

Abstract
Short interspersed elements (SINEs) make up a significant fraction of total DNA in mammalian genomes, providing a rich substrate for chromosomal rearrangements by SINE-SINE recombinations. Proliferation of mammalian SINEs is mediated primarily by LINE1 (L1) non-LTR retrotransposons that preferentially integrate at DNA sequence targets with average length ~15 bp and containing conserved endonucleolytic nicking signals at both ends. We report that sequence variations in the first of the two nicking signals, represented by a 5′TT-AAAA consensus sequence, affect the position of the second signal thus leading to target site duplications (TSDs) of different lengths. The length distribution of TSDs appears to be affected also by L1-encoded enzyme variants, since targets with the same 5′ nicking site can be of different average length in different mammalian species. Taking this into account, we re-analyzed the second nicking site and found that it is larger and includes more conserved sites than previously appreciated, with a consensus of 5′ANTNTN-AA. We also studied potential involvement of the nicking sites in stimulating recombinations between SINE elements. We determined that SINE elements retaining TSDs with perfect 5′TT-AAAA nicking sites appear to be lost relatively rapidly from the human and rat genomes, and less rapidly from dog. We speculate that the introduction of single-strand DNA breaks induced by recurring endonucleolytic attacks at these sites, combined with the ubiquitousness of SINEs, may significantly promote recombination between repetitive elements, leading to the observed losses. At the same time new L1 subfamilies may be selected for “incompatibility” with pre-existing targets. This provides a possible driving force for the continual emergence of new L1 subfamilies which, in turn, may affect selection of L1-dependent SINE subfamilies.
Keywords: non-LTR retrotransposons, recombination, SINE integration targets
INTRODUCTION
Non-LTR (long terminal repeat) retrotransposons such as human Alu elements and LINEs (long interspersed elements) have proved remarkably successful at colonizing mammalian genomes (Deininger et al. 2003). Transposition-competent LINEs are autonomous elements ~6–8 kb long, that encode an endonuclease/reverse transcriptase protein, and an RNA-binding domain (Ostertag and Kazazian 2001). They make up around 20% of the genome in human (Lander et al. 2001). Alu elements are members of the SINE (small interspersed elements) family of repetitive elements, which are non-autonomous and rely on the activity of LINEs for transposition. They comprise over 10% of the human genome, with around 1.2 million copies in total (Lander et al. 2001). Alu elements are ~300 bp long and consist of two similar monomers that derived from 7SL RNA, which is a major component of the ribosome-interacting signal recognition particle. Numerous subfamilies of Alu have been identified, corresponding to waves of transpositional activity in the past. The AluJ subfamily includes elements that transposed at least 60 million years (Myr) ago, while AluS subfamilies were active between 60 and 20 Myr ago (Kapitonov and Jurka, 1996). The relatively young AluY subfamilies have proliferated in the past 20 Myr, and continue to do so, along with their corresponding LINEs (Brouha et al., 2003, Salem et al., 2003). Recent subfamilies are highly (>99%) similar to their consensus sequences, and even old AluJ elements are typically >80% similar. Other L1-dependent mammalian SINE elements analyzed in this paper include rodent B1 and B2 elements (Krayev et al. 1980, Krayev et al. 1982, Kramerov et al. 1985, Okada and Ohshima, 1995) as well as rodent BC1/ID (Milner et al. 1984), and dog SINEC elements (Coltman and Wright, 1994). BC1 descended from tRNA and is thought to be a progenitor to the oldest of at least four known ID subfamilies. The remaining ID subfamilies descended from ID-related master genes (Kim et al., 1994). Like human Alu elements, B1 is derived from 7SL RNA, whereas B2 and SINEC elements are tRNA-derived. Like Alu elements, B1 and B2 elements also include a number of subfamilies (Quentin, 1989; Kass et al. 1997). Dog SINEC elements are composed of two major subfamilies, the older SINEC1 and younger SINEC2, and a number of sub(sub)families (Jurka and Gentles, Repbase Update, release 9.11 December 2004).
Integration of non-LTR retrotransposons such as mammalian L1 and related SINE elements occurs at DNA targets of varying length determined by two conserved nicking sites (Jurka, 1997; Cost and Boeke, 1998; Klonowski and Jurka, 1996). The first nicking site is relatively well conserved at six consensus nucleotides 5′TT-AAAA, where the hyphen indicates the preferred position for enzymatic nicking. Some variants of the first nicking site, particularly those differing by a single base substitution, are relatively well represented among the integration targets (e.g. TT-AGAA or TT-AAGA). The second nicking site is less conserved and occurs at variable positions relative to the first site. Apart from a limited sequence conservation, factors affecting the second cleavage site are poorly understood. Recently, a new class of 3′-truncated tRNA-derived SINEs, termed “tailless retropseudogenes” was identified (Schmitz et al., 2004). These tailless SINEs lack a poly-A tail, and exhibit non-random integration preferences. Although their mechanism of retrotransposition is unknown, it is believed that L1 may be involved.
Alu elements have played an undoubted role in remodeling the human genome during evolution; both directly by transposition, and by providing a substrate for unequal homologous recombination producing chromosomal rearrangements and deletions (e.g. Brosius, 2005; Jurka, 2004). In particular, Alu elements are enriched at the boundaries of segmental duplications in the human genome, and likely played a key role in their generation (Bailey et al. 2003). Similar observations have been made for mouse B1 and B2 elements (Jurka et al. 2005). Alu elements are GC-rich and although they integrate preferentially into GC-poor regions of the genome, they are more likely to be retained in GC-rich regions, and it is in these regions that they are most prominent (Pavlicek et al. 2001). LINEs on the other hand tend to be under-represented in GC-rich regions, and are frequently truncated during integration, or by subsequent mutation and rearrangement. Since gene density and GC-density are correlated in vertebrate genomes, Alu elements are frequent in gene-rich genomic regions. Consequently they have been implicated in the etiology of an increasing number of human diseases (reviewed in Kolomietz et al. 2002, Deininger and Batzer 1999, Kapitonov et al. 2004). Recent examples include elucidation of the role of Alu elements in promoting instability at the BRCA1 tumor suppressor locus (Pavlicek et al. 2004), and Alu-mediated gene rearrangements in low copy repeats of the 22q11 region of the human genome, which is associated with a variety of disease syndromes (Babcock et al. 2003). Alu-Alu recombinations are also involved in rare deletions in the Smith-Magenis syndrome region at 17p11.2 (Shaw and Lupski, 2005).
