

Abstract
Acute μ and κ opioids activate the ERK/MAPK phosphorylation cascade that represents an integral part of the signaling pathway of growth factors in astrocytes. By this cross-talk, opioids may impact neural development and plasticity among other basic neurobiological processes in vivo. The μ agonist, [D-ala2, mephe4, gly-ol5]enkephalin (DAMGO), induces a transient stimulation of ERK phosphorylation, whereas κ agonist, U69,593, engenders sustained ERK activation. Here we demonstrate that acute U69,593 and DAMGO stimulate ERK phosphorylation by utilization of different secondary messengers and protein kinase C (PKC) isoforms upstream of the growth factor pathway. Immortalized astrocytes transfected with either antisense calmodulin (CaM), a mutant μ opioid receptor that binds CaM poorly or a dominant negative mutant of PKCε were used as a model system to study μ signaling. Evidence was gained to implicate CaM and PKCε in DAMGO stimulation of ERK. DAMGO activation of PKCε and/or ERK was insensitive to selective inhibitors of Ca2+ mobilization, but it was blocked upon phospholipase C inhibition. These results suggest a novel mechanism wherein, upon DAMGO binding, CaM is released from the μ receptor and activates phospholipase C. Subsequently, phospholipase C generates diacylglycerides that activate PKCε. In contrast, U69,593 appears to act via phosphoinositide 3-kinase, PKCζ, and Ca2+ mobilization. These signaling components were implicated based on studies with specific inhibitors and a dominant negative mutant of PKCζ. Collectively, our findings on acute opioid effects suggest that differences in their mechanism of signaling may contribute to the distinct outcomes on ERK modulation induced by chronic μ and κ opioids.
Although both μ and κ opioids stimulate the MAPK1 phosphorylation cascade via growth factor receptor transactivation, their effect on the extracellular signal-regulated kinase (ERK) varies in duration in immortalized astrocytes (1). The μ agonist, [D-ala2, mephe4, gly-ol5] enkephalin (DAMGO), induces a transient activation of ERK that dissipates within 30 min, whereas that of the κ ligand U69,593 persists for several hours. In addition, chronic μ opioids inhibit growth factor-induced ERK activation, whereas chronic U69,593 does not. Examples of differential signaling mechanisms by subtypes of G protein-coupled receptors (GPCRs) are replete in the literature and are consistent with their possession of distinct functions (e.g. see Refs. 2 and 3). In studies of another astrocytic model system, rat C6 glioma cells, these differences in chronic μ and κ opioid regulation of ERK activity correlated well with their actions on mitogenesis, consistent with other evidence indicating that the ERK member of the MAPK family is implicated in cell proliferation (4).
The mechanisms that occur in the GPCR branch of the heterologous pathway by which μ and κ opioids signal to ERK/MAPK have not been studied in detail in comparison with the better understood steps downstream in the growth factor phase. Since opioid signaling to ERK may underlie basic mechanisms related to neuroplasticity as well as to cell proliferation, the early phase of this signaling pathway was investigated. PKC is an integral component of most GPCR signaling pathways to ERK/MAPK (5). Accordingly, PKC was found to be an early signaling component in the opioid pathway to ERK (6-8). Nevertheless, little is known about the actual isoforms involved in this pathway.
GPCR-mediated ERK activation via EGF receptor transactivation (9) is one of a number of cell- and GPCR-specific variations of this type of cross-talk that were detected (5, 10). In earlier studies, PKC was found to play a role in directly activating Raf-1 and thereby circumventing involvement of the receptor tyrosine kinases (11). Recently, PKC was also implicated in the activation of the MMP/ADAMs that are responsible for the release of extracellular membrane-bound EGF-like ligands, which trigger EGF receptor transactivation (12).
Although the phosphoinositide kinases (PI3Ks) are found in many growth factor signaling pathways, the γ isoform can also mediate GPCR signaling to ERK upstream of the growth factor pathway in some cells (13-15). The Gβγ subunit of heterotrimeric GTP binding proteins activates PI3Kγ, which then acts on the 3′-OH position of phosphatidyl myo-inositol lipids to release different phosphorylated lipid products as secondary messengers. The initial evidence for its implication in the GPCR phase of the ERK signaling pathway arose from PI3K enzyme activity and co-immunoprecipitation studies in cultured Swiss 3T3 cells and human myeloid-derived cells (16, 17). Subsequently, selective PI3K inhibitors (LY294002 and wortmannin) have been used to implicate this signaling component. Opioid stimulation of ERK is wortmannin-sensitive in COS-7 but not in C6 glioma cells, wherein MOR is overexpressed (18).
In prior investigations with HEK293 cells, we obtained evidence to suggest that the μ opioid pathway leading to EGF receptor and ERK activation featured the Ca2+-binding protein, CaM, as a secondary messenger and PKC as an intermediate (7). This work was prompted by the discovery of the ability of CaM to bind to GPCRs such as MOR, dopamine, vasopressin, and the metabotropic glutamate receptor (19-23). In the case of the metabotropic glutamate receptor subtypes 5 and 7, interaction with CaM was Ca2+-dependent, but MOR binding to CaM was shown to be at least partially Ca2+-independent (20). Direct evidence that CaM binding to MOR initiates signaling to ERK was obtained by using cells stably transfected with wild type human MOR or with a mutant MOR (K273A) that was shown to bind CaM poorly and coupled more efficiently to G protein in the original studies by Sadee and co-workers (7, 20). Here the roles of PKC isoforms, PLC, PI3K, and CaM in μ and κ opioid activation of ERK were assessed in astrocytes.
EXPERIMENTAL PROCEDURES
Reagents—Chemicals were purchased from Sigma with the following exceptions: DAMGO-trifluoroacetate was obtained from Multiple Peptide Systems (San Diego, CA); U69,593 was from NIDA Drug Supply (Research Triangle, NC); EGF (human, recombinant) was from Invitrogen; Dulbecco's modified Eagle's medium and fetal bovine serum were from ATCC (Manassas, VA); phorbol 12-myristate 13-acetate (PMA), CaM inhibitors N-(6-aminohexyl)5-chloro-1-naphthalenesulfonamide and fluphenazine, PLC inhibitor U73122, PI3K inhibitors LY294002 and wortmannin, tyrphostin AG1478, and protein kinase C inhibitors bisindolylmaleimide I (GFX) and Gö6983 were from Calbiochem; Protein G plus A-agarose suspension was purchased from Oncogene Research Products (Cambridge, MA). The following antibodies (Abs) were purchased: anti-phospho-ERK Ab from Cell Signaling Technology (Beverly, MA); CaM and EGFR (sheep polyclonal or mouse monoclonal) Abs from Upstate (Charlottesville, VA); and ERK, PKC isoform, and actin Abs from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell Cultures—Rat cortical astrocytes (CTX TNA2, ATCC) were established from cultures of primary type 1 astrocytes from 1-day-old rat brain frontal cortex. The cultures were originally transfected with a DNA construct containing the oncogenic early region of SV40 under the transcriptional control of the human glial acidic fibrillary protein promoter (24). The astrocytes have the phenotypic characteristics of type 1. The cell line was maintained in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum at 37°C in a humidified atmosphere of 95% air, 5% CO2 for up to 20-35 passages. For all experiments in which endogenous MOR or KOR were activated, early passage (passages 3-10) cells were grown in 6-well plates at least overnight to adhere well to the plate surface. Later passage (passages 10-35) cells were used for MOR/KOR transfection experiments. Optimal starvation of cultures was achieved in Dulbecco's modified Eagle's medium devoid of serum for 24 h. In all assays, agonists or inhibitors were delivered in serum-free media.
