoblotted with anti-pY463 (pY463), stripped and re-probed with anti-PKD (HA-PKD). Total lysates were immunoblotted with anti-Abl or anti-Src. (D) HeLa cells were either left untreated or stimulated with H2O2 for the indicated times. Endogenous PKD was immunoprecipitated (anti-PKD) and immunoblotted with anti-pY463, stripped and re-probed with anti-PKD. (E) Cells transiently transfected with HA-PKD (WT) or HA-PKD.Y463F mutant were either treated with H2O2 (left panel) or co-transfected with active Abl (middle panel) or active Src (right panel). PKD was immunoprecipitated (anti-HA) and activity was measured by immune-complex kinase assay (substrate phosph.). Protein expression was evaluated by immunoblotting immunoprecipitates (PKD) with anti-PKD, or total cell lysates with anti-Abl or anti-Src. (F) Cells were transiently transfected with HA-tagged PKD (WT), PKD.Y463E or PKD.Y463Q, and PKD activity measured as in (E). All results are typical of three independent experiments.
To demonstrate phosphorylation of PKD at Y463 more directly, we developed a phospho-specific antibody against the peptide epitope surrounding this residue. Transfected PKD.Y463F was not detected with the pY463 phospho-antibody under any conditions. Co-transfection with active Src or active Abl induced phosphorylation of PKD at Y463 (Figure 3C). Similarly, stimulation of cells with H2O2 also induced phosphorylation of endogenous PKD at Y463 (Figure 3D).
The Y463F allele was partially resistant to activation by expression of both active Src and Abl, and by H2O2 treatment (Figure 3E). Interestingly, mutation of Tyr463 to a Glu (Y463E), predicted to mimic the net effect of phosphorylation, resulted in a constitutively active PKD as judged by in vitro kinase assays (Figure 3F). As a control, the activity of a Y463Q mutant was unchanged from wild-type PKD. Therefore, activation of PKD mediated by Src–Abl in response to oxidative stress occurs through phosphorylation of Y463 in the PH domain.
Oxidative stress induces NF-κB activation via Src and Abl
Since H2O2-induced activation of NF-κB is enhanced by expression of PKD (Figure 1A), we analyzed the contribution of Y463 phosphorylation to this event. H2O2 or PV stimulation of cells expressing a wild-type Src allele resulted in increased NF-κB activity when compared with control cells (Figure 4A and B). Conversely, cells expressing kinase-inactive Src showed a reduced response. Similarly, expression of constitutively active Src (Y527F) increased NF-κB activation in the absence of H2O2 or PV (Figure 4B). NF-κB activation was blocked when activated Abl (p120 v-Abl) was used in combination with the Abl inhibitor STI-571 (Figure 4C). NF-κB activation induced by H2O2 and PV was potentiated in cells expressing wild-type Abl (Figure 4D; Supplementary figure S2), and STI-571 blocked NF-κB activation also in response to H2O2 (Figure 4E). These data demonstrate that cells subjected to oxidative stress induce the activation of NF-κB by a pathway that requires Src and Abl.

Fig. 4. Src–Abl-mediated activation of PKD induces NF-κB. (A–E) Cells were transfected with the indicated constructs and pre-treated with STI-571 (5 µM for C, 20 µM for E) 6 h after transfection. Cells were also treated with PV (75 µM) or H2O2 (250 nM) 8 h after transfection as indicated. NF-κB–luciferase reporter gene assays were carried out. Western immunoblot analysis was carried out in each case to reveal equivalent expression of the transfected proteins (see Supplementary figure S3). All results are typical of three independent experiments.
Phosphorylation of PKD at Y463 is sufficient to stimulate NF-κB activation
We next sought to evaluate the contribution of PKD Y463 phosphorylation in the activation of NF-κB. As already shown in Figure 1A, expression of wild-type PKD potentiated NF-κB activity in cells stimulated with H2O2. This increase was not observed when the PKD Y463F mutant was expressed (Figure 5A), suggesting that Y463 phosphorylation is required for efficient NF-κB activation. Neither STI-571 nor PKD.Y463F inhibited TNFα-mediated NF-κB activation, suggesting that the Abl–PKD pathway upstream of NF-κB is restricted to oxidative-stress signaling (Supplementary figure S5). We also took a gain-of-function approach to more conclusively demonstrate a role for PKD.Y463 in controlling NF-κB activation. The PKD.Y463E mutant shows constitutive protein kinase activity (Figure 3F) and as predicted, also potently induced NF-κB activation in unstimulated HeLa cells (Figure 5B). Thus, PKD tyrosine phosphorylation at Y463 is linked to activation of NF-κB.