Recently it has been proposed that revisiting of Alu targets by L1 endonuclease may stimulate homologous recombination between Alu elements (Babcock et al. 2003). In this paper we analyze elimination of targets associated with SINE subfamilies from different species, as well as evolving target profiles of L1 endonucleases, and discuss the results in the light of the preceding hypothesis. We also re-analyzed the second endonucleolytic nicking site and determined a refined consensus sequence of 5′ANTNTN-AA (N denoting any nucleotide). In comparison to the previous consensus sequence (Jurka, 1997), the new one includes three more conserved bases indicating strong preferences of the nicking enzyme(s).
METHODS
Complete assembled genome sequences were downloaded from the UCSC Genome Browserfor human (Build 34 assembly, July 2003, http://genome.ucsc.edu); Rattus norvegicus (June 2003 assembly; Rat Genome Sequencing Consortium, 2004), and Canis familiaris (July 2004 freeze, UCSC version canFam1; Broad Institute of MIT and Harvard, and Agencourt Bioscience). Genomes were screened for SINE elements using Censor version 4.2 (Jurka et al. 1996; available at http://www.girinst.org), with the “-filter -nosimple” arguments for classification. Human genome sequences were screened against all Alu subfamilies, using consensus sequences contained in Repbase (Jurka, 2000); rat sequences were screened against rat SINE elements (Rat Genome Sequencing Consortium, 2004)); and the dog genome was screened against newly derived consensus sequences for the SINEC1_CF and SINEC2_CF families (SINEC1A_CF, SINEC1B1_CF, SINEC1B2_CF, SINEC1C1_CF, SINEC1C2_CF, SINEC1D_CF, SINEC1_CF, SINEC2A1_CF, SINEC2A2_CF; Repbase Update, December 2004).
Resulting Censor maps were cleaned by eliminating elements shorter than 90% of the full consensus sequence length for each subfamily. In addition, elements were removed that did not begin at the first position of the consensus sequence. Flanking repeats of the remaining elements were identified by aligning the 20 bp immediately 5′ of the repeat element, to the 100 bp region of sequence 3′ from the element, using an implementation of the Smith-Waterman alignment algorithm. These parameters were chosen to maximize sensitivity by allowing for polymorphism in the length of 3′ poly-A tails, while eliminating spurious alignments by considering only 20 bp immediately 5′ of the repeat element. After identification of flanking repeats, the original nicking sites were inferred as the hexamer starting 2 bp upstream of the 5′ flanking repeat. The secondary nicking sites were assumed to coincide with the 3′ ends of TSDs. For each repeat element, the target hexamer, flanking repeat length, number of mismatches between 5′ and 3′ repeats, and similarity of the repeat to its consensus sequence were recorded for further analysis. Figure 1 illustrates these features for a SINE that is integrated in the genome. The degree of similarity between repeat copies and their consensus sequence was determined by alignment of the core domains of the elements, excluding poly-A tails and microsatellites. In addition, because CpG dinucleotides mutate at a much faster rate than non-CpG dinucleotides due to methylation-deamination-mutation, these were excluded from the calculation of similarities.
Figure 1.
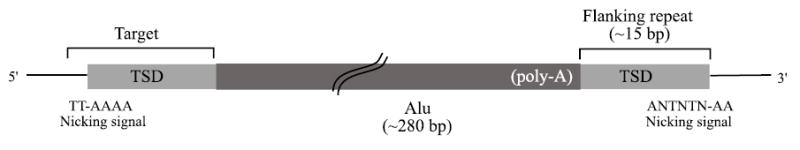
Flanking repeats were identified by alignment of 5′ and 3′ sequences immediately adjacent to repeat elements. Here, an Alu is shown integrated in the genome, and is flanked by TSDs (target site duplications). The first nicking site is inferred as the hexamer starting 2 bp 5′ of the upstream TSD (TT-AAAA in the example shown). As indicated, the TT dinucleotides are part of the first nicking site, but not part of the TSD. The second nicking site is located at the 3′ terminal end of the TSD. The flanking repeat (TSD) lengths are distributed with median 15 bp in most cases (see text).
Comparisons of distributions, as discussed in the Results, were performed using Kolmogorov-Smirnov (K-S) non-parametric test statistics, in R (http://www.r-project.org). The K-S p-value is not exactly computable in the case of discrete distributions. However, the estimated p-values are over-estimates (ie. the true values are more significant than indicated). We validated this by computing exact p-values for the discrete distributions with random permutations added. In all cases, the p-values obtained were lower than those presented in Results, as expected.
Data on putative deletions between the human and Pan troglodytes genomes were downloaded from the UCSC genome browser annotation database. The deletions identified therein are based on alignment of the chimpanzee November 2003 genome assembly to the human July 2003 assembly.
RESULTS
Frequency of identified targets in different species
We analyzed proportions of SINEs with intact target site duplications (TSDs) in the human, rat and dog genomes. Flanking repeats, and associated target sequences, were identified for 376 069 Alu elements (1/3 of the genomic total), from all major Alu families and subfamilies. Predictably, the younger the subfamily, the higher the proportion of identified TSDs, since over time TSDs will be lost through mutation, or recombination between repeats. In rat, targets were identified for 134 956 elements from the most common SINE subfamilies, specifically B2_Rat1, B2-Rat2, B2_Rat3, B2_Rat4, B2_Rn, B2_Rn1, B2_Rn2, BC1_Rn, ID_Rn1, and ID_Rn2 (Rat Genome Sequencing Consortium, 2004). B1 is no longer active in the rat lineage, although it continues to proliferate in the mouse genome, and most rat B1 elements have mutated significantly together with their targets. On the other hand, ID-like elements, with the exception of BC1, appear to be relatively inactive in mouse, but actively proliferate in rat. B2 elements are active in both species and are included in our comparative analysis later on. In dog, we focused on abundant SINEC1 and SINEC2 families and their subfamilies listed in Repbase Update, identifying 485 623 TSDs.
Approximately 1/3 of human Alu elements (376069 of ~1.2 million in total) have an identifiable target site duplication, and almost 86% of these have a flanking repeat that is longer than 10 bp. 82 465 Alu elements with an identified target (22%) have perfect flanking repeats longer than 10 bp, with no mismatches between the 5′ and 3′ copies. 59 814 of the 134 956 rat targets identified have a perfect flanking repeat >10 bp (44% of elements with an identified target, nearly twice as high as in human). In dog, the corresponding figure is 166 401 perfect repeats (34% of identifiable targets, intermediate between human and rat). These results reflect a relatively large fraction of young SINEs in rat, followed by dog and human SINEs. Species-specific differences are accounted for in the following analyses.