Transient and Stable Transfections—Cells were transiently transfected with pcDNA3 (for mock transfections), rat or human MOR, human K273A-MOR, rat KOR (in pcDNA3 or pCMV-neo expression vectors), or dnPKCε or dnPKCζ. The aforementioned cDNAs of MORs, KOR, dnPKCε, and dnPKCζ were kind gifts from Drs. W. Sadee, H. Akil, P. Blumberg, and S. Gutkind, respectively). Transfections were conducted using 1-2 μg of cDNA and 3-6 μl of FuGENE 6 following the manufacturer's instructions. After 24-48 h of incubation, transfection medium was replaced with serum-free medium for an additional 24 h. The efficiency of transfection was determined to be 9 ± 1% (n = 6) by in situ identification of cells expressing β-galactosidase. Negative controls included untransfected and/or mock-transfected cells.
Since the ATCC immortalized rat cortical astrocytes under investigation are transfectants selected with G418, we used zeocin (400 μg/ml) as a selection marker to prepare rat wild type MOR and/or antisense CaM stable transfections. Six clones were generated with stably transfected wild type MOR, and their MOR binding and DAMGO activation of ERK were assessed. The stable transfectants show a direct correlation between DAMGO binding to MOR and the extent of DAMGO-induced ERK phosphorylation. Stably transfected MOR clones activate ERK to a greater extent and have higher MOR binding levels than cells with endogenous MOR. DAMGO activation of ERK assays was performed only with clones containing 100-200 fmol/mg protein, a MOR concentration routinely found in rat forebrain. They gave on average 5-fold stimulation of ERK phosphorylation, whereas that of transiently transfected or untransfected cells was about 3-fold higher than basal levels in these studies.
Antisense CaM stable transfectants were also generated, and 11 clones displayed CaM levels ranging from 20 to 90% lower than control immortalized astrocytes as determined by Western blotting with a CaM Ab (Upstate). Immunoblotting was performed following the manufacturer's instructions.
Cell Membrane Preparation—Cells were harvested and homogenized by gentle disruption in a “cell cracker” as described (25). A membrane fraction (P20) was prepared from cell homogenates by sedimenting at 20,000 × g for 20 min. A mixture containing 10 μg/ml leupeptin, 2 μg/ml pepstatin A, 200 μg/ml bacitracin, and 1 mM phenylmethylsulfonyl fluoride was added to the 50 mM Tris buffer, pH 7.4, during the preparation of this membrane fraction. These P20membrane preparations were used in MOR binding and phospho-PKC immunoblotting experiments.
MOR Binding—P20 membranes (300-500 μg/ml) were incubated with 1-5 nM 3H-labeled DAMGO (35 Ci/mmol) at room temperature for 1 h. Nonspecific binding was determined in the presence of 1-10 μM DAMGO. Reactions were terminated by the addition of cold Tris buffer to the tubes followed by rapid filtration over GF/B filters in a Brandel cell harvester (Brandel Inc., Gaithersburg, MD). Filters were washed twice with cold 50 mM Tris-HCl, pH 7.4, buffer and then counted. Total and specific binding were calculated in fmol of MOR/mg of protein for each MOR-transfected cell line.
PKC Isoform Translocation and Activation—Membrane and cytosolic fractions were isolated from rat astrocytes using the method of Krotova et al. (26) with some modifications. Briefly, cells treated with opioids or vehicle were washed several times with PBS and collected by scraping in the presence of 5 mM Tris-HCl buffer, pH 7.4, containing 5 mM EGTA, 2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 50 μg/ml aprotinin, and 50 μg/ml leupeptin. Cell suspensions were sonicated and were spun at 1000 × g for 5 min, to remove unbroken cells, cell debris, and nuclear pellet and then centrifuged at 100,000 × g for 60 min at 4°C. The supernatant was used as the cytosolic fraction, whereas the pellet was resuspended in the above Tris buffer and used as the membrane fraction. Levels of PKCε and PKCζ were measured by immunoblotting with the corresponding Abs (27) in both cytosolic and membrane fractions from rat astrocytes. PKCε and PKCζ activation was also measured in P20 membrane fractions from rat astrocytes by immunoblotting, applying phospho-PKC∈ (Ser729; Santa Cruz Biotechnology) and phospho-PKCζ (Thr410/403; Cell Signaling Technology, Beverly, MA) Abs and following the manufacturers′ instructions.
ERK Assay—ERK phosphorylation was measured by immunoblotting as described (7). Briefly, cells were treated first with different inhibitors and then with DAMGO or U69,593 as described in each figure legends. Cells were then washed with PBS and lysed with buffer containing 20 mM HEPES, 10 mM EGTA, 40 mM β-glycerophosphate, 2.5 mM MgCl2, 2 mM sodium vanadate, 1% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, and 20 μg/ml leupeptin. Cell lysates were centrifuged at 14,000 × g for 20 min at 4°C, and protein concentration of the supernatants was determined. Samples (10-20 μg of protein/lane) were separated by 10% SDS-PAGE. Proteins were blotted on Immobilon P™ polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA). Nonspecific sites were blocked with 5% milk in Tris-buffered saline plus 0.2% Tween 20 (TBST). Blots were then washed three times with TBST and incubated with anti-phospho-ERK Ab, diluted 1:2000 in TBST for at least 15 h at 4°C. After three washes with TBST, blots were incubated with 1:2000 diluted goat anti-mouse horseradish peroxidase-conjugated IgG (Sigma) for 1 h at room temperature. For assurance of equivalent total ERK protein per lane, representative blots were stripped (0.2 M glycine, pH 2.5, 60 min at room temperature) and exposed to ERK Ab, followed by goat antirabbit horseradish peroxidase-conjugated IgG. Bands were visualized using an ECL chemiluminescence detection system (Amersham Biosciences) and exposure to Classic Blue sensitive x-ray film (Molecular Technologies, St. Louis, MO). Band intensities were determined by densitometric analysis using a Kodak DC120 digital camera, Kodak ds 1D version 3.0.2 software (Scientific Imaging Systems, New Haven, CT), and NIH ImageJ, version 1.32j.