Fig. 5. PKD is required for H2O2-stimulated NF-κB activation. (A and B) Cells were transfected with the indicated constructs and stimulated with H2O2 6 h after transfection. NF-κB–luciferase reporter gene assays were carried out. Western immunoblot analysis was carried out in each case to reveal equivalent expression of the transfected proteins (see Supplementary figure S4). (C) HeLa cells stably transfected with the NF-κB–luciferase reporter and β-gal were transiently transfected with the indicated RNAi duplexes (RNAi Luc, luciferase RNAi duplex; RNAi PKD, PKD RNAi duplex) or as a negative control, single-stranded PKD RNAi (RNAi control). After the transfection protocol, cells were either left untreated, or stimulated with 0.5 µM H2O2 for 9 h as indicated, and NF-κB luciferase assays were carried out. A portion of the total lysates was used for immunoblotting using anti-PKD (PKD) or anti-tubulin as control. (D) HeLa cells were transfected with PKD or PKD* together with pSUPER or pSUPER.PKD RNAi (ratio 1:2). After 24 h, cells were transfected with NF-κB–luciferase and β-gal reporter constructs. Forty-eight hours after the initial transfection, cells were treated for 16 h with 0.5 µM H2O2 and NF-κB luciferase assays were carried out.
To more directly demonstrate a role for endogenous PKD in the activation of NF-κB, we used RNAi (RNA interference) to reduce endogenous PKD expression. An RNAi duplex from the N-terminus of human PKD was specific to PKD as judged by a BLAST search of the GenBank human EST database. A luciferase gene RNAi duplex was used as a positive control (RNAi Luc), whereas single-stranded PKD RNAi was used as a negative control (RNAi control). A reduction in PKD expression was observed only in cells transfected with the PKD RNAi duplex (Figure 5C). As expected, the luciferase RNAi duplex blunted NF-κB luciferase induction in cells treated with H2O2, and a similar reduction was also observed in cells transfected with the PKD RNAi duplex, but not in cells transfected with single-stranded PKD RNAi (Figure 5C). We also took advantage of a recently described plasmid-based RNAi expression system, pSUPER (Brummelkamp et al., 2002). Transient expression of pSUPER.PKD RNAi also significantly reduced PKD expression with a concomitant reduction in H2O2-induced NF-κB activation (Figure 5D). This reduction in PKD levels and NF-κB activation was not observed when a mutant PKD allele containing silent nucleotide mutations in the RNAi target sequence (PKD*; Figure 5D) was co-transfected with pSUPER.PKD. These results demonstrate that endogenous PKD is required for efficient NF-κB activation in response to H2O2.
The Src–Abl–PKD pathway activates IKKβ leading to IκBα degradation
To further define the mechanism by which PKD signals to NF-κB, we investigated the activation of the IKK complex. We first examined the kinetics of PKD activation and tyrosine phosphorylation in response to H2O2 and PV. A peak of PKD protein kinase activity and tyrosine phosphorylation was detected between 5 and 10 min of H2O2 stimulation (Figure 6A). In response to PV, this response was delayed slightly, peaking between 10 and 30 min (Figure 6B). To determine IKKβ activation, endogenous IKKβ kinase activity was measured in an immune-complex kinase assay using GST–IκBα as substrate. The kinetics of IKKβ activation were identical to those for PKD, with maximal IKKβ activity at 10 min following H2O2 stimulation, but delayed slightly in response to PV (Figure 6C). As a control, IKKβ activation was more rapid in TNFα-stimulated cells. Finally, we examined the degradation of IκBα in these cells. Again, the kinetics of IκBα degradation in response to H2O2 and PV were identical to those of PKD and IKKβ activation (Figure 6D). Thus, stimulation of cells with either H2O2 or PV leads to activation of NF-κB via the classical IKKβ activation and IκBα degradation pathway.

Fig. 6. Kinetics of activation of PKD, IKKβ and IκBα degradation. (A) Cells were stimulated with H2O2 (10 µM) and the activity of PKD was measured by immune complex kinase assay (top panel). In a separate experiment, tyrosine phosphorylation of PKD was evaluated (pY-PKD, middle panel). Protein expression was evaluated by stripping and reprobing the immunoblots with anti-PKD (PKD, bottom panel). (B) Cells were stimulated with PV (75 µM) or PMA (100 nM), and PKD activity (top panel), tyrosine phosphorylation (middle panel) and protein (bottom panel) was evaluated as in (A). (C) Cells were stimulated with 10 µM H2O2, PV (75 µM) or TNFα (50 ng/ml), and endogenous IKK complex immunoprecipitated with anti-IKKγ antibody. An immune-complex kinase assay using GST–IκBα was carried out. (D) Cell lysates generated from the experiment described in (C) were immunoblotted with anti-IκBα antibody, the position of IκBα is shown. All panels are representative of three independent experiments.
The Src–Abl–PKD signaling pathway activates NF–κB via IKKβ
The above data demonstrate a correlation between PKD activation, IKKβ activation and IκBα degradation. We next directly investigated the contribution of Src, Abl and PKD in the regulation of IKK. Both active Src and active Abl, and importantly, active PKD.Y463E were able to induce IKK activation when compared with control cells (Figure 7A). In response to many stimuli, IKKβ is phosphorylated at S177 and S181 in the activation loop leading to kinase activation and subsequent phosphorylation of IκBα. Active Src, active Abl and active PKD induced phosphorylation of IKKβ at S181 in vivo (Figure 7B). The consequence of this event is degradation of IκBα, as judged by immunoblotting endogenous IκBα in cells transfected with the above active alleles (Figure 7C, top panel). The degradation of IκBα mediated by Abl was blocked when the PKD.Y463F mutant was co-expressed, when compared with the wild-type PKD (Figure 7C, lower panel). These data indicate that the Src–Abl–PKD pathway activates IKKβ leading to IκBα degradation. This led us to investigate whether PKD might be a direct IKKβ kinase. However, we have thus far been unable to demonstrate significant direct phosphorylation of IKKβ by purified, recombinant PKD in vitro (our unpublished data). This is despite the fact that H2O2 treatment of cells resulted in the recruitment of PKD to the IKK complex, as revealed by co-immunoprecipitation of IKKβ with PKD (Figure 7D). Phosphorylation of Y463 is not required for this association because the PKC.Y463F mutant was also found in a complex with IKKβ in H2O2-stimulated cells.