Figure 2 shows the frequencies of the most commonly identified perfect TSDs over 10 bp long, flanking young human, rat and dog SINE elements that are ≥98% identical to their consensus sequences (8412 human Alu elements, 28 517 rat SINEs, 56 036 dog SINEs). Expectedly, TSDs containing TT-AAAA nicking sites represent the largest group of targets: 20% in human and dog, and 23% in rat. Targets with TT-AAGA are also frequent, particularly in rat where they make up nearly 13% of all identified targets. While all targets that comprise a single substitution of A for G from within the consensus TT-AAAA feature prominently, the ones with G replacing the third A are notably more frequent than the others.
Figure 2.
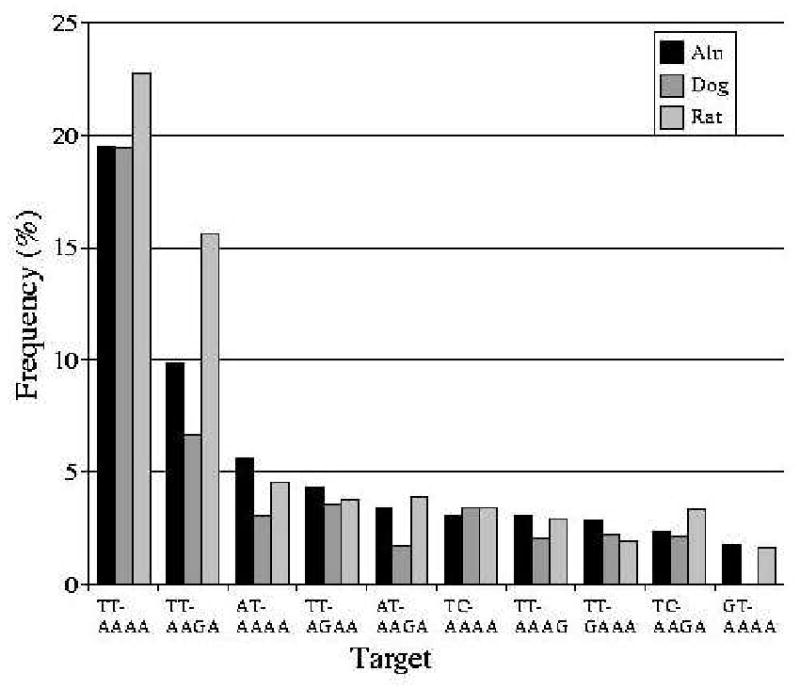
Proportions of different target types in young SINE elements less than 2% diverged from their consensus, that have a flanking repeat of >10 bp length, with no mismatches between 5′ and 3′ repeat copies.
Species-specific relationship between the primary nicking signals and the length of TSDs
We studied lengths of TSDs associated with different SINE subfamilies from the human, rodent and dog genomes, and containing variants of the first consensus nicking signal TT-AAAA. Figure 3 shows the detailed length distributions of TSDs for human AluY and AluYa5 elements, rat BC1/ID and B2 elements, dog C1D and C2A1 elements, and mouse B1 and B2 elements. All presented TSDs are derived from variants of targets containing single-base A→G substitutions relative to the consensus hexanucleotide nicking signal TT-AAAA. The distributions of TSD lengths are clearly distinct from each other, and in several cases are not simply single peaks. While SINE flanking direct repeat lengths are typically in the range of 14–16 bp long, the distributions can peak at lower values. Some of the differences are species-specific and SINE family-specific. TSDs derived from TT-AGAA- and TT-GAAA-type targets are the longest in human Alu elements, with modes of 16 bp and 15 bp, respectively. The longest TT-AAGA-derived TSDs are in young dog SINEC2A1 elements (mode 16 bp). TT-AAAG-type targets produce a characteristic bimodal distribution of TSDs whereas the TT-AGAA targets tend to form unimodal distributions. For example, TT-AGAA TSD lengths have modes of 14 and 15 bp in dog SINEC1D and SINEC2A1, and 16 bp in human. This probably reflects both a continuum of enzyme variants coded by L1 subfamilies within the species, and more distinct differences between L1 enzymes from different species. Stochastic variationsre in enzyme selection of target sites could contribute additional variation to the distributions of TSD lengths. By and large, the length differences between the same types of targets in different species are within 1–2 bp. Length differences between different types of targets within or between species can be larger.
Figure 3.
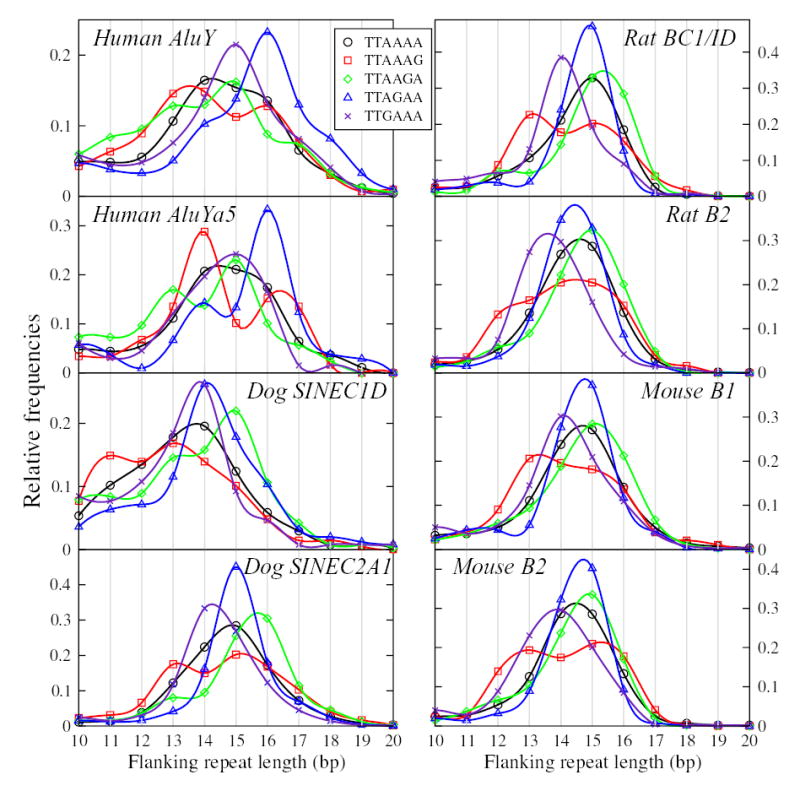
Distribution of lengths of flanking repeats for TT-AAAA targets, and variants involving a single A→G change (the most common target types observed). In each panel, the horizontal axis shows flanking repeat length, while the vertical axis shows relative frequency. The plots have been smoothed to facilitate visual comparisons. Significance of differences between distributions (evaluated by Kolmogorov-Smirnov tests) is tabulated in Tables 1 and 2, and in supplementary table S1, as discussed in the text.