EGFR Immunoprecipitation—The EGFR immunoprecipitation protocol followed a previously described procedure (7). Briefly, serum-starved cells were administered DAMGO or U69,593 (0.1 μM, 3-5 min). Cultures were lysed by using a modified radioimmune precipitation buffer containing 50 mM Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mM Na3VO4, 1 mM NaF. Cell lysates of 0.8-1.2 mg of protein (diluted to ∼1 μg/μl) were used. EGFR was immunoprecipitated by adding 5-10 μg of either a mouse monoclonal or sheep polyclonal anti-EGFR Ab (Upstate) to the lysates and incubating overnight at 4°C. This step was followed by the addition of a 50-μl suspension of protein G plus A-Sepharose beads per sample and incubation for 3-4 h at 4°C. The beads were washed three times with PBS, resuspended in SDS loading buffer, and boiled for 5 min before SDS-PAGE. Proteins were blotted on Immobilon P™ polyvinylidene difluoride membranes and tested with Tyr(P) Ab (Cell Signaling) and peroxidase-conjugated mouse secondary Ab. Bands were visualized using a chemiluminescence detection system as described above.
Protein Assay and Statistical Analysis—Protein concentrations were determined by the Bradford method (28) with bovine serum albumin (1 mg/ml) as a standard. Statistical determinations were made by t test analysis (version 2.01; GraphPad Software, Inc.). Data are expressed as the mean ± S.E.
RESULTS
μ and κ Opioid-induced ERK Phosphorylation Is Mediated by Different PKC Isoforms—Previously, both time and dose dependence plots for DAMGO and U69,593 were obtained for cortical type 1 immortalized astrocytes to optimize conditions for ERK activation by these opioids (Ref. 1 and data not shown). Selective μ (CTAP) and κ (nor-binaltorphimine) opioids inhibited DAMGO and U69,593, respectively. Although PKC mediates the activation of ERK by all opioid receptor subtypes in a number of cell lines, including C6 glioma cells (4, 6, 7), initial experiments with the cortical astrocytes revealed that the general PKC inhibitor, GFX, blocked μ opioid-induced ERK phosphorylation but not that by κ (Fig. 1A). To determine whether the opioids under investigation here use different PKC isoforms in this signaling pathway than those in previously studied cells, we assessed the effects of another PKC inhibitor, Gö6983, which complements the PKC isoform selectivity profile of GFX (Fig. 1, B and C). GÖ6983 abolished U69,593-induced ERK phosphorylation without affecting that of three different μ opioid agonists, DAMGO, morphine, and endomorphin.
FIG. 1.

Differential effects of PKC inhibitors, GFX and Gö6983, on μ and κ opioid agonist stimulation of ERK phosphorylation as determined by immunoblotting assays. A, GFX abolishes DAMGO but not U69,593 stimulation of ERK phosphorylation. Astrocytes were pretreated with GFX (0.1 μM, 30 min) before adding DAMGO (0.1 μM, 5-10 min) or U69,593 (0.1 μM, 10 min). Endogenous MOR activity was measured in DAMGO-treated cells, whereas transiently transfected KOR astrocytes were treated with U69,593. A representative immunoblot is shown. n = 4-7 experiments. *, significantly greater than controls (p < 0.05). #, significantly less than DAMGO alone (p < 0.05). B, Gö6983 did not alter μ opioid agonist stimulation of ERK phosphorylation. MOR stably transfected astrocytes were pretreated with Gö6983 (0.1 μM, 30 min) before treatment with DAMGO (0.1 μM), endomorphin (0.1-1 μM), or morphine (0.5 μM) for 5 min. A representative immunoblot is shown. n = 3-6 experiments. *, significantly greater than controls (p < 0.05). **, significantly greater than controls (p < 0.01). C, Gö6983 abolished U69,593 stimulation of ERK phosphorylation. Astrocytes transiently transfected with KOR were pretreated with Gö6983 (0.1 μM, 30 min) before treatment with U69,593 (0.1 μM,10 min). A representative immunoblot is shown. n = 6-12 experiments. *, significantly greater than controls (p < 0.05). #, significantly less than U69,593 alone (p < 0.05).
Of the ∼11 known isozymes of PKC, GFX inhibits all of the conventional isoforms (α, β1, β2, γ) that are DAG- and Ca2+-dependent and several novel isoforms (δ, ε) that are DAG-dependent but do not require Ca2+ (29). Whereas Gö6983 is also selective for conventional PKC isoforms, it inhibits PKCδ and the atypical PKCζ that is insensitive to Ca2+ and DAG (29). On the basis of the isoform selectivity profiles of the two PKC inhibitors, we tested DAMGO for its ability to activate PKCε. Both PKCε membrane translocation (Fig. 2A) and phosphorylation assays (Fig. 2B) showed that the μ opioid activated this PKC. Time course studies on DAMGO-induced PKCε phosphorylation suggested that it peaks at 3 min, and by 5 min it is reduced to basal levels (data not shown). Transfection of astrocytes with a dominant negative mutant of PKCε attenuated DAMGO-induced phosphorylation of both PKCε (Fig. 2B) and ERK (Fig. 2C). Alternately, U69,593 signaling to ERK was not affected by the expression of dnPKCε in astrocytes (11 ± 1.4 in control cells versus 12 ± 1.5 in dnPKCε-expressing cells, n = 4). The data implicate PKCε in μ opioid signaling to ERK.
FIG. 2.
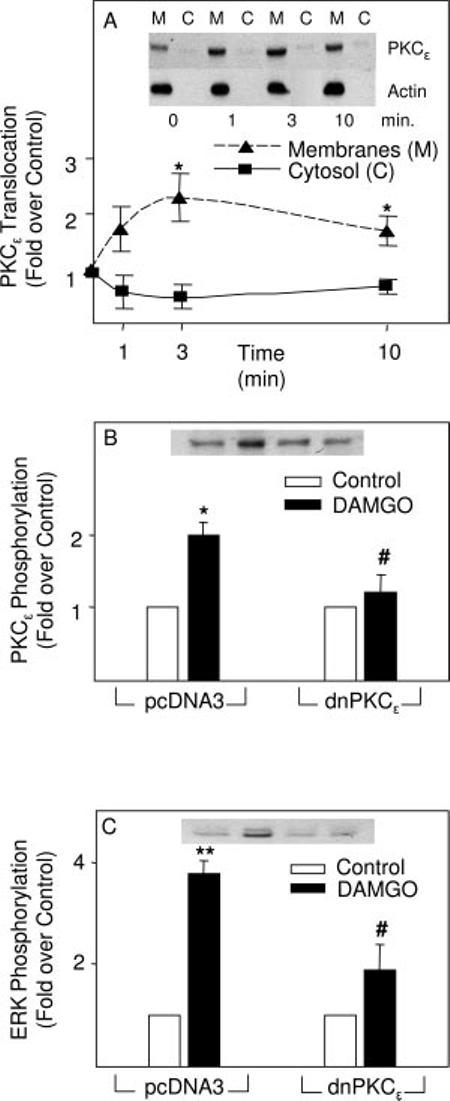
DAMGO activation of PKCε as determined by immunoblotting assays. A, DAMGO induced translocation of PKCε into the membrane fraction of astrocytes. Cells stably transfected with MOR were treated with DAMGO (0.1 μM) for 1, 3, or 10 min before sonication and preparation of membrane and cytosolic fractions by differential centrifugation. Representative immunoblots using a PKCε Ab and an actin Ab (loading control) are shown. n = 2-3 experiments. *, significantly greater than controls (p < 0.05.). B, DAMGO-induced PKCε phosphorylation was abolished by dnPKCε. Astrocytes stably transfected with MOR were transiently transfected with either an empty vector (pcDNA3) or dnPKCε and were treated with DAMGO (0.1 μM, 3 min). A representative immunoblot is shown. n = 3-4 experiments. *, significantly greater than controls (p < 0.05.). #, significantly less than DAMGO alone (p < 0.05). C, DAMGO induction of ERK phosphorylation was attenuated by dnPKCε. Astrocytes stably transfected with MOR were transiently transfected with either an empty vector (pcDNA3) or dnPKCε and were treated with DAMGO (0.1 μM, 3 min). A representative immunoblot is shown. n = 3 experiments. **, significantly greater than controls (p < 0.01). #, significantly less than DAMGO alone (p < 0.05).