Fig. 7. The Src–Abl–PKD signaling pathway activates NF-κB via the IKK complex. (A) Cells were transiently co-transfected with active Abl (v-Abl p120), active Src, active PKD.Y463E or vector alone, and endogenous IKKβ immunoprecipitated. An immune-complex kinase assay using GST–IκBα was performed. Total cell lysates were also used for western immunoblot analysis to reveal equivalent expression of the transfected or endogenous proteins, as indicated (IKKβ, HA-PKD, v-Abl and Src.Y527F). (B) Cells were transiently co-transfected with either vector alone, active Abl, active Src, active PKD.Y463E and wild-type IKKβ. IKKβ was immunoprecipitated and immunoblotted with IKKβ anti-pS181 (pS-IKKβ). Total lysate was used to evaluate equivalent expression of IKKβ in all lanes (bottom panel). (C) Cells were transfected with either vector alone, wild-type PKD (WT), active PKD (PKD Y463E), active Src or active Abl (top panel), or with wild-type PKD (WT) or PKD.Y463F in combination with active Abl or vector alone (lower panel). Lysates were immunoblotted with anti-IκBα. (D) PKD, PKC.Y463F and IKKβ were transiently expressed and cells were treated with 10 µM H2O2 for 10 min. IKKβ was immunoprecipitated. Immunoprecipitates were immunoblotted with antibodies against PKD, stripped and re-probed against the immunoprecipitated protein. (E and F) Cells were transfected with the NF-κB–luciferase and β-gal plasmids and the indicated plasmids [active Abl, v-Abl p120; wild-type (WT) PKD or PKD.Y463E; wild-type (WT) IKKβ or IKKα; dominant-negative (DN) IKKα or IKKβ; empty vector] and NF-κB–luciferase reporter assays were carried out. Total cell lysates were also used for western immunoblot analysis to reveal equivalent expression of the transfected proteins (Supplementary figure S7). All results are typical of three independent experiments.
To further define the contribution of PKD in the activation of the IKK complex, we used a mutational strategy to implicate either IKKα or IKKβ in the regulation of NF-κB activation. Co-expression of constitutively active v-Abl with wild-type IKKα induced a small increase in NF-κB transcriptional activity, which was not significantly different from that induced by a kinase inactive, dominant-negative IKKα allele (Figure 7E). However, co-expression of v-Abl with wild-type IKKβ led to a striking increase in NF-κB activity, and this was not observed with a corresponding kinase-inactive, dominant-negative IKKβ mutant. Therefore, Src–Abl-mediated NF-κB activation is mediated primarily through IKKβ.
Next, we investigated the role of PKD in controlling NF-κB activation via IKKβ. Wild-type PKD or PKD.Y463E were co-expressed alone or in combination with the wild-type or dominant-negative IKKα and IKKβ, and NF-κB transcriptional activity was evaluated. As shown in Figure 7F, PKD.Y436E induced a significant increase in NF-κB activity when expressed either alone or in combination with either IKKα wild type or dominant negative. In addition, co-expression of PKD.Y463E with wild-type IKKβ further increased NF-κB activity when compared with Y463E alone. As in Figure 7E, the dominant-negative IKKβ allele was without effect. These data point to an important role for PKD in controlling NF-κB activation by acting through IKKβ, and suggest that phosphorylation of Y463 is necessary for this event.
Phosphorylation of PKD at Y463 mediates increased cellular survival in response to oxidative stress
Finally, to understand the relevance of PKD-mediated NF-κB activation, we investigated the contribution of this pathway in oxidative-stress-induced cell death. Stable HeLa cell transfectants were generated expressing either wild-type PKD or the inactive mutant Y463F. Two clones expressing each of the two alleles were picked and found to express transfected PKD at levels similar to the endogenous protein (Figure 8A). Interestingly, cells over-expressing wild-type PKD were less sensitive to increasing concentrations of oxidative-stress-induced cell death when compared with control cells stably transfected with empty vector alone (Figure 8B). More strikingly, the two transfectants overexpressing the PKD.Y463F mutant were significantly more sensitive to oxidative-stress-induced death when compared with the wild-type PKD-expressing cells (Figure 8C), or control cells; for example, at 0.8 µM H2O2, 37% (clone 1/9, ± 4.1% SEM) and 30% (clone 1/11, ± 2% SEM) cell viability was observed in control cells, but this was decreased to 5% (clone 4/2, ± 1.3% SEM) and 6% (clone 4/7, ± 1.2% SEM) in cells expressing PKD.Y463F.