To evaluate the significance of the differences between distributions of TSD lengths, we performed Kolmogorov-Smirnov (K-S) non-parametric comparisons between them. Significance was estimated (a) for each nicking site, comparing between different repeat types; and (b) for each repeat type, comparing differences between nicking sites. Results are presented in Table 1 for the nicking site TT-AAAG, comparing repeats; and Table 2 compares different targets for AluYa5 elements. These examples were selected because they represent the “worst” (least significant) differences, primarily due to small sample sizes. Taking a threshold of p<0.05 it can be seen from Table 1 that for TT-AAAG targets, the AluY and AluYa5 distributions are not significantly different. Nor are B2/AluY, B2/AluYa5, B2/BC1, BC1/AluYa5, AluYa5/SINEC2A1, or B2/SINEC2A1. Other repeats with TT-AAAG targets do show statistically significant differences between their TSD length distributions. In Table 2, the comparisons for different targets for AluYa5 show that the distributions are significantly different between TT-AAAA/TT-AAGA, TT-AAAA/TT-AGAA, TT-AAGA/TT-AGAA, and TT-AGAA/TT-GAAA; but not for other pairwise comparisons. The complete set of K-S test statistics for all possible comparisons of the distributions shown in Fig. 3 are listed in Supplementary Table 1. For rat BC1 elements for example, all differences between TSD length distributions are significant, with p<0.002.
Table 1.
p-values for differences between distributions of TSD lengths for TTAAAG targets, comparing between species/repeat type. Lower values indicate higher significance (i.e. greater certainty that the distributions are different). Bold face indicates p<0.05.
| AluY | AluYa5 | BC1 | B2 | SINEC1D | SINEC2A1 | |
|---|---|---|---|---|---|---|
| AluY | - | 0.178 | 5.35.10−3 | 0.618 | 9.26.10−8 | 1.15.10−3 |
| AluYa5 | - | 0.864 | 0.544 | 7.45.10−6 | 0.393 | |
| BC1 | - | 0.262 | 4.55.10−5 | 0.151 | ||
| B2 | - | 3.56.10−9 | 7.95.10−3 | |||
| SINEC1D | - | 9.77.10−15 | ||||
| SINEC2A1 | - |
Table 2.
p-values for differences between distributions of TSD lengths of different target types, for AluYa5 elements. Lower values are more significant. Bold face indicates p<0.05. The high p-values are largely due to the small sample size available for AluYa5 elements.
| TTAAAA | TTAAAG | TTAAGA | TTAGAA | TTGAAA | |
|---|---|---|---|---|---|
| TTAAAA | - | 1.000 | 1.69.10−3 | 1.94.10−3 | 1.000 |
| TTAAAG | - | 0.393 | 0.178 | 0.957 | |
| TTAAGA | - | 7.79.10−7 | 0.996 | ||
| TTAGAA | - | 9.66.10−3 | |||
| TTGAAA | - |
Second endonucleolytic nicking site
To investigate the second nicking site for human-specific L1, we extracted identical TSDs at least 14 bp long, which started with TT-AAAA nicking sites. The selection of TSDs 14 bp or longer is justified below. We analyzed the 9 bp long 3′ terminal portion of each TSD, and determined frequencies of bases at each position and in the immediate 3′-adjacent regions. The frequencies were compared to expected frequencies based on the composition calculated for the 109 bp DNA segments that included the 9 bp TSD fragments and their 3′ adjacent 100 bp regions. All adjacent regions containing any known repeats from the human section of Repbase Update were eliminated. This eliminates multiple repeat insertions near the same locus, which could lead to spurious matches between flanking regions, and confusion over which element inserted there first. We evaluated significance by calculating χ2 values at each position, as shown in Fig. 4. The base composition at each of the nine terminal positions, and an additional four 3′ flanking positions is also shown, in the second panel of the figure. Nucleotides are significantly over-represented at a particular position for χ2>16.27 (p<0.001). The second nicking site is less strongly conserved than the first, but conforms to the consensus 5′ANTNTN-AA (N is any nucleotide). The alternating ANTNTN pattern is strongly evident in Fig. 4. We also analyzed all TSDs at least 14 bp long irrespective of the first nicking signal. The results were indistinguishable from those in Fig. 4 (data not shown). It was necessary to examine TSDs at least 14 bp long, as this is the minimum size which can accommodate complete versions of both nicking sites. Shorter TSDs would have overlapping signals (see Fig. 1)
Figure 4.
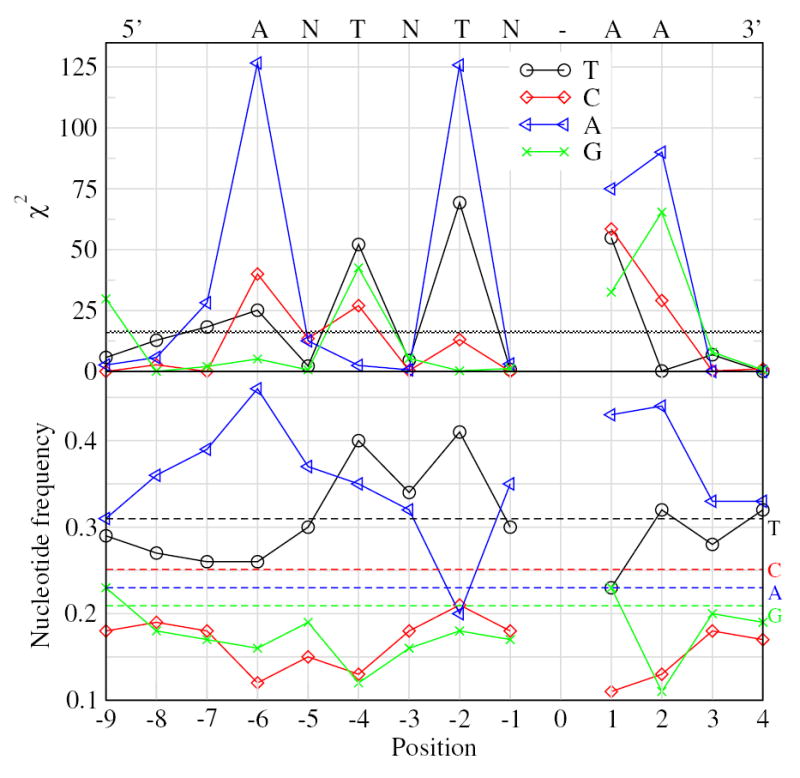
Determination of the consensus sequence for the second endonucleolytic nicking sequence. The top panel shows χ2 values at the indicated positions relative to the nicking site (see Jurka, 1997 for detailed description of methodology). χ2 values above the horizontal graded line at χ2=16.27 are significant at the p=0.001 level. Nucleotide composition at each position relative to the predicted nicking site is shown in the bottom panel. At positions −1, −3, and −5, no nucleotide is significantly over-represented (“N”). At positions +1, +2, and −6 “A” is unambiguous. “T” has the highest χ2 at positions −2 and −4. Horizontal lines in the bottom panel show the mean composition of the 3′ flanking regions of the elements studied, identified by nucleotide letter on the right-side axis.
