


Abstract
The high-mobility-group (HMG) domain containing transcription factor Sox10 is an important regulator of various processes including the development of neural crest cells and glial cells. Target gene promoters contain multiple Sox10-binding sites, which either support monomeric or cooperative, dimeric binding. The latter is unusual for Sox proteins and might contribute to functional specificity of Sox10. We find that specific amino acid residues in a conserved region immediately preceding the HMG domain of Sox10 are required for cooperative binding. These residues cooperate with the HMG domain during dimeric binding in a manner dependent on specific determinants within the first two α-helices of the HMG domain. Cooperativity of DNA binding is surprisingly refractory to changes in the overall conformation of the DNA-bound dimer. Whereas maintenance of cooperativity is essential for full activation of the promoter of the myelin protein zero target gene, dimer-dependent conformational changes such as the exact bending angle introduced into the promoter appear to be less important, shedding new light on the architectural function of Sox proteins.
INTRODUCTION
Over the last years, a large number of different Sox proteins has been identified (1,2). All members of this family of transcription factors share a highly related high-mobility-group (HMG) box as DNA-binding domain and are capable of recognizing a 7 bp consensus DNA element (A/T)(A/T) CAA(A/T)G. Upon binding to the minor groove, a strong bend is introduced into DNA (3). This has led to the hypothesis that Sox proteins not only function as classical transcriptional activators, but might also exert architectural functions by shaping the three-dimensional structure of target gene promoters with bound transcription factors in a manner similar to other HMG domain proteins (4). Differences within the HMG box and sequence similarities outside this region allow classification of Sox proteins in subgroups A–J (1).
Given the similarity in their DNA-binding domain it is an intriguing question how functional specificity of Sox proteins is generated. One mechanism involves the temporally and spatially defined pattern of expression during development. The class E protein Sox10, for example, is expressed in neural crest cells and various derivatives as well as cells of the oligodendroglial lineage in the central nervous system (5). This effectively restricts Sox10 function to these cells. Accordingly, Sox10-deficient mice exhibit defects in several neural crest-derived cell types (such as melanocytes, cells of the enteric nervous system and peripheral glia) as well as a failure of oligodendroglia to terminally differentiate and produce myelin (6–9). Several genes that code for myelin proteins such as the genes for protein zero, myelin basic protein, proteolipid protein and connexin-32 are under direct transcriptional control of Sox10 (9–11).
The identification of natural target genes for Sox10 has revealed another mechanism which might contribute to specificity. Many target gene promoters contain binding sites that allow cooperative binding of two Sox10 molecules (9–11). These Sox10 dimers can only be detected on DNA, not in solution. For at least one of the target gene promoters (i.e. the promoter of the gene for myelin protein zero) cooperative binding has been shown to be essential for full target gene activation because the dimeric site present in this promoter (C/C′) could not be replaced by a monomeric Sox10 recognition site (11). Cooperative binding is dependent upon a region of the Sox10 protein immediately preceding the DNA-binding HMG domain. This domain has no detectable influence on binding of monomeric Sox10 to DNA and is conserved only in the two other closely related class E proteins, Sox8 and Sox9 (12). Accordingly, many other Sox proteins fail to cooperatively bind to dimeric sites in Sox10 target gene promoters (12). Here, requirements for and consequences of dimeric binding were studied as potential causes for functional specificity of Sox10.
MATERIALS AND METHODS
Plasmids
The pCMV5-based expression vector for full-length rat Sox10 (amino acids 1–466), the isolated HMG domain, and the C-terminally truncated MIC variant (consisting of amino acids 1–189) have been described before (5,11,13). The pCMV5-based expression vector for the C-terminally truncated rat Sox11 (consisting of amino acids 1–129) (14) was generated in an analogous manner by deleting all sequences behind the HMG domain. Chimeras between the C-terminally shortened versions of Sox10 and Sox11 (CH11α3, CH11α23, CH11α123, see Fig. 2A) as well as alanine substitution mutants in the conserved domain preceding the Sox10 HMG domain (Sox10 aa1–aa5, see Fig. 1B) were generated by PCR-directed mutagenesis. The triple alanine substitution aa1 (C71, I72, R73 → A) was introduced into both the MIC variant and full-length Sox10; all other alanine substitutions were generated only in the context of the MIC variant. The alanine-117 to valine mutation (see Fig. 1B) occurred spontaneously during PCR-directed mutagenesis.
Figure 2.