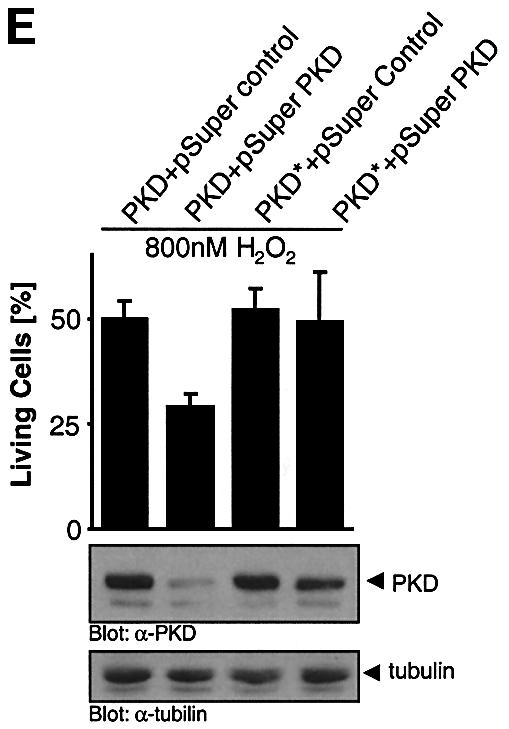
Fig. 8. PKD Y463 phosphorylation mediates cellular survival in response to oxidative stress. (A) Two independent clones of HeLa cells stably expressing wild-type PKD (WT, clones 2/18 and 2/1) or the Y463F mutant (clones 4/2 and 4/7), or stably transfected with empty vector (pcDNA3 clones 1/9 and 1/11) were immunoprecipitated with anti-PKD to reveal expression of transfected PKD (HA-PKD) and endogenous PKD (PKD). (B and C) Stable clones were treated with H2O2 in a dose-dependent manner. After 16 h, the percentage of surviving cells was determined. (D) HeLa cells (left panel) and HEK293E cells (right panel) were transiently transfected with the PKD RNAi duplex or control Luc RNAi. After transfection, cells were treated with 0.8 µM H2O2 for 16 h, after which the percentage of surviving cells was determined. A portion of the total cell lysate from the same experiment was used for immunoblotting with anti-PKD and anti-tubulin. All results are representative of three independent experiments. (E) HeLa cells were transfected with PKD or PKD* together with pSUPER or pSUPER.PKD RNAi (ratio 1:2). Forty-eight hours after the initial transfection, the cells were treated for 16 h with 0.8 µM H2O2 and cell survival assays were carried out.
We speculated that the presence of the endogenous PKD in these transfectants imparts a significant survival advantage under conditions of oxidative stress. To circumvent this problem, we took advantage of the PKD RNAi to selectively reduce endogenous PKD and evaluate survival under conditions of oxidative stress. Both HeLa and HEK293E cells were significantly more sensitive to oxidative-stress-induced cell death when transfected with the PKD RNAi duplex compared with cells transfected with the control RNAi (Figure 8D). Finally, transfection of pSUPER.PKD RNAi significantly reduced PKD levels, with a concomitant 50% reduction in cell survival in response to H2O2 (Figure 8E). Moreover, the PKD* mutant allele containing silent mutations in the RNAi recognition sequence was not silenced by pSUPER.PKD, and was able to efficiently protect from H2O2-induced cell death.
Discussion
Collectively, the above data provide evidence for a signaling pathway in which tyrosine phosphorylation and activation of PKD is relayed to the IKK complex and NF-κB, increasing cellular survival under conditions of oxidative stress. The key observations are as follows: first, low doses of H2O2 induce both the tyrosine phosphorylation of PKD and the activation of NF-κB, whereas other stimuli, which do not lead to PKD tyrosine phosphorylation, do not induce NF-κB; secondly, oxidative-stress-induced tyrosine phosphorylation of PKD at Y463 in the PH domain is mediated by the Src–Abl tyrosine kinase signaling pathway; thirdly, induction of NF-κB by PKD occurs through activation of IKKβ and degradation of IκBα; and finally, tyrosine phosphorylation and activation of PKD mediates increased cellular survival under conditions of oxidative stress (Figure 9). Activation of NF-κB by H2O2 and oxidative stress has been reported in many cell types, and occurs in response to exposure of cells to H2O2 (Li and Karin, 1999). However, activation of NF-κB by oxidative stress is not observed in all cell types, suggesting that NF-κB activation by H2O2 may not occur through a common mechanism, but rather require multiple signaling pathways converging upstream of NF-κB. One such pathway includes the tyrosine phosphorylation of the inhibitory IκBα protein. Such a pathway has been reported to exist in stimulated T cells and been shown to require the tyrosine kinase Lck (Imbert et al., 1996). In response to oxidative stress of HeLa cells, however, we have failed to detect tyrosine phosphorylation of IκBα (our unpublished data). Moreover, in T cells, tyrosine phosphorylation of IκBα does not require activation of the IKK complex and does not lead to IκBα degradation, whereas our data demonstrate that both IKKβ activation and IκBα degradation are a consequence of H2O2 stimulation. Therefore, our studies indicate that there exists a separate pathway that mediates NF-κB activation in response to ROS and which requires the activation of IKKβ.