The TSD length appears to be correlated with sequence variations at the first nicking signal, which is suggestive of target site selection by L1 encoded enzymes. Therefore a target which is selected by one enzyme variant may not be selected by another variant if the conserved bases of the second nicking signal are at the wrong distance from the first site. In other words, the combination of first/second nicking site, together with the separation between them, may match a characteristic preference of different L1 variants. The presence of such enzymatic variants may be of biological significance, as discussed later.
Target elimination
We studied time-dependent changes in the relative proportions of the most abundant SINE element targets containing a TT-AAAA nicking site. Figure 5 shows the fraction of SINEs, which have perfect flanking repeats (i.e. no mismatches between 5′ and 3′ copies) of 15 bp or more, that have a preserved TT-AAAA site, as a function of their difference from their consensus sequence. Calculating this fraction relative to SINEs with a perfect flank, rather than to all SINEs, allows for the fact that targets are less likely to be identifiable for older SINE elements, due to mutation. Grouping elements according to their divergence from the respective consensus sequences naturally accounts for variation in the divergence rates of SINEs depending on their genomic location (see also the legend to Figure 5).
Figure 5.
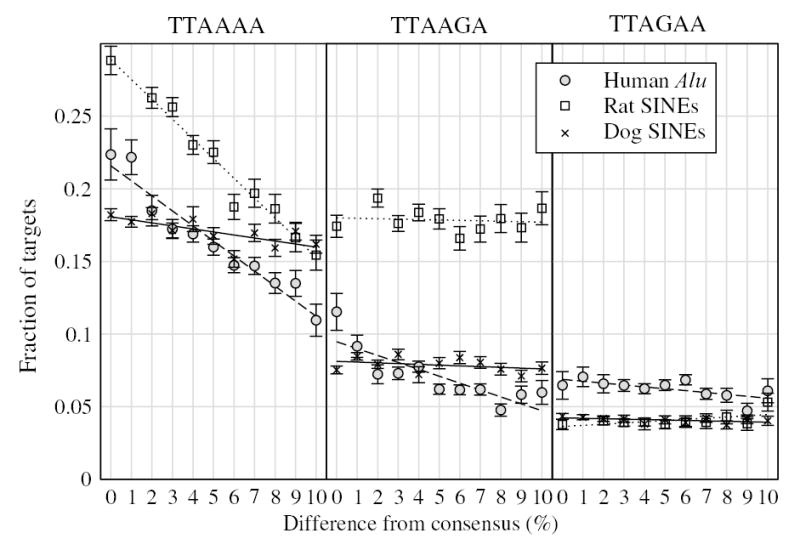
| Human | Rat | Dog | |
|---|---|---|---|
| TTAAAA | −1.04±0.09 | −1.36±0.08 | −0.21±0.07 |
| TTAAGA | −0.48±0.10 | −0.03±0.09 | −0.05±0.05 |
| TTAGAA | −0.13±0.04 | 0.08±0.04 | −0.02±0.01 |
Figure 5 also shows linear best-fits to the data, with the regression parameters indicated in the figure legend. The downward slope of the TT-AAAA line in Alu, rat and dog indicates progressively lower frequency of occurrence of these targets in more diverged (older) repeat elements. The rate of loss is highest in rat, and lowest in dog. The ratio between mean rates of loss for human:rat:dog is 5.0:6.5:1. Net decline of TT-AAAA targets is 51% in human Alu elements, 46% in rat SINEs, and only 11% in dog SINEs. This strong relative decline for TT-AAAA contrasts with the behaviour of targets with other nicking sites. The second most common nicking site (TT-AAGA) contains a single A to G mutation at the third A position. While it exhibits an initial decline in the Alu elements studied (see figure 5 for Alu elements with divergence from consensus <2%), there is apparently no significant decline in rat or dog elements, (fluctuations at higher divergence values are due to reduced sample sizes for more diverged SINEs). Next most frequent overall among A-G variants is TT-AGAA, which show no decline over time, although the initial fraction of SINEs with this target is much lower than TT-AAAA or TT-AAGA. The results remain unaltered if we impose a less strict requirement, of only a 10 bp perfect intact flanking repeat.
Constraining the flanking repeats to be identical copies of at least 15 nucleotides makes it unlikely that both repeats have mutated, since this would require simultaneous identical mutations at the same position in the 5′ and 3′ copies of the repeat. Under the simple approximation that all single base mutations are equally probable, the chance that simultaneous identical mutations have occurred in 5′ and 3′ flanks of length 15 bp is around 10−4 for an Alu that has diverged by 20% from its consensus sequence. However, simply considering the fraction of targets of TT-AAAA type as a function of difference from consensus leaves open the possibility that the decline in TT-AAAA in more diverged elements is simply due to mutation of one or both T’s, since they are not part of the flanking repeat (Fig. 1). Therefore, we also considered loss of TT-AAAA from among all TT-NNNN targets, to verify if TT-AAAA is lost more rapidly than other TT-NNNN targets. If loss of TT-AAAA is due simply to mutation of TT dinucleotides, we would expect that other targets starting with TT- would be lost at a comparable rate.
The result of one such comparison is shown in Figure 6, which compares the ratio of TT-AAAA to TT-AGAA targets as a function of similarity of SINE elements to their consensus. To compare the rate of loss between species, the plots have been normalized relative to the initial ratio between TT-AAAA and TT-AGAA (see legend to Figure 6). In human and rat, there is a steep decline in the relative abundance of TT-AAAA compared to TT-AGAA. Initially, in SINEs that are 100% identical to their consensus, TT-AAAA targets are 3.4 times as frequent as TT-AGAA in Alu elements, 4.2 times as frequent in dog SINEs, and 7.6 times as frequent in rat SINEs. As shown in the figure, by the time that SINE elements have diverged by 10% from the consensus, the ratio between TT-AAAA and TT-AGAA in Alu elements has declined to slightly over 50% of it’s starting value, and in rat has declined to around 38%. In contrast, there appears to have been almost no drop off in dog SINEs, indicating that TT-AAAA and TT-AGAA targets decline at the same (low) rate in this species. A similar pattern emerges when alternative targets of type TT-NNNN are considered. The sample sizes examined for 15 bp flanking repeats for TT-NNNN targets were: 14 925 Alu elements, 18 898 rat SINEs, and 48 820 dog SINEs. For 10 bp flanking repeats, these numbers become 32 278, 31 649, and 75 857 respectively, but the above results are essentially unchanged regardless of whether 10 bp or 15 bp repeats are considered. Thus TT-AAAA targets are clearly lost at a faster rate than other targets beginning with TT.