Mapping of regions important for cooperative Sox10 binding within the Sox10 HMG domain. (A) Schematic representation of chimeric proteins generated by combining regions from the MIC variant of Sox10 and a C-terminally truncated form of Sox11 (Sox11ΔC). α1, α2 and α3 mark the three α-helices contained within the HMG domain of Sox proteins. Sox10-derived sequences are depicted as open boxes, Sox11-derived sequences as gray boxes. (B) Expression of all chimeras was verified by western blots of extracts from transfected COS cells with polyclonal antisera against Sox10 and Sox11 as indicated. Numbers indicate size of molecular weight markers in kDa. (C) Electrophoretic mobility shift assays with nuclear extracts from transfected COS cells expressing the proteins shown in (A). Oligonucleotides C/C′ and C′mut [in which C is mutated so that cooperative binding is lost, see Peirano and Wegner (12)] were used as probes as indicated below the lanes. (D) Electrophoretic mobility shift assays with nuclear extracts from transfected COS cells expressing the chimeric protein CH11α123 and the isolated Sox10 HMG domain alone or in combination as indicated above the lanes. –, extract from mock-transfected COS cells; m, bound monomer; d, bound dimer.
Figure 1.

Mapping of amino acid residues important for cooperative binding outside the Sox10 HMG domain. (A) Sequence comparison of the region immediately preceding the HMG domain between Sox10 (rat), Sox9 and Sox8 (both mouse). Conserved residues are marked with an asterisk. Residues altered during mutagenesis are boxed and their positions are listed above the Sox10 sequence. The numbers on the right indicate the position of the last residue shown for each Sox protein. (B) Schematic representation of Sox10 mutants aa1–aa5 generated by alanine substitution of indicated residues. All mutations were introduced into the MIC variant of Sox10 (corresponding to amino acids 1–189). Replacement of alanine-117 by valine occurred spontaneously during PCR-directed mutagenesis. Sox10 aa2* combines the aa2 mutation and the alanine-to-valine exchange at position 117. (C) Expression of Sox10 proteins (truncated at residue 189) was verified by western blots of nuclear extracts from transfected COS cells with a polyclonal antiserum against Sox10. Numbers indicate size of molecular weight markers in kDa. (D) Electrophoretic mobility shift assays with the dimeric binding site C/C′ (for sequence see Fig. 4A) as probe and extracts from transfected COS cells expressing the proteins shown in (B). (E) Electro phoretic mobility shift assay with the monomeric binding site B as probe [for sequence, see Peirano et al. (11)] and extracts from transfected COS cells expressing wild-type Sox10 (wt) or Sox10 aa1 (aa1) as truncated versions. Increasing amounts (2-, 5-, 20-, 50-, 100-fold molar excess) of unlabeled site B were added as competitor (comp). –, extract from mock-transfected COS cells; m, bound monomer; d, bound dimer.
The luciferase reporter plasmid –435 P0 luc, which contains the proximal part of the rat P0 promoter (positions –435 to +48), including the two high-affinity Sox10-binding sites B and C/C′, has been described before (11). Site-directed mutagenesis was employed to increase the spacing between the C and C′ half-sites by an additional 2–5 bp in the context of this promoter. Wild-type C/C′ from the P0 promoter and mutants with increased spacing (see Fig. 4A) were also introduced between XbaI and SalI sites of pBEND2 (15) and used as probes in electrophoretic mobility shift assays.
Figure 4.