Fig. 9. Model for the NF-κB activation pathway induced by oxidative stress. Oxidative stress induces the transcriptional activity of NF-κB by activating the Src–Abl signaling pathway, leading to phosphorylation of PKD at Y463 in the PH domain. Oxidative stress also leads to activation of PKCs, which mediate phosphorylation of the PKD activation-loop residues S738/742 in the catalytic kinase domain. These two events result in a fully-active kinase, which mediates activation of IKKβ and degradation of IκBα, leading to the activation of NF-κB. This pathway contributes to the survival of cells exposed to oxidative stress.
In addition to NF-κB activation, it is well established that oxidative stress of cells increases intracellular tyrosine phosphorylation. Although activation of Src and Src-like kinases, as well as Abl, is known to occur in response to oxidative stress, the precise mechanism is unclear. One potential mechanism is inactivation of protein tyrosine phosphatases, and a recent study showed that stimulation of cells with H2O2 results in the reversible oxidation of cysteine residues in several protein tyrosine phosphatases leading to their inactivation (Meng et al., 2002). Pervanadate stimulation of cells also results in activation of Src and Abl, and consistent with the model above, induces tyrosine phosphorylation of PKD and activation of NF-κB. Our studies have also highlighted a hierarchy in the signaling pathway leading to tyrosine phosphorylation of PKD in response to H2O2. Because Src can potentiate Abl protein kinase activity in response to H2O2, whereas kinase-inactive Abl blocks Src-mediated PKD tyrosine phosphorylation, this indicates that Src is upstream of Abl in the PKD activation pathway (Figure 9).
Interestingly, stimulation of HeLa, HEK293E and NIH 3T3 cells with other known activators of PKD, such as bradykinin or PDGF, did not result in NF-κB activation. Importantly, these agonists did not lead to tyrosine phosphorylation of PKD, indicating that the specific phosphorylation of Y463 in the PKD PH domain mediates NF-κB activation. While it is possible that other signaling pathways mediate NF-κB activation in cells subjected to oxidative stress, two pieces of evidence point to a critical requirement for PKD and Y463 phosphorylation. First, the gain-of-function mutant Y463E was by itself able to mediate NF-κB activation, specifically through activation of IKKβ. Secondly, the inactive Y463F mutant blocked NF-κB activation in response to H2O2. This indicates that this pathway is both necessary and sufficient to mediate NF-κB activation under conditions of oxidative stress, but not when cells are stimulated with other agonists that do not induce tyrosine phosphorylation of PKD at Y463.
Previous studies have shown that activation of PKD requires the input of a PKC-dependent pathway (Zugaza et al., 1996). PKCs mediate activation of PKD by inducing the phosphorylation of the activation-loop residues S738/S742 in the catalytic domain (Waldron et al., 2001), and recent studies point to PKCε as a direct upstream kinase for PKD (Brandlin et al., 2002). Because deletion of the PKD PH domain results in constitutive kinase activity, it is likely that it exerts negative regulation on the kinase domain. It is possible that phosphorylation of Y463 serves to release this autoinhibition allowing S738/S742 phosphorylation. Consistent with this, the PKD Y463E mutant is constitutively phosphorylated at S738/S742 in vivo (our unpublished data). Because maximal PKD activity is absolutely dependent on activation-loop phosphorylation, this raises the question as to the mechanism of S738/S742 phosphorylation in response to oxidative stress. Because PKCs have been shown to be activated by ROS (Konishi et al., 1997), it is tempting to speculate that under conditions of oxidative stress, two independent pathways merge at the level of PKD to stimulate full kinase activation; one requiring Src–Abl-mediated phos phorylation of Y463, and a second, parallel pathway requiring PKC-dependent activation-loop phosphorylation (Figure 9).
How do these results fit with other models for NF-κB activation? The signaling pathways leading to NF-κB activation mediated by pro-inflammatory cytokines are well understood. For example, death receptor signaling through the TNF-receptor leads to NF-κB activation via Traf2/RIP/NIK complexes, whereby the NF-κB-inducing kinase NIK plays a crucial role in the activation of IKKβ (Delhase et al., 1999). In response to oxidative stress however, NIK does not appear to be involved, since it does not potentiate NF-κB activation when co-expressed with wild-type PKD (Supplementary figure S6). Similarly, the lack of a role of IKKα in mediating PKD-dependent NF-κB activation is reminiscent of the pathway leading to NF-κB activation stimulated by inflammatory cytokines, where IKKα activity is also not required for the stimulation of the IKK complex. Although another NIK-independent pathway leading to NF-κB activation involving PI 3-K and Akt/PKB-mediated phosphorylation of IKKβ was described recently (Romashkova and Makarov, 1999), in our studies we found that Akt/PKB did not play a significant role in inducing NF-κB in HeLa cells (Supplementary figure S6).
However, regulation of NF-κB by the PKD pathway in response to oxidative stress shares several similarities with other pathways known to activate this transcription factor. PKD acts upstream of NF-κB at the level of IKK complex, and specifically through IKKβ. Both PKD activity (and tyrosine phosphorylation), IKKβ activation and IκBα degradation displayed similar kinetics in either H2O2- or PV-stimulated cells. More importantly, the Src–Abl–PKD pathway mediates IKKβ phosphorylation and activation resulting in the degradation of IκBα, essentially recapitulating the classical NF-κB activation pathway. In response to oxidative stress, PKD can also be found in a complex with IKKβ. The precise nature of this interaction remains to be determined, since PKD does not appear to directly phosphorylate any of the components of the IKK complex as judged by in vitro kinase assays. Therefore, it is possible that PKD functions by regulating one of the putative upstream kinases involved in IKK complex activation. Similarly, it is possible that in addition to controlling PKD activation, Y463 phosphorylation may serve as a docking site for interacting proteins that directly control IKK regulation. Future studies will address these issues.