μ Opioid-induced PKCε and ERK Phosphorylation Is Mediated by PLC but Not PI3K—There are reports that PKCε can be activated by either DAG, which is normally generated by PLC hydrolysis of phosphatidylinositide (30), or by PI3K (29). To address this question, the phorbol ester PMA, which mimics DAG by binding at its activation site on some PKCs, was tested and found to stimulate PKCε phosphorylation to about the same extent as DAMGO (Fig. 3A). This issue was further examined by subjecting astrocytes to chronic PMA, which down-regulates PKCs that contain a DAG binding site. Accordingly, overnight treatment of astrocytes with PMA abolished DAMGO- but not U69,593-induced ERK phosphorylation (Fig. 3B). Evidence that PLC generates the DAG in question was obtained by demonstrating that a specific inhibitor of this enzyme, U73112, attenuated DAMGO-induced phosphorylation of both PKCε (Fig. 3A) and ERK (Fig. 3C). Moreover, selective inhibitors of PI3K, wortmannin and LY294002, had no effect on μ opioid-induced ERK phosphorylation (Fig. 3C).
FIG. 3.
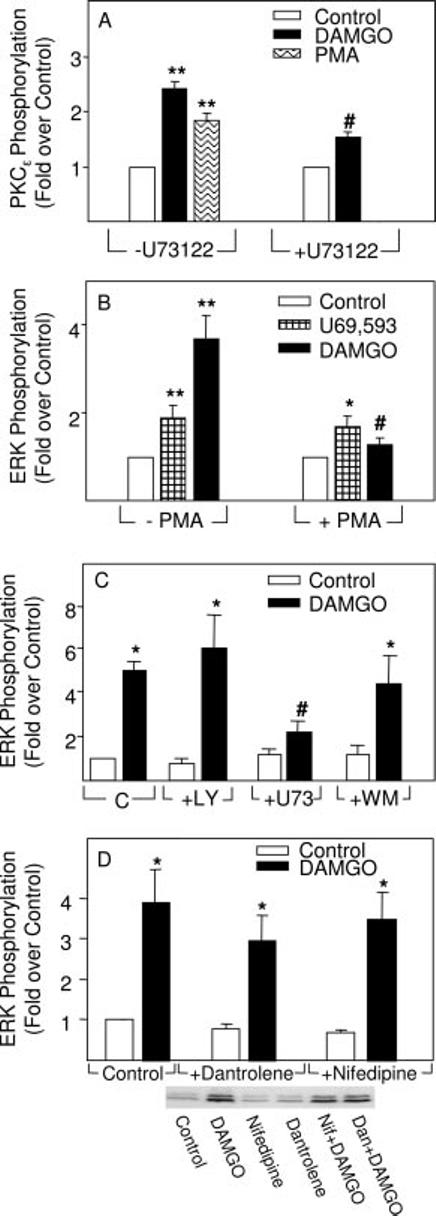
Differential effects of PLC inhibitor (U73122), phorbol ester (PMA), PI3K inhibitors (wortmannin and LY294002), or Ca2+ mobilization inhibitors (dantrolene and nifedipine) on DAMGO stimulation of PKCε and/or ERK phosphorylation as determined by immunoblotting assays. A, DAMGO induced PKCε phosphorylation by a mechanism attenuated by U73122, a selective inhibitor of PLC. Astrocytes stably transfected with MOR were pretreated with 10 μM U73122 for 30 min before treatment with 0.1 μM DAMGO for 3 min. In some experiments, cells were treated with PMA (1 μM, 3 min) as a positive control for stimulation of PKCε phosphorylation. n = 4-5 experiments. **, significantly greater than controls (p < 0.01). #, significantly less than DAMGO alone (p < 0.05). B, effect of chronic PMA on U69,593 or DAMGO stimulation of ERK phosphorylation. Astrocytes transiently transfected with KOR or stably transfected with MOR were pretreated with PMA (1 μM) overnight and treated with 0.1 μM U69,593 for 10 min or 0.1 μM DAMGO for 5 min, respectively. n = 4-5 experiments. *, significantly greater than controls (p < 0.05). **, significantly greater than controls (p < 0.01). #, significantly less than DAMGO alone (p < 0.05). C, differential effects of U73122 (U73), wortmannin (WM), and LY294002 (LY) on DAMGO stimulation of ERK phosphorylation. Astrocytes stably transfected with MOR were pretreated with U73122 (10 μM), wortmannin (1 μM), or LY294002 (10 μM) for 30 min before treatment with 0.1 μM DAMGO for 5 min. n = 3-4 experiments. *, significantly greater than controls (p < 0.05). #, significantly less than DAMGO alone (p < 0.05). D, DAMGO induction of ERK phosphorylation was unaffected by an inhibitor of microsomal Ca2+ release (dantrolene) or L-type Ca2+ channels (nifedipine). Astrocytes were pretreated for 30 min with 1 μM dantrolene (Dan) or 1 μM nifedipine (Nif) before treatment with 0.1 μM DAMGO for 5 min, respectively. A representative immunoblot is shown. n = 5-10 experiments. *, significantly greater than controls, p < 0.05.
As a member of the novel PKC isoform family, PKCε should be Ca2+-independent. Accordingly, we measured the intermediacy of Ca2+ mobilization and discovered that μ activation of ERK was not blocked by nifedipine, an L-type Ca2+ channel inhibitor, or by dantrolene, an inhibitor of microsomal Ca2+ release (Fig. 3D).
CaM Is a Secondary Messenger in μ Opioid-induced ERK Phosphorylation—How is PLC activated to generate DAG? There is evidence to suggest that CaM can activate PLCβ (31). In prior studies using HEK293 cells, we discovered that the μ opioid pathway leading to growth factor transactivation featured CaM as a second messenger and PKC as an intermediate (7). Thus, we took three different approaches to implicate CaM in astrocytes. CaM antagonists, N-(6-aminohexyl)5-chloro-1-naphthalenesulfonamide and fluphenazine, were shown to attenuate DAMGO stimulation of ERK phosphorylation (data not shown). However, these antagonists are not completely selective. A second approach entailed the use of astrocytes stably transfected with CaM antisense cDNA. For the antisense experiments shown here a clone was adopted that displayed 55% less CaM than basal levels (n = 3; see inset in Fig. 4A). Astrocytes transfected with CaM antisense and transiently transfected with MOR were treated with DAMGO or, in some cases, morphine and were found to be incapable of stimulating ERK activity (Fig. 4A). However, CaM antisense clones transiently transfected with KOR retained their ability to mediate κ opioid activation of ERK (Fig. 4B). Astrocytes of similar passage number were also transiently transfected with MOR or KOR and used as controls in these experiments.