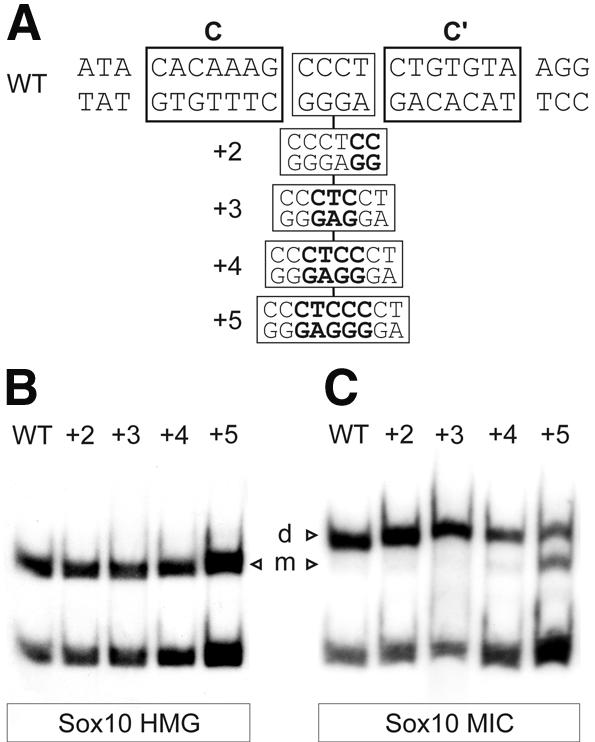
Spacing requirements for cooperative binding of Sox10. (A) Sequence of oligonucleotide probes with location of site C and site C′ indicated by boxes. Sequences inserted between site C and site C′ in the spacing mutants are indicated. (B and C) Electrophoretic mobility shift assays with extracts from transfected COS cells expressing the HMG domain (B) or the MIC variant (C) of Sox10. Oligonucleotides C/C′ (WT) and its spacing variants (+2, +3, +4, +5) were used as probes as indicated above the lanes. m, bound monomer; d, bound dimer.
Cell culture, transfections, luciferase assays, western blots and electrophoretic mobility shift assays
Tet-On N2A neuroblastoma cells capable of doxycycline-dependent induction of Sox10 expression (11) were maintained, transfected in quadruplicates, treated with doxycycline and harvested for luciferase assays as described. A stable cell line capable of inducibly expressing the Sox10 aa1 mutant (C71, I72, R73 → A) was generated and used for transfections in an analogous manner. Data are presented as reported previously (11).
COS cells were maintained in DMEM supplemented with 10% fetal calf serum. Forty-eight hours after transfection (10 µg of DNA per 10 cm plate) with DEAE-dextran (500 µg/ml) and subsequent chloroquine treatment, cells were harvested and extracts were prepared as described (16). Polyclonal rabbit antisera directed against Sox10 or Sox11 (1:3000 dilutions) served as primary antibodies and horse-radish-peroxidase-coupled protein A as secondary detection reagent in western blots using the ECL detection system (5,14).
For electrophoretic mobility shift assays, 0.5 ng of 32P-labeled probe (for oligonucleotides see Fig. 4A, for restriction fragments from pBEND2 see Fig. 5A) were incubated with a COS cell extract for 20 min on ice in a 20 µl reaction mixture as described using poly(dG–dC) as unspecific competitor (12). Samples were loaded onto native 4% polyacrylamide gels and electrophoresed in 0.5× TBE (45 mM Tris/45 mM boric acid/1 mM EDTA, pH 8.3) at 120 V for 1.5 h. Gels were dried and exposed for autoradiography.
Figure 5.

Bending properties of Sox10. (A) C/C′ and its spacing variants were inserted into the multiple cloning site of the pBEND2 vector and retrieved with flanking sequences using the indicated restriction enzymes (fragments 1–5). (B) Electrophoretic mobility shift analyses of Sox10 protein expressed in transiently transfected COS cells with fragments 1–5 (see A) containing either C/C′ (WT) or various spacing mutants (+2, +3, +4, +5). The +5 mutant was on a separate gel. (C) Summary of bending angles ± SEM obtained in three separate experiments.
RESULTS
Amino acid residues outside the HMG domain of Sox10 determine cooperative binding ability
The gene for the myelin protein zero (P0) was the first identified Sox10 target gene (11,12). Within its promoter, it contains a binding site for Sox10 dimers (C/C′), consisting of a medium-affinity (C) and a low-affinity (C′) 7 bp recognition site in a tail-to-tail arrangement with four separating base pairs. Neither fits the consensus for Sox-binding sites with C containing one, and C′ containing two mismatches (Fig. 4A).
We have previously shown that cooperative binding of Sox10 to C/C′ requires a region of 40 amino acids immediately preceding the HMG domain of Sox10 (12). This region is strongly conserved between all class E proteins (Fig. 1A). To determine which residues are critical for cooperative binding, several Sox10 mutants were generated in which three conserved consecutive amino acid residues were replaced by alanines (Fig. 1B). The mutations were introduced into the MIC variant of Sox10 (consisting of amino acids 1–189) and expressed by transient transfection in COS cells. All mutants were generated in approximately similar amounts and primarily localized to the nucleus as evident from western blot analysis and immunohistochemistry (Fig. 1C and data not shown). Mutant Sox10 aa3 in which the two non-conserved residues asparagine-95 and glycine-96 of the dimerization domain were substituted by alanines served as a control.