In summary, we have shown that in response to oxidative stress, a pathway exists leading to NF-κB activation and enhanced cell survival. A key component of this pathway is the PKD serine/threonine kinase, which mediates IKKβ/NF-κB activation when phosphorylated at Y463. Under conditions of oxidative stress, which induce cell death, this pathway provides a survival advantage.
Materials and methods
Cell culture
HeLa, HEK293E, NIH 3T3 and MDA-MB-435 cell lines were from the American Type Culture Collection and maintained in high glucose (low glucose for MDA-MB-435) DMEM supplemented with 10% fetal bovine serum. Stable HeLa clones were obtained by transfection with empty vector, HA-PKD, or HA-PKD.Y463F cloned into pcDNA3. Stable NF-κB-reporter HeLa cell clones were obtained by transfection with pcDNA3 empty vector, NF-κB–luc and pCS2-(n)β-galactosidase (β-gal).
Antibodies, reagents and purified proteins
The anti-Abl, anti-PKD, anti-IKKα, anti-IKKβ, anti-IKKγ, anti-tubulin or anti-NIK antibodies were from Santa Cruz (Santa Cruz, CA), anti-IκBα and anti-phospho-IKKα/IKKβ (pSer181) from Cell Signaling Technologies (Beverly, MA), anti-Src and anti-phosphotyrosine (4G10) from Upstate Biotechnology (Waltham, MA). Anti-HA was purified from the 12CA5 hybridoma. Anti-pY463 antibody was a polyclonal antibody raised against the epitope TGSRYpYKEIPL (amino acids 458–468 in human PKD). The antibody was purified against immobilized phospho-peptide and non phospho-peptide (ResGen/Invitrogen Corporation, Huntsville, AL). STI-571 was from Novartis. All other inhibitors and phosphatases were obtained from Biomol (Plymouth Meeting, PA). TNF was a gift from H.Wajant (University of Stuttgart, Germany). Pervanadate was prepared by mixing 1 ml of a 20 mM sodium orthovanadate with 330 µl of 30% H2O2 (Fisher Scientific, Suanee, GA) for 10 min at room temperature, yielding a 15 mM solution of PV and H2O2. Residual H2O2 was inactivated by a 15 min incubation with 10 µl catalase (8 U/mg, Sigma). The PKD-specific substrate peptide used was AALVRQMSVAFFFK. Recombinant PKD was expressed in insect cells after infection with baculovirus harboring HA-tagged PKD in pFAST-Bac (Invitrogen Life Technologies) and purified on a Ni-NTA affinity column. Purified active Src was from Upstate Biotechnology and purified Abl kinase fragment was from Calbiochem (La Jolla, CA).
Expression plasmids
All PKD expression plasmids are based on an N-terminal HA-PKD in pcDNA3, derived from full-length human PKD (generously provided by F.J.Johannes), using PCR with the following primer pairs: 5′-GCGGGATCCATGTATCCTTATGATGTTCTTGATTATGCTAGCG CCCCTCCGGTCCTG-3′ and 5′-GCGCTCGAGTCAGAGGATGCTG ACACGCTC-3′. Mutagenesis was carried out by PCR using QuikChange (Stratagene, La Jolla, CA) for the following mutants: PKD.Y463F, 5′-GACACAGGAAGCAGGTACTTCAAGGAAATTCCTTTATCT-3′ and 5′-AGATAAAGGAATTTCCTTGAAGTACCTGCTTCCTGTGTC-3′; PKD.Y463Q, 5′-GACCACGGAAGCAGGTACCAAAAGGAAATTC TTTTATCT-3′ and 5′-AGATAAAGGAATTTCCTTTTGGTACCTG CTTCCTGTGTC-3′; PKD.Y463E, 5′-GACACAGGAAGCAGGTAC GAAAAGGAAATTCTTTTATCT-3′ and 5′-AGATAAAGGAATTT CCTTTTCGTACCTGCTTCCTGTGTC-3′; and PKD* (PKD.L187L. G189G) 5′-GATCACTGTGGAGAAATGCTCTGGGGCCTGGTAC GTCAAGGTCTT-3′ and 5′-AAGACCTTGACGTACCAGGCCCCA GAGCATTTCTCCACAGTGATC-3′.
Constructs were verified by DNA sequencing. All other plasmids have been described previously; wild-type and dominant-negative IKKα and IKKβ (Yamaoka et al., 1998), wild-type and kinase-inactive Src (Mukhopadhyay et al., 1995), v-Abl and SrcY527F (Engelman and Rosenberg, 1990) and wild-type Abl (Smith et al., 1999). Superfect (Qiagen, Valencia, CA) was used for transient transfections. Cells were stimulated or harvested 24 h after transfection.