Figure 6.
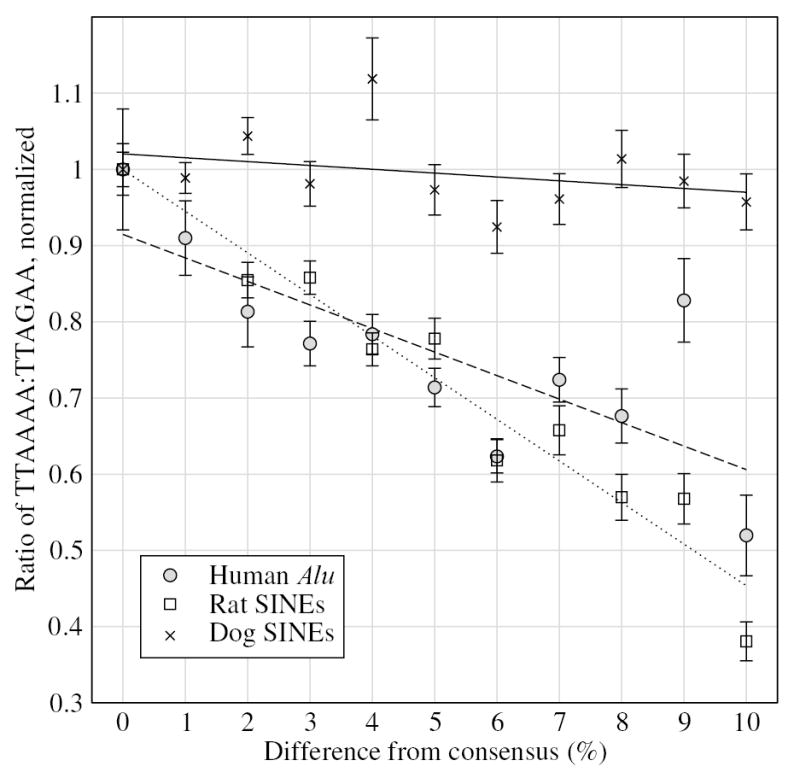
TT-AAAA target loss relative to TT-AGAA. Plots show the ratio of TT-AAAA to TT-AGAA targets in human, dog and rat, normalized to be 1 at 0% divergence from consensus. (ie. if the ratio of TT-AAAA to TT-AGAA targets is rd for repeats which are d% diverged from their consensus, then the figure shows rd/r0. The initial ratios between TT-AAAA and TT-AGAA are respectively 3.4:1, 4.2:1 and 7.6:1 in human, dog, rat.
The flanking repeat length is constrained, we consider only perfect repeats in identifying these targets, and control for possible variations of the initial TT dinucleotide. Hence the rapid loss of targets with the canonical TT-AAAA nicking site cannot be attributed purely to mutation. The most likely explanation is that SINE elements with intact TT-AAAA sites are eliminated from the genome more rapidly than SINE elements with other nicking sites such as TT-AGAA (see discussion).
DISCUSSION
There are two conserved nicking signals that determine target site duplications flanking non-LTR retrotransposons. The second consensus nicking signal (5′ANTNTN-AA) weakly resembles the first nicking signal (5′TTTT-AA), as previously suggested (Szak et al., 2002). Despite these interesting similarities, the second signal appears to be longer and shows less overall conservation than the first one, with alternating conserved and nonconserved bases, although the 3′AA located outside the TSDs is strongly supported. The L1 ORF2 encodes a reverse transcriptase domain and an N-terminal APE-type endonuclease domain (Weichenrieder et al., 2004; Martinez et al. 1996; Feng et al. 1996). To date, only this one endonucleolytic domain has been established to be encoded by L1 elements. However, given the two different nicking signals and the characteristic inter-nick distance it cannot be ruled out that they reflect an enzymatic recognition pattern, which is difficult to reconcile with a single endonucleolytic domain model. Therefore, we propose a three-factor target recognition model involving enzymatic recognition of both nicking signals and of the distance between them. The model does not predict whether the second nick is created by the same protein (as in the case of R1Bm retrotransposon endonuclease; Feng et al. 1998), or a different protein interacting with both nicking sites, separated by a specific distance. However, it implies that targets recognized by one protein variant(s) may not necessarily be recognized by other variant(s) if the nicking signals are not located at the variant-specific distance. If the model is correct, it would suggest a much higher difference in target specificity between proteins derived from different L1 subfamilies than previously thought. This may have practical implications for designing L1-based vectors, which can be directed to a smaller subgroup of targets than virus-based vectors.
It has been suggested that endonucleolytic enzymes revisiting 5′ targets associated with integrated SINE elements can trigger SINE-SINE recombination (Babcock et al. 2003), which can be damaging to the host, and hence re-visiting of targets could be selected against. Based on the three-factor model, the changing inter-nick distances within and between species may indicate that newly emerging L1variant subfamilies are selected for their incompatibility with targets used by the preceding subfamilies. This model may also provide a rationale for the generation of different L1 subfamilies which can in turn affect selection of the associated SINE subfamilies.
Until recently, it was largely thought that double-strand DNA breaks were required to initiate homologous recombination (Szostak et al. 1983). Ironically, most original models proposed for recombination involved single-strand nicks (Holliday, 1964; Meselson and Radding, 1975). Recently however, there has been a renewed interest in recombination initiated by single-strand nicks following the discovery that RAG proteins involved in V (D)J recombination can create such breaks, inducing recombination (Lee et al. 2004). Indeed, mutated versions of the RAG proteins, that produce mainly single-strand breaks, seem to promote even more promiscuous recombination than the wild-type RAG proteins, which primarily produce double-strand breaks. In addition to creating nicks in DNA, LINE retrotransposition extends the length of the broken strand during reverse transcription of its own RNA, or the RNA of a SINE element. Thus L1 activity might provide opportunities for single-strand invasion of homologous repetitive sequence, and provide a substantial region of homology, increasing the chance for recombination to occur.
If L1-stimulated SINE-SINE recombination takes place, then it should lead to faster disappearance of SINEs with perfect TSDs. As shown in Figs. 5 and 6, the fraction of TTAAAA-containing targets declines in humans and rodents over time. It is much weaker in less abundant targets beginning with TTAAGA and TTAGAA. We propose that the decline could be a result of target-stimulated recombination. Figure 7 indicates how this might proceed between two repeats of the same type. Recombination stimulated by L1 nicking at one of the elements leads to the removal of one 5′, and one 3′ target, leaving a composite remnant with non-matching 5′ and 3′ flanks and the resulting recombination product no longer has detectable flanking repeats (see also Martignetti and Brosius, 1993; Kass et al., 1995).