FIG. 4.

Evidence implicating CaM in μ but not κ opioid agonist stimulation of EGFR and/or ERK phosphorylation as determined by immunoblotting assays. A, CaM antisense abolished DAMGO-induced ERK phosphorylation. Astrocytes were stably transfected with antisense CaM cDNA and shown to have reduced CaM levels by immunoblotting. Control cells and antisense CaM (anti-CaM) clones were transiently transfected with MOR cDNA and treated with DAMGO (0.1 μM, 5 min) or PMA (10 μM, 5 min) before measuring ERK phosphorylation. Representative immunoblot is shown. n = 4-9 experiments. *, significantly greater than controls (p < 0.05). #, significantly less than agonist alone (p < 0.05). Inset, representative immunoblot showing levels of CaM in equivalent amounts of protein from lysates of control and antisense CaM cells. B, CaM antisense did not alter U69,593 stimulation of ERK phosphorylation. Control cells and antisense CaM-transfected clones were transiently transfected with KOR cDNA. Cells were treated with U69,593 (0.1 μM, 10 min) before measuring ERK phosphorylation. A representative immunoblot is shown. n = 4-7 experiments. *, significantly greater than controls (p < 0.05). C, CaM antisense abolished DAMGO- and morphine-induced EGFR phosphorylation. Control cells and antisense CaM-transfected clones were transiently transfected with MOR cDNA. Cells were treated with DAMGO or morphine (0.1 μM, 1 min) before measuring EGFR phosphorylation by immunoprecipitation with EGFR Ab and immunoblotting with phospho-Tyr Ab. n = 4-9 experiments. *, significantly greater than controls (p < 0.05). #, significantly less than agonist alone (p < 0.05). D, DAMGO modulation of ERK phosphorylation in wild type MOR and mutant K273A MOR-transfected rat astrocytes. Cells were transiently transfected with either wild type human MOR or mutant human K273A MOR cDNA. Cultures were treated with DAMGO (0.1 μM, 5 min) before immunoblotting. Representative immunoblot is shown. n = 8 experiments. *, significantly greater than controls (p < 0.05). #, significantly less than wild type MOR (p < 0.05).
In some cells, CaM has been implicated in the modulation of growth factor-induced ERK activation. This can occur either by a direct interaction with Ras-Raf (32) or EGFR itself (33). Although CaM kinases have been implicated in activation of MAPKs (34), some have also been shown to phosphorylate EGFR and reduce its response to EGF in CHO cells (35). Earlier studies revealed that opioid modulation of ERK activation was insensitive to CaM kinase inhibitor, KN93, in astrocytes.2 Since DAMGO-induced EGFR phosphorylation was blocked in the CaM antisense clones, the CaM mediation occurs at a step upstream of PKCε activation (Fig. 4A) and of EGFR transactivation (Fig. 4C). Finally, astrocytes, transfected with wild type MOR or with the mutant MOR (K273A) that binds CaM poorly, gave results similar to those with HEK293 cells (7). Namely, μ opioid activation of ERK is attenuated in astrocytes expressing mutant MOR (K273A) by about 40% when compared with wild type MOR (Fig. 4D). In light of our transfection efficiency of 9 ± 1% and basal phospho-ERK levels, this inhibition is appreciable.
κ Opioid-induced ERK Phosphorylation Is Mediated by PKCζ and PI3K—On the basis of the selectivity profiles of the PKC inhibitors used in Fig. 1, the PKCζ isoform may mediate κ opioid signaling to ERK. Accordingly, U69,593 was found to induce PKCζ phosphorylation (Fig. 5A). PKCζ was detected in astrocytes by immunoblotting, but U69,593 did not induce its translocation (data not shown), consistent with other findings on its activation (30). Interestingly, U69,593 stimulation of PKCζ activity continued for at least 60 min (7.6 ± 2.9-fold over control, p < 0.05, n = 4), suggesting that there is a sustained activation of this PKC isoform parallel with that of ERK. Moreover, the transfection of dnPKCζ into astrocytes resulted in the attenuation of chronic (2 h) U69,593 stimulation of ERK phosphorylation (data not shown).
FIG. 5.
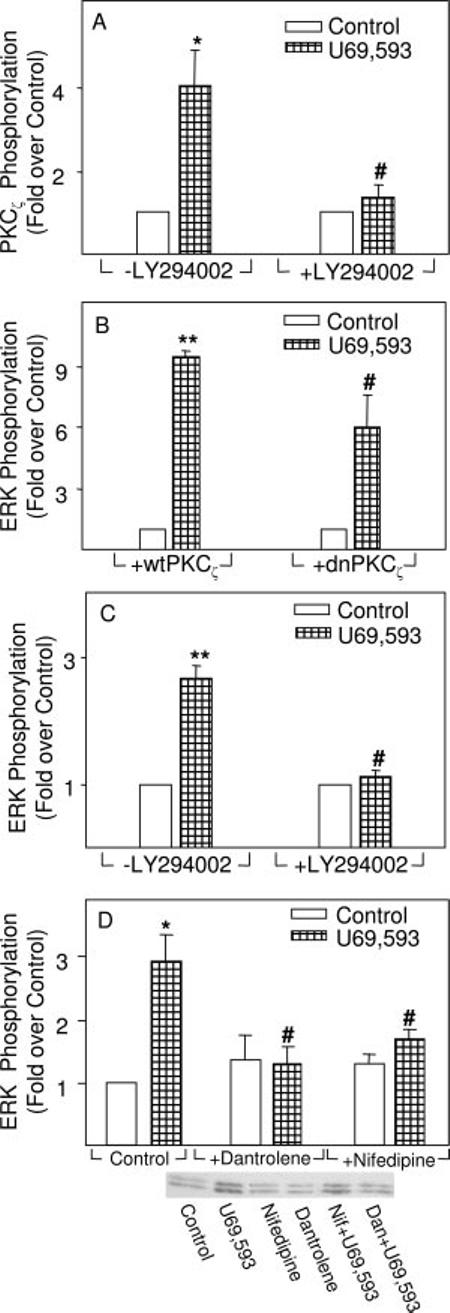
Evidence implicating PI3K, PKCζ and Ca2+ mobilization in U69,593 stimulation of ERK phosphorylation as determined by immunoblotting assays. A, U69,593 induced PKCζ phosphorylation by a mechanism blocked by the PI3K inhibitor, LY294002. Astrocytes transiently transfected with KOR were pretreated with LY294002 (10 μM) for 30 min before treatment with 0.1 μM U69,593 for 5 min. *, significantly greater than controls (p < 0.05). #, significantly less than U69,593 alone (p < 0.05). B, astrocytes transfected with dnPKCζ attenuated U69,593 stimulation of ERK phosphorylation. Cells transiently transfected with KOR cDNA were cotransfected with either wild type (wt) or dnPKCζ. Astrocytes were treated with U69,593 (0.1 μM, 10 min), and ERK phosphorylation was measured. n = 5-6 experiments. **, significantly greater than controls (p < 0.01). #, significantly less than U69,593 alone, p < 0.05. C, U69,593 stimulation of ERK phosphorylation was abolished by LY294002. Cells transiently transfected with KOR were exposed to LY294002 (10 μM) for 30 min before treatment with 0.1 μM U69,593 for 10 min. n = 5-6 experiments. **, significantly greater than controls (p < 0.01). #, significantly less than U69,593 alone (p < 0.05). D, U69,593 stimulation of ERK phosphorylation was abolished by an inhibitor of microsomal Ca2+ release (dantrolene) or of L-type Ca2+ channels (nifedipine). Astrocytes were pretreated for 30 min with 1 μM dantrolene (Dan) or 1 μM nifedipine (Nif) before treatment with U69,593 (0.1 μM) for 10 min. A representative immunoblot is shown. n = 5-10 experiments. *, significantly greater than controls (p < 0.01). #, significantly less than U69,593 alone (p < 0.05).