Immunoblotting and immunoprecipitation
Cells were lysed in lysis buffer (50 mM Tris–HCl pH 7.4, 1% Triton X-100, 150 mM NaCl, 5 mM EDTA pH 7.4) plus Protease Inhibitor Cocktail (Sigma-Aldrich, St Louis, MO), and lysates were used either for immunoblot analysis or proteins of interest were immunoprecipitated by a 1 h incubation with the respective antibody (2 µg) followed by a 30 min incubation with protein A/G–agarose (Santa Cruz). Immune complexes were washed five times with TBS (50 mM Tris–HCl pH 7.4, 150 mM NaCl), and resolved by SDS–PAGE or subjected to in vitro kinase assays.
PKD assays
After immunoprecipitation (anti-PKD for endogenous and anti-HA for transfected PKD) and washing, 20 µl kinase buffer (50 mM Tris–HCl pH 7.4, 10 mM MgCl2 and 2 mM dithiothreitol) was added to the precipitates, and the kinase reaction was carried out for 20 min after adding 10 µl of kinase substrate mix (150 µM PKD-specific substrate peptide, 50 µM ATP, 10 µCi [γ-32P]ATP in kinase buffer). To terminate, the samples were centrifuged and the supernatants spotted onto P81 phosphocellulose paper (Whatman, Clifton, NJ). The papers were washed three times with 0.75% phosphoric acid, once with acetone, dried, and activity was determined by liquid scintillation counting.
Abl and IKKβ protein kinase assays
Twenty microliters of kinase buffer (50 mM Tris–HCl pH 7.4, 10 mM MgCl2, 2 mM dithiothreitol) was added to immunoprecipitates, and the reaction was carried out for 30 min by addition of 10 µl kinase mix [4 µg GST–CRK (Abl substrate) or GST–IκBα amino acids 5–55 (IKKβ substrate), 50 µM ATP, 10 µCi [γ-32P]ATP in kinase buffer]. To terminate, 30 µl of 2× SDS sample buffer was added and the samples were resolved by SDS–PAGE. The gels were dried and analyzed on a Molecular Imager (Bio-Rad, Hercules, CA).
RNAi
To silence the expression of PKD, the following oligonucleotides were used: 5′-AUGCUGUGGGGGCUGGUACdTdT-3′ and 5′-GUACCAG CCCCCACAGCAUdTdT-3′ (nucleotides 496–516 in human PKD). The annealed oligonucleotide duplex was transfected into cells using TransIT-TKO (Mirus, Madison, WI). Cells were seeded in 24-well plates and transfected at 30% confluency. Transfection efficiency was controlled using a Cy3-labeled Luciferase GL2 duplex (Dharmacon, Lafayette, CO). Experiments were performed 72 h after initial transfection of the RNAi. The pSUPER.PKD RNAi vector was constructed by ligation of the following oligonucleotide pair to pSUPER (Brummelkamp et al., 2002): 5′-GATCCCCATGCTGTGGGGGCTGGTACTTCAAGAGAGTACC AGCCCCCACAGCATTTTTTGGAAA-3′ and 5′-AGCTTTTCCAAA AAATGCTGTGGGGGCTGGTACTCTCTTGAAGTACCAGCCCCC ACAGCATGGG-3′. Cells were transfected with pSUPER.PKD RNAi or pSUPER in combination with PKD or PKD* using the TransIT HeLa reagent. Twenty-four hours after the initial transfection, cells were again transfected with reporter genes using Superfect. Forty-eight hours after the initial transfection cells were stimulated for 16 h with H2O2 and reporter gene assays or cell survival assays were carried out.
Reporter gene assays
Cells were transiently co-transfected with an NF-κB-reporter (NF-κB–luc, 5 µg), 1 µg pCS2-(n)β-gal and the protein(s) of interest (1 µg) using Superfect (Qiagen). Twenty-four hours after transfection, assays for luciferase and β-gal activity were performed on total cell lysates and measured on a luminometer. Luciferase activity was normalized to the β-gal activity. For reporter gene assays where synthetic RNAi duplexes were used, a HeLa clone stably transfected with the NF-κB reporter and the β-gal reporter [pCS2-(n)β-gal] was used. Experiments were performed 72 h after initial transfection of the RNAi duplex and the cells were treated with H2O2 for 9 h.
Cell survival assays
Cells were seeded in 96-well plates in cell culture media and after 24 h treated with H2O2 for 16 h. Experiments using RNAi were performed 72 h after initial transfection of the synthetic RNAi duplex and the cells were treated with H2O2 for 16 h. Cells were then washed twice with PBS and stained for 15 min with a crystal-violet solution (0.5% crystal-violet in 20% methanol). Plates were washed, dried and optical density measured (550 nm) on an ELISA reader.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
The authors would like to thank Heike Döppler for excellent techni cal assistance, and A.Israel, J.Brugge, B.Schaffhausen, B.Mayer, N.Rosenberg, F.-J.Johannes, R.Van Etten, T.Maniatis and R.Agami for generously providing expression plasmids, and H.Wajant for providing TNF. We also thank Elizabeth Lipscomb for advice with the RNAi experiments, E.Buchdunger (Novartis Inc.) for providing STI-571, and members of the Toker laboratory for insightful discussions. This work was supported by grants from the National Institutes of Health (CA 75134; A.T.) and from the Deutsche Forschungsgemeinschaft (STO 439/1–1; P.S.).