Figure 7.
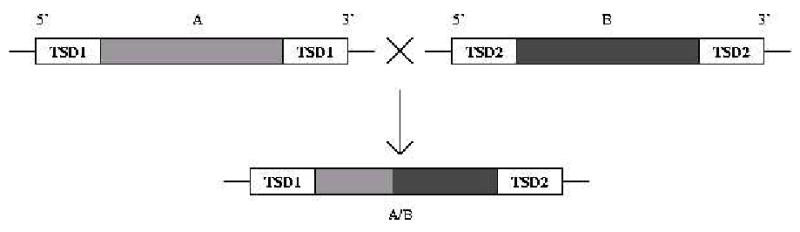
Loss of perfect targets by recombination between two similar SINEs. Each sequence is flanked by a perfect repeat, which is different for the two elements. Recombination results in loss of the 3′ repeat copy of SINE A, and 5′ copy of SINE B. At the same time, the resulting composite element is no longer flanked by a repeat, since the 5′ and 3′ flanking sequences come from SINE A, and B respectively.
No significant decline in target proportions was observed in dog. However, dog is also different in terms of germ line-specific retrotransposition and elimination of SINEs. Based on the relative abundance of young SINE elements on chromosome X, retrotransposition of SINEs in dog appears to proceed mostly in the female germline (data not shown). In contrast, SINE retrotransposition in humans and rodents occurs in male germ lines (Jurka et al. 2004, 2005). It has been proposed that elevated nicking of SINE integration targets by L1-encoded endonuclease, combined with faster replication rate in male than in female germlines could contribute to loss of newly inserted elements through a replication slippage-like process (Jurka, 2004). The female-driven retrotransposition in dog may indicate a lack of significant expression of L1 elements in dog male germ lines, reducing the rate of male-driven loss of SINEs. This may be a major factor responsible for the observed slow rate of target elimination.
In principle, it should be possible to detect directly the occurrence of nicking-stimulated recombinations by comparison of closely related genome sequences. In practice, this is difficult. For example, human diverged from the chimpanzee Pan troglodytes around 6 Myr ago (Goodman 1999). However, AluS and AluJ proliferated >20 Myr ago, while AluY was most active in the past 20 Myr. Thus we would anticipate that many TT-AAAA targets have been removed either prior to the human-chimpanzee divergence, or that they have been lost from both lineages. Either case would render the deletion event undetectable by pairwise genome comparisons. Nevertheless, we examined AluY targets in relation to putative deletions in human relative to chimpanzee. The reverse comparison is not possible due to the frequency of sequencing gaps in chimpanzee. Of 108 274 AluY elements in the chimpanzee genome, 13 263 (12.2%) have an intact TT-AAAA target. There are 1 954 AluYs in chimp which overlap the end of a region that putatively has been lost from human (UCSC human-chimpanzee deletion data). Of these, 21.6% have an intact TT-AAAA target. Thus AluYs associated with possible deletions between human and chimp are more likely to have a perfect target than AluYs across the whole genome (21.6% vs. 12.2%), which is consistent with our hypothesis.
An alternative explanation for the observed decrease in the proportion of TT-AAAA targets in older repeats is that there has been a change in nicking site preference of the L1 machinery over time. If in the past TT-AAAA was not the most favoured integration site, then this would account for the reduction in its observed proportion in older repeats. It is difficult to exclude this possibility unequivocably. Nevertheless, several lines of evidence suggest that a shift in nicking site preference does not account for the observed results. Firstly, despite the decrease in TT-AAAA targets, this is still the predominant single hexamer even for old SINEs. There is no indication that in the past, an alternative nicking site was more frequent than TT-AAAA. Secondly, there is a similar pattern of loss of TT-AAAA targets in both the human and rat lineages. Thus, any explanation based on shifts in target site preferences would have to posit the same systematic shift in both rat and human. Finally, the loss of perfect targets begins immediately in younger Alu elements such as AluY and subfamilies, and shows a nearly linear relation to the amount of divergence from the consensus sequence. It is hard to account for this in terms of a shift in enzyme preferences, which would have to be continuous (and presumably ongoing), and shift seamlessly from subfamily to subfamily. The most parsimonious explanation would seem to be that the targets are actually lost along with their associated repeat copy.
Acknowledgments
We would like to thank Vladimir Kapitonov and Adam Pavlicek for discussions on the manuscript, and anonymous referees for suggested changes. This work was supported by National Institutes of Health grant 2 P41 LM006252-07A1.
References
- Babcock M, Pavlicek A, Spiteri E, Kashork CD, Ioshikhes I, Shaffer LG, Jurka J, Morrow BE. Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res. 2003;13:2519–2532. doi: 10.1101/gr.1549503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Liu G, Eichler EE. An Alu transposition model for the origin and expansion of human segmental duplications. Am J Hum Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentolila S, Bach JM, Kessler JL, Bordelais I, Cruaud C, Weissenbach J, Panthier JJ. Analysis of major repetitive DNA sequences in the dog (Canis familiaris) genome. Mamm Genome. 1999;10:699–705. doi: 10.1007/s003359901074. [DOI] [PubMed] [Google Scholar]
- Brosius, J. 2005. Echoes from the past - are we still in an RNP world ? Cytogenet. Genome Res., In press. [DOI] [PubMed]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA. 2003;100:5280–5. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltman DW, Wright JM. Can SINEs: a family of tRNA-derived retroposons specific to the superfamily Canoidea. Nucleic Acids Res. 1994;22:2726–30. doi: 10.1093/nar/22.14.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Boeke JD. Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochem. 1998;37:18081–18093. doi: 10.1021/bi981858s. [DOI] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- Deininger PL, Moran JV, Batzer MA, Kazazian HH., Jr Mobile elements and mammalian genome evolution. Curr Opin Genet Dev. 2003;13:651–8. doi: 10.1016/j.gde.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–91. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- Feng Q, Schumann G, Boeke JD. Retrotransposon R1Bm endonuclease cleaves the target sequence. Proc Natl Acad Sci U S A. 1998;95:2083–8. doi: 10.1073/pnas.95.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M. The genomic record of humankind’s evolutionary roots. Am J Hum Genet. 1999;64:31–39. doi: 10.1086/302218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. A mechanism for gene conversion in fungi. Genet Res Camb. 1964;5:282–304. doi: 10.1017/S0016672308009476. [DOI] [PubMed] [Google Scholar]
- Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci USA. 1997;94:1872–7. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–20. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Jurka J. Evolutionary impact of human Alu repetitive elements. Curr Opin Genet Dev. 2004;14:603–8. doi: 10.1016/j.gde.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Jurka J, Klonowski P. Integration of retroposable elements in mammals: selection of target sites. J Mol Evol. 1996;43:685–9. doi: 10.1007/BF02202117. [DOI] [PubMed] [Google Scholar]
- Jurka J, Klonowski P, Dagman V, Pelton P. CENSOR - a program for identification and elimination of repetitive elements from DNA sequences. Comput Chem. 1996;201:119–21. doi: 10.1016/s0097-8485(96)80013-1. [DOI] [PubMed] [Google Scholar]
- Jurka J, Klonowski P, Trifonov EN. Mammalian retroposons integrate at kinkable DNA sites. J Biomol Struc, Dyn. 1998;15:717–721. doi: 10.1080/07391102.1998.10508987. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kohany O, Pavlicek A, Kapitonov VV, Jurka MV. Duplication, coclustering, and selection of human Alu retrotransposons. Proc Natl Acad Sci U S A. 2004;101:1268–72. doi: 10.1073/pnas.0308084100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J., Kohany, O., Pavlicek, A., Kapitonov, V. V., and Jurka, M. V. 2005. Clustering, duplication and chromosomal distribution of mouse SINE retrotransposons. Cytogenet. Genome Res. In press. [DOI] [PubMed]
- Jurka J, Krnjajic M, Kapitonov VV. Active Alu elements are passed primarily through paternal germlines. Theor Popul Biol. 2002;61:519–530. doi: 10.1006/tpbi.2002.1602. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. The age of Alu subfamilies. J Mol Evol. 1996;42:59–65. doi: 10.1007/BF00163212. [DOI] [PubMed] [Google Scholar]
- Kapitonov, V.V., Pavlicek, A. and Jurka, J. 2004. Anthology of Human Repetitive DNA. In Meyers, R.A. (ed) Encyclopedia of Molecular Cell Biology and Molecular Medicine. Wiley-VCH, Vol. 1, pp. 251–305.