Acute U69,593 activation of ERK was attenuated by a dnPKCζ but not by wild type PKCζ transfected into astrocytes (Fig. 5B). In contrast, DAMGO stimulation of ERK proved to be insensitive to this mutant, since the stimulation observed in its presence did not differ from that in the absence of dnPKCζ (data not shown). Since PKCζ is an atypical isoform, it should be PMA-insensitive. This is consistent with the data seen in Fig. 3B, wherein chronic PMA inhibits DAMGO but not U69,593 activation of ERK.
PKCζ has been identified as a downstream target of PI3K and its activation can result in ERK phosphorylation (36, 37). Consistent with this possibility, the PI3K inhibitor, LY294002, attenuated U69,593-induced PKCζ phosphorylation (Fig. 5A). Accordingly, LY294002 also inhibited U69,593-induced ERK phosphorylation (Fig. 5C).
Nifedipine, a selective inhibitor of L-type Ca2+ channel influx, and dantrolene, a blocker of release of intracellular Ca2+ stores, attenuated U69,593-induced ERK activation (Fig. 5D). Since PKCζ activation is not Ca2+-dependent, these results suggest that the location of the Ca2+ mobilization step in this pathway is downstream of PKCζ. There is ample precedence in the literature for this signaling sequence (see “Discussion”).
PKC, PI3K, and PLC have also been implicated in growth factor signaling to ERK in some cells, raising the question of the location of these three signaling components in this heterologous pathway. To examine this issue further, exogenous EGF stimulation of ERK phosphorylation was assayed and found to be unaffected in astrocytes pretreated with 0.1 μM Gö6983, 0.1 μM GFX, 10 μM U73122, 1 μM wortmannin, or 10 μM LY294002 for 30 min before 5-min treatment with 10 or 100 ng/ml EGF. EGF induced phospho-ERK levels >10-fold higher than basal levels (p < 0.01, n = 3-4 experiments). Taken together with the data shown in Figs. 3A, 4C, and 5A, the results suggest that μ opioid-induced PKCε phosphorylation via CaM and PLC and κ opioid activation of PKCζ via PI3K occur in the GPCR branch of the pathway of the astrocytes.
DISCUSSION
The data presented here reveal that the μ opioid agonist, DAMGO, activates ERK/MAPK by a mechanism that differs appreciably from that of the κ opioid agonist, U69,593 in the GPCR phase of the signaling pathway in immortalized rat cortical type 1 astrocytes. In fact, KOR signaling often served as a negative control for MOR signaling in this investigation and vice versa. The results on μ opioid signaling support a novel mechanism, in which upon binding to MOR, DAMGO induces the release of CaM from this receptor. CaM may then activate PLC, generating DAG that binds to PKCε, leading to its phosphorylation. PKCε can then signal to an ADAM/MMP, which, on the basis of previous studies, can cleave membraneanchored EGF-type ligands, thereby initiating EGF receptor transactivation and ultimately activation of the MAPK phosphorylation cascade (1, 7, 9). Interlocking evidence supports this sequence. In addition to DAMGO-induced time-dependent translocation of PKCε from the cytosol to membranes, PKCε phosphorylation at Ser729 in the C-terminal hydrophobic motif was demonstrated to be induced by both DAMGO and PMA, establishing activation of this PKC isoform (38). Since this phosphorylation is blocked by the PLC inhibitor U73122, PLC should be upstream of PKCε in the pathway. Based on the acute and chronic PMA modulation of PKCε and ERK activities observed here, a DAG may activate this PKC isoform and may be derived from PLC action. Since DAMGO-induced ERK phosphorylation is abolished by a dnPKCε, this isoform is required for activation of ERK. The antisense CAM and K273A MOR mutant data are consistent with earlier findings of MOR signaling to ERK in HEK293 cells that support the secondary messenger role of CaM (7). Finally, there is evidence to show that PLCβ isoforms are activated by CaM and are regulated by chronic opioids such as morphine (31, 39).
In contrast to the μ opioid signaling pathway to ERK, our working hypothesis is that U69,593-induced ERK activation entails Gβγ transduction of PI3Kγ. PI3Kγ can then generate phosphatidylinositol 3,4,5-triphosphate that activates PKCζ, which in turn mobilizes Ca2+. Implication of PI3Kγ is based on the sensitivity of the pathway to the selective inhibitor, LY294002, and the fact that it is the primary activator of PKCζ (40). This mechanism is consistent with data indicating that opioid receptor-mediated ERK activation is mediated by PI3K in certain cells (2, 18, 41). Moreover, a PI3K pathway to ERK mediated by MOR and the nociceptin orphan opioid receptor proved insensitive to GFX and chronic PMA (which depletes PKC levels) in Chinese hamster ovary cells (2). The involvement of PKCζ is also based on the ability of U69,593 to phosphorylate this isoform at Thr410 in the activation loop of this isoform (42). In addition, U69,593 activation of ERK is attenuated by the dnPKCζ. Finally, studies that show that the inhibitors of PKC, PLC, and PI3K used in the studies on acute μ and κ opioid activation of ERK failed to block EGF stimulation of ERK phosphorylation support the theory that the mechanisms delineated occur in the GPCR phase of this heterologous signaling pathway. The Ca2+ dependence of the κ pathway in immortalized astrocytes differs from μ opioid signaling to ERK, which uses the novel, Ca2+-independent PKCε isoform in this cell line. This κ pathway also varies from μ and κ opioid-induced ERK activation in rat C6 glioma cells, wherein conventional PKC isoforms appear to be signaling components on the basis of the inhibition of opioid-induced phosphatidylinositide turnover and/or ERK activation by GFX, chronic phorbol esters, and antagonists of intracellular and extracellular Ca2+ mobilization via microsomal stores and L-type Ca2+ channels, respectively (8, 18). All PKCs have been reported to be abundantly expressed in the transformed C6 cell line but not in primary glial cultures, which only express specific isoforms. Since PKCζ is Ca2+-independent, we believe that the mobilization of this ion, which is required in the κ opioid-signaling in immortalized astrocytes, is downstream of this atypical isoform. Moreover, it has been shown that PKCζ can activate Ca2+ channels (43, 44). Since our previous studies have also implicated ADAM/MMP and EGFR transactivation in the KORmediated pathway, it is possible that the ADAM/MMP isoform involved is Ca2+-dependent. ADAM-10, which mediates GPCR-induced EGFR transactivation, has been shown to be Ca2+-dependent in fibroblasts (45, 46). Of course, the cell type-based diversity of signaling pathways restricts the import of such precedence. Accordingly, there may be other roles for Ca2+ in these cells. As seen in neuronal model systems that appear to differ considerably from astrocytes in their GPCR-ERK heterologous signaling, the Ca2+-dependent focal adhesion kinase, Pyk2, is an integral signaling component downstream of PKC in the GPCR pathway (8, 47, 48). In addition, high extracellular Ca2+ concentrations are required for persistent PKCζ activation in human platelets (49). This raises the question of whether sustained ERK activation induced by chronic U69,593 may be due to the high Ca2+ levels that maintain the persistent PKCζ signaling.