References
- Beraud C., Henzel,W.J. and Baeuerle,P.A. (1999) Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-κB activation. Proc. Natl Acad. Sci. USA, 96, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandlin I., Eiseler,T., Salowsky,R. and Johannes,F.J. (2002) PKCµ regulation of the JNK pathway is triggered via PDK1 and PKCε. J. Biol. Chem., 277, 45451–45457. [DOI] [PubMed] [Google Scholar]
- Brummelkamp T.R., Bernards,R. and Agami,R. (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–553. [DOI] [PubMed] [Google Scholar]
- Delhase M., Hayakawa,M., Chen,Y. and Karin,M. (1999) Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science, 284, 309–313. [DOI] [PubMed] [Google Scholar]
- Engelman A. and Rosenberg,N. (1990) bcr/abl and src but not myc and ras replace v-abl in lymphoid transformation. Mol. Cell. Biol., 10, 4365–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias T. and Rozengurt,E. (1998) Protein kinase D activation by mutations within its pleckstrin homology domain. J. Biol. Chem., 273, 410–416. [DOI] [PubMed] [Google Scholar]
- Imbert V. et al. (1996) Tyrosine phosphorylation of IκB-α activates NF-κB without proteolytic degradation of IκB-α. Cell, 86, 787–798. [DOI] [PubMed] [Google Scholar]
- Johannes F.J., Prestle,J., Eis,S., Oberhagemann,P. and Pfizenmaier,K. (1994) PKCµ is a novel, atypical member of the protein kinase C family. J. Biol. Chem., 269, 6140–6148. [PubMed] [Google Scholar]
- Konishi H., Tanaka,M., Takemura,Y., Matsuzaki,H., Ono,Y., Kikkawa,U. and Nishizuka,Y. (1997) Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl Acad. Sci. USA, 94, 11233–11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Bharti,A., Mishra,N.C., Raina,D., Kharbanda,S., Saxena,S. and Kufe,D. (2001) Targeting of the c-Abl tyrosine kinase to mitochondria in the necrotic cell death response to oxidative stress. J. Biol. Chem., 276, 17281–17285. [DOI] [PubMed] [Google Scholar]
- Li N. and Karin,M. (1999) Is NF-κB the sensor of oxidative stress? FASEB J., 13, 1137–1143. [PubMed] [Google Scholar]
- Li Z.W., Chu,W., Hu,Y., Delhase,M., Deerinck,T., Ellisman,M., Johnson,R. and Karin,M. (1999) The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor κB activation and prevention of apoptosis. J. Exp. Med., 189, 1839–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews S.A., Rozengurt,E. and Cantrell,D. (2000) Protein kinase D. A selective target for antigen receptors and a downstream target for protein kinase C in lymphocytes. J. Exp. Med., 191, 2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng T.C., Fukada,T. and Tonks,N.K. (2002) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell, 9, 387–399. [DOI] [PubMed] [Google Scholar]
- Mercurio F. and Manning,A.M. (1999) NF-κB as a primary regulator of the stress response. Oncogene, 18, 6163–6171. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D., Tsiokas,L., Zhou,X.M., Foster,D., Brugge,J.S. and Sukhatme,V.P. (1995) Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature, 375, 577–581. [DOI] [PubMed] [Google Scholar]
- Peters R.T. and Maniatis,T. (2001) A new family of IKK-related kinases may function as IκB kinase kinases. Biochim. Biophys. Acta, 1471, M57–M62. [DOI] [PubMed] [Google Scholar]
- Romashkova J.A. and Makarov,S.S. (1999) NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature, 401, 86–90. [DOI] [PubMed] [Google Scholar]
- Smith J.M., Katz,S. and Mayer,B.J. (1999) Activation of the Abl tyrosine kinase in vivo by Src homology 3 domains from the Src homology 2/Src homology 3 adaptor Nck. J. Biol. Chem., 274, 27956–27962. [DOI] [PubMed] [Google Scholar]
- Van Lint J., Rykx,A., Maeda,Y., Vantus,T., Sturany,S., Malhotra,V., Vandenheede,J.R. and Seufferlein,T. (2002) Protein kinase D: an intracellular traffic regulator on the move. Trends Cell Biol., 12, 193–200. [DOI] [PubMed] [Google Scholar]
- Waldron R.T. and Rozengurt,E. (2000) Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J. Biol. Chem., 275, 17114–17121. [DOI] [PubMed] [Google Scholar]
- Waldron R.T., Rey,O., Iglesias,T., Tugal,T., Cantrell,D. and Rozengurt,E. (2001) Activation loop Ser744 and Ser748 in protein kinase D are transphosphorylated in vivo. J. Biol. Chem., 276, 32606–32615. [DOI] [PubMed] [Google Scholar]
- Yamaoka S., Courtois,G., Bessia,C., Whiteside,S.T., Weil,R., Agou,F., Kirk,H.E., Kay,R.J. and Israel,A. (1998) Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell, 93, 1231–1240. [DOI] [PubMed] [Google Scholar]
- Zugaza J.L., Sinnett-Smith,J., Van Lint,J. and Rozengurt,E. (1996) Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J., 15, 6220–6230. [PMC free article] [PubMed] [Google Scholar]