- Kass DH, Batzer MA, Deininger PL. Gene conversion as a secondary mechanism of short interspersed element (SINE) evolution. Mol Cell Biol. 1995;15:19–25. doi: 10.1128/mcb.15.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass DH, Kim J, Rao A, Deininger PL. Evolution of B2 repeats: the muroid explosion. Genetica. 1997;99:1–13. doi: 10.1007/BF02259494. [DOI] [PubMed] [Google Scholar]
- Kim J, Martignetti JA, Shen MR, Brosius J, Deininger PL. Rodent BC1 RNA gene as a master gene for ID element amplification. Proc Natl Acad Sci USA. 1994;91:3607–3611. doi: 10.1073/pnas.91.9.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E, Meyn MS, Pandita A, Squire JA. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes, Chrom Canc. 2002;35:97–112. doi: 10.1002/gcc.10111. [DOI] [PubMed] [Google Scholar]
- Kramerov DA, Tillib SV, Ryskov AP, Georgiev GP. Nucleotide sequence of small polyadenylated B2 RNA. Nucleic Acids Res. 1985;13:6423–37. doi: 10.1093/nar/13.18.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krayev AS, Kramerov DA, Skryabin KG, Ryskov AP, Bayev AA, Georgiev GP. The nucleotide sequence of the ubiquitous repetitive DNA sequence B1 complementary to the most abundant class of mouse fold-back RNA. Nucleic Acids Res. 1980;8:1201–15. doi: 10.1093/nar/8.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krayev AS, Markusheva TV, Kramerov DA, Ryskov AP, Skryabin KG, Bayev AA, Georgiev GP. Ubiquitous transposon-like repeats B1 and B2 of the mouse genome: B2 sequencing. Nucleic Acids Res. 1982;10:7461–75. doi: 10.1093/nar/10.23.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- Martignetti JA, Brosius J. BC200 RNA: A neural RNA polymerase III product encoded by a monomeric Alu element. Proc Natl Acad Sci USA. 1993;90:11563–11567. doi: 10.1073/pnas.90.24.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Olivares M, Lopez MC, Alonso C. Do non-long terminal repeat retrotransposons have nuclease activity? Trends Biochem Sci. 1996;21:283–285. [PubMed] [Google Scholar]
- Meselson MS, Radding CM. A general model for genetic recombination. Proc Natl Acad Sci USA. 1975;72:358–361. doi: 10.1073/pnas.72.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner RJ, Bloom FE, Lai C, Lerner RA, Sutcliffe JG. Brain-specific genes have identifier sequences in their introns. Proc Natl Acad Sci U S A. 1984;81:713–7. doi: 10.1073/pnas.81.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, N. and Ohshima, K. 1995. Evolution of tRNA-derived SINEs, in Maraia, R. J. (ed.): The impact of short interspersed elements (SINEs) on the host genome, pp. 61–79 (R. G. Landes, Austin TX)
- Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–38. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- Pavlicek A, Jabbari K, Paces J, Paces V, Hejnar J, Bernardi G. Similar integration but different stability of Alus and LINEs in the human genome. Gene. 2001;276:39–45. doi: 10.1016/s0378-1119(01)00645-x. [DOI] [PubMed] [Google Scholar]
- Pavlicek A, Noskov VN, Kouprina N, Barrett JC, Jurka J, Larionov V. Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum Mol Genet. 2004;13:2737–51. doi: 10.1093/hmg/ddh301. [DOI] [PubMed] [Google Scholar]
- Quentin Y. Successive waves of fixation of B1 variants in rodent lineage history. J Mol Evol. 1989;28(4):299–305. doi: 10.1007/BF02103425. [DOI] [PubMed] [Google Scholar]
- Rat Genome Sequencing Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Salem AH, Kilroy GE, Watkins WS, Jorde LB, Batzer MA. Recently integrated Alu elements and human genomic diversity. Mol Biol Evol. 2003;20:1349–61. doi: 10.1093/molbev/msg150. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Churakov G, Zischler H, Brosius J. A novel class of mammalian-specific tailless retropseudogenes. Genome Res. 2004;14:1911–1915. doi: 10.1101/gr.2720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- Smit AF. The origin of interspersed repeats in the human genome. Curr Opin Genet Dev. 1996;9:657–663. doi: 10.1016/s0959-437x(96)80030-x. [DOI] [PubMed] [Google Scholar]
- Szak ST, Pickeral OK, Makalowski W, Boguski MS, Landsman D, Boeke JD. Molecular archeology of L1 insertions in the human genome. Genome Biol. 2002;3:research0052.1–0052.18. doi: 10.1186/gb-2002-3-10-research0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- Weichenrieder O, Repanas K, Perrakis A. Crystal structure of the targeting endonuclease of the human LINE-1 retrotransposon. Structure. 2004;12:975–986. doi: 10.1016/j.str.2004.04.011. [DOI] [PubMed] [Google Scholar]