Thus, profound differences in acute μ and κ opioid signaling to ERK in the GPCR phase of the pathway include involvement of different PKC isoforms. It appears that various PKC isoforms may possess specific roles. For example, PKCδ has been implicated in growth and tissue remodeling, whereas there is evidence to suggest a role for PKCζ in proliferation via ERK (40, 50-55). Finally, PKCε has been implicated in cell differentiation and can play a role in inhibitory actions in this process (29, 56, 57).
Do these acute μ and κ opioid signaling variations impact on the divergent chronic actions of these two subtypes? Certainly, PKC isoforms have been implicated in opioid receptor desensitization as well as repression induced by chronic morphine (39, 58, 59). The chronic κ opioid pathway features sustained, PKCζ-dependent ERK activation in immortalized astrocytes but more transient activation in COS-7 and C6 cells in which different PKC isoforms could well be involved as suggested by inhibitor studies (1, 8, 60). In contrast, acute μ opioid signaling to ERK is transient, and chronic μ opioid agonists have no effect but cause a 30-40% inhibition of mitogen-activated ERK in COS-7, C6 cells, and immortalized rat astrocytes (1, 4, 60, 61). The duration of MAPK activation is an essential factor for its role in gene regulation (62). Thus, it is important to determine whether PKC isoforms may be responsible for the transient actions of μ and the sustained activation of ERK by chronic κ opioids.
Footnotes
This work was supported in part by National Institutes of Health Grant DA05412. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: MAPK, mitogen activated protein kinase; Ab, antibody; ADAM, a disintegrin and metalloprotease; CaM, calmodulin; DAG, diacylglycerol; DAMGO, [D-ala2,mephe4,glyol5]enkephalin; dnPKC, dominant negative PKC; EGF, epidermal growth factor; EGFR, EGF receptor; ERK, extracellular signal-regulated kinase; GPCR, G protein-coupled receptor; KOR, κ opioid receptor; MMP, matrix metalloprotease; MOR, μ opioid receptor; PI3K, phosphoinositide-3 kinase; PLC, phospholipase C; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate.
M. M. Belcheva, A. L. Clark, P. D. Haas, J. S. Serna, J. W. Hahn, A. Kiss, and C. J. Coscia, unpublished observations.
REFERENCES
- 1.Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ. Mol. Pharmacol. 2003;64:1391–1401. doi: 10.1124/mol.64.6.1391. [DOI] [PubMed] [Google Scholar]
- 2.Hawes BE, Fried S, Yao XR, Weig B, Graziano MP. J. Neurochem. 1998;71:1024–1033. doi: 10.1046/j.1471-4159.1998.71031024.x. [DOI] [PubMed] [Google Scholar]
- 3.Olivares-Reyes JA, Jayadev S, Hunyady L, Catt KJ, Smith RD. Mol. Pharmacol. 2000;58:1156–1161. doi: 10.1124/mol.58.5.1156. [DOI] [PubMed] [Google Scholar]
- 4.Bohn LM, Belcheva MM, Coscia CJ. J. Neurochem. 2000;74:574–581. doi: 10.1046/j.1471-4159.2000.740574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belcheva MM, Coscia CJ. Neurosignals. 2002;11:34–44. doi: 10.1159/000057320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukuda K, Kato S, Morikawa H, Shoda T, Mori K. J. Neurochem. 1996;67:1309–1316. doi: 10.1046/j.1471-4159.1996.67031309.x. [DOI] [PubMed] [Google Scholar]
- 7.Belcheva MM, Szucs M, Wang DX, Sadee W, Coscia CJ. J. Biol. Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- 8.Bohn LM, Belcheva MM, Coscia CJ. J. Neurochem. 2000;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daub H, Weiss FU, Wallasch C, Ullrich A. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 10.Shah BH, Catt KJ. Trends Neurosci. 2004;27:48–53. doi: 10.1016/j.tins.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- 12.Pierce KL, Tohgo A, Ahn S, Field ME, Luttrell LM, Lefkowitz RJ. J. Biol. Chem. 2001;276:23155–23160. doi: 10.1074/jbc.M101303200. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 14.Hawes BE, Luttrell LM, Vanbiesen T, Lefkowitz RJ. J. Biol. Chem. 1996;271:12133–12136. doi: 10.1074/jbc.271.21.12133. [DOI] [PubMed] [Google Scholar]
- 15.Duckworth BC, Cantley LC. J. Biol. Chem. 1997;272:27665–27670. doi: 10.1074/jbc.272.44.27665. [DOI] [PubMed] [Google Scholar]
- 16.Stephens L, Eguinoa A, Corey S, Jackson T, Hawkins PT. EMBO J. 1993;12:2265–2273. doi: 10.1002/j.1460-2075.1993.tb05880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumagai N, Morii N, Fujisawa K, Nemoto Y, Narumiya S. J. Biol. Chem. 1993;268:24535–24538. [PubMed] [Google Scholar]
- 18.Belcheva MM, Haas PD, Tan Y, Heaton VM, Coscia CJ. J. Pharmacol. Exp. Ther. 2002;303:909–918. doi: 10.1124/jpet.102.038554. [DOI] [PubMed] [Google Scholar]
- 19.Bofill-Cardona E, Kudlacek O, Yang Q, Ahorn H, Freissmuth M, Nanoff C. J. Biol. Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- 20.Wang DX, Sadee W, Quillan JM. J. Biol. Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- 21.Minakami R, Jinnai N, Sugiyama H. J. Biol. Chem. 1997;272:20291–20298. doi: 10.1074/jbc.272.32.20291. [DOI] [PubMed] [Google Scholar]
- 22.Nickols HH, Shah VN, Chazin WJ, Limbird LE. J. Biol. Chem. 2004;279:46969–46980. doi: 10.1074/jbc.M407351200. [DOI] [PubMed] [Google Scholar]
- 23.Nakajima Y, Yamamoto T, Nakayama T, Nakanishi S. J. Biol. Chem. 1999;274:27573–27577. doi: 10.1074/jbc.274.39.27573. [DOI] [PubMed] [Google Scholar]
- 24.Radany EH, Brenner M, Besnard F, Bigornia V, Bishop JM, Deschepper CF. Proc. Natl. Acad. Sci. U. S. A. 1992;89:6467–6471. doi: 10.1073/pnas.89.14.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belcheva M, Barg J, Rowinski J, Clark WG, Gloeckner CA, Ho A, Gao XM, Chuang DM, Coscia C. J. Neurosci. 1993;13:104–114. doi: 10.1523/JNEUROSCI.13-01-00104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krotova KY, Zharikov SI, Block ER. Am. J. Physiol. 2003;284:L1037–L1044. doi: 10.1152/ajplung.00308.2002. [DOI] [PubMed] [Google Scholar]
- 27.Brandlin I, Eiseler T, Salowsky R, Johannes FJ. J. Biol. Chem. 2002;277:45451–45457. doi: 10.1074/jbc.M205299200. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. Anal. Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 29.Chow JYC, Uribe JM, Barrett KE. J. Biol. Chem. 2000;275:21169–21176. doi: 10.1074/jbc.M002160200. [DOI] [PubMed] [Google Scholar]
- 30.Chen CC. FEBS Lett. 1993;332:169–173. doi: 10.1016/0014-5793(93)80506-p. [DOI] [PubMed] [Google Scholar]
- 31.McCullar JS, Larsen SA, Millimaki RA, Filtz TM. J. Biol. Chem. 2003;278:33708–33713. doi: 10.1074/jbc.M301940200. [DOI] [PubMed] [Google Scholar]
- 32.Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- 33.San Jose E, Benguria A, Geller P, Villalobo A. J. Biol. Chem. 1992;267:15237–15245. [PubMed] [Google Scholar]
- 34.Enslen H, Tokumitsu H, Stork PJS, Davis RJ, Soderling TR. Proc. Natl. Acad. Sci. U. S. A. 1996;93:10803–10808. doi: 10.1073/pnas.93.20.10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Countaway JL, Nairn AC, Davis RJ. J. Biol. Chem. 1992;267:1129–1140. [PubMed] [Google Scholar]
- 36.Mansat-De Mas V, Hernandez H, Plo I, Bezombes C, Maestre N, QuilletMary A, Filomenko R, Demur C, Jaffrezou JP, Laurent G. Blood. 2003;101:1543–1550. doi: 10.1182/blood-2002-05-1585. [DOI] [PubMed] [Google Scholar]
- 37.Canto C, Suarez E, Lizcano JM, Grino E, Shepherd PR, Fryer LGD, Carling D, Bertran J, Palacin M, Zorzano A, Guma A. J. Biol. Chem. 2004;279:12260–12268. doi: 10.1074/jbc.M308554200. [DOI] [PubMed] [Google Scholar]
- 38.Cenni V, Doppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A. Biochem. J. 2002;363:537–545. doi: 10.1042/0264-6021:3630537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakrabarti S, Liu NJ, Gintzler AR. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13686–13691. doi: 10.1073/pnas.2335885100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirai T, Chida K. J. Biochem. (Tokyo) 2003;133:1–7. doi: 10.1093/jb/mvg017. [DOI] [PubMed] [Google Scholar]
- 41.Polakiewicz RD, Schieferl SM, Gingras AC, Sonenberg N, Comb MJ. J. Biol. Chem. 1998;273:23534–23541. doi: 10.1074/jbc.273.36.23534. [DOI] [PubMed] [Google Scholar]
- 42.Bandyopadhyay G, Sajan MP, Kanoh Y, Standaert ML, Quon MJ, Reed BC, Dikic I, Farese RV. J. Biol. Chem. 2001;276:35537–35545. doi: 10.1074/jbc.M106042200. [DOI] [PubMed] [Google Scholar]
- 43.Swartz KJ, Merritt A, Bean BP, Lovinger DM. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- 44.Liu K, Hsiung S, Adlersberg M, Sacktor T, Gershon MD, Tamir H. J. Neurosci. 2000;20:1365–1373. doi: 10.1523/JNEUROSCI.20-04-01365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan Y, Shirakabe K, Werb Z. J. Cell Biol. 2002;158:221–226. doi: 10.1083/jcb.200112026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagano O, Murakami D, Hartmann D, De Strooper B, Saftig P, Iwatsubo T, Nakajima M, Shinohara M, Saya H. J. Cell Biol. 2004;165:893–902. doi: 10.1083/jcb.200310024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 48.Zwick E, Wallasch C, Daub H, Ullrich A. J. Biol. Chem. 1999;274:20989–20996. doi: 10.1074/jbc.274.30.20989. [DOI] [PubMed] [Google Scholar]
- 49.Baldassare JJ, Henderson PA, Burns D, Loomis C, Fisher GJ. J. Biol. Chem. 1992;267:15585–15590. [PubMed] [Google Scholar]
- 50.Berra E, Diazmeco MT, Dominguez I, Municio MM, Sanz L, Lozano J, Chapkin RS, Moscat J. Cell. 1993;74:555–563. doi: 10.1016/0092-8674(93)80056-k. [DOI] [PubMed] [Google Scholar]
- 51.Corbit KC, Foster DA, Rosner MR. Mol. Cell. Biol. 1999;19:4209–4218. doi: 10.1128/mcb.19.6.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fedorov YV, Jones NC, Olwin BB. Mol. Cell. Biol. 2002;22:1140–1149. doi: 10.1128/MCB.22.4.1140-1149.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosh PM, Bedolla R, Mikhailova M, Kreisberg JI. Can. Res. 2002;62:2630–2636. [PubMed] [Google Scholar]
- 54.Steinberg SF. Biochem. J. 2004;384:449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chou MM, Hou WM, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Cur. Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 56.Brodie C, Bogi K, Acs P, Lazarovici P, Petrovics G, Anderson WB, Blumberg PM. Cell Growth Differ. 1999;10:183–191. [PubMed] [Google Scholar]
- 57.Mangoura D. J. Neurosci. Res. 1997;50:391–401. doi: 10.1002/(SICI)1097-4547(19971101)50:3<391::AID-JNR5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 58.Sweitzer SM, Wong SME, Tjolsen A, Allen CP, Mochly-Rosen D, Kendig JJ. Pain. 2004;110:281–289. doi: 10.1016/j.pain.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 59.Narita M, Mizoguchi H, Suzuki T, Dun NJ, Imai S, Yajima Y. J. Biol. Chem. 2001;276:15409–15414. doi: 10.1074/jbc.M009716200. [DOI] [PubMed] [Google Scholar]
- 60.Belcheva MM, Vogel Z, Ignatova E, Avidor-Reiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ. J. Neurochem. 1998;70:635–645. doi: 10.1046/j.1471-4159.1998.70020635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belcheva MM, Wong YH, Coscia CJ. Cell. Signal. 2000;12:481–489. doi: 10.1016/s0898-6568(00)00095-4. [DOI] [PubMed] [Google Scholar]
- 62.Shah BH, Farshori MP, Jambusaria A, Catt KJ. J. Biol. Chem. 2003;278:19118–19126. doi: 10.1074/jbc.M212932200. [DOI] [PubMed] [Google Scholar]