As expected, the Sox10 aa3 control exhibited the same strongly cooperative dimeric binding as wild-type Sox10 (Fig. 1D). Dimeric binding was also undisturbed in the Sox10 aa4 mutant in which serine-77, glutamine-78 and valine-79 were replaced by alanines, as well as in the Sox10 aa5 mutant that carried alanines instead of valine-88, proline-89 and methionine-90. The Sox10 aa2 mutant on the other hand exhibited slight, but significant, changes in its binding behavior. Although dimeric binding was observed after alanine substitution of tyrosine-83, aspartate-84 and tryptophane-85 in this mutant, the relative rate of dimeric versus monomeric binding was altered in favor of an increased monomeric binding. This effect was even more pronounced in the Sox10 aa1 mutant (cysteine-71, isoleucine-72 and arginine-73 to alanines, Fig. 1B). For this mutant, binding of two Sox10 molecules could hardly be observed and most likely represents independent binding of two Sox10 molecules to the probe rather than cooperative binding of a dimer (Fig. 1D). Such an assumption is supported by the altered mobility of the dimer band for the Sox10 aa1 mutant. When the Sox10 aa1 mutant was bound to a classic monomer site such as site B from the P0 promoter (11), it behaved very similar to wild-type Sox10 protein as evidenced by the fact that equal amounts of cold competitor DNA were needed to suppress binding of either protein to the radioactive probe (Fig. 1E). The aa1 mutant thus possesses a binding affinity for monomeric sites comparable to wild-type and exhibits a specific loss of cooperative binding ability.
Specific determinants within the HMG domain are required for cooperative binding of Sox10
During generation of the Sox10 aa2 mutant, we obtained a clone in which alanine-117 in the HMG domain was changed to valine by a PCR artifact. When the resulting Sox10 aa2* mutant was analyzed in electrophoretic mobility shift assays for its binding to C/C′, we noticed the almost complete failure to bind cooperatively to this site (Fig. 1D). The effect on cooperative binding was much more pronounced in the Sox10 aa2* mutant than in the Sox10 aa2 mutant. This led us to assume that in addition to the conserved region in front of the HMG domain, the HMG domain itself might contribute to cooperative binding ability.
We have previously shown that several proteins from other classes of the Sox family of transcription factors do not exhibit cooperative binding to C/C′ (12). One of these Sox proteins is the class C Sox11 (14). In accord with previous findings for the full-length Sox11, a truncated Sox11 with the C-terminal part of the protein missing (Fig. 2A and B) failed to bind cooperatively to C/C′ (Fig. 2C).
To analyze the contribution of the HMG domain to cooperative binding, we generated chimeric proteins between Sox10 and Sox11 sequences (Fig. 2A). The structure of the HMG domain of Sox proteins had previously been shown by NMR studies to consist of three α-helices, connecting turns and additional regions at either ends (3,17). To keep these α-helices intact, transitions from Sox10 to Sox11 sequences were in the connecting turns between the helices (Fig. 2A). As evident from western blot analyses, all chimeras were detected by antisera directed against Sox10 and Sox11 (Fig. 2B). Correlating with the relative contribution of Sox10 and Sox11, chimeras strongly detected by the Sox10 antiserum were weakly detected by the Sox11 antiserum and vice versa.
Complete replacement of the HMG domain of Sox10 by the corresponding region from Sox11 led to a protein that, similar to wild-type Sox11, failed to show cooperative binding (CH11α123 in Fig. 2C). Only binding of monomers was observed indicating that the conserved region in front of the HMG domain of Sox10 does not cooperate with all HMG domains during dimeric binding, but requires specific residues within the HMG domain. A protein that contained the N-terminal HMG-box sequences and α-helix 1 of Sox10, but α-helix 2, α-helix 3, and the C-terminal HMG-box sequences of Sox11 (CH11α23 in Fig. 2A and B) already exhibited a substantial increase in dimeric binding relative to the construct in which the complete HMG domain was derived from Sox11 (Fig. 2C). This argues that helix 1 of the HMG domain and preceding sequences carry important determinants for dimeric binding. Full recovery of dimeric binding was, however, only observed when N-terminal sequences as well as α-helices 1 and 2 were Sox10-derived (CH11α3 in Fig. 2C). Helix 3 from Sox11 on the other hand was able to replace the corresponding region of Sox10 indicating that specificity requirements for helix 3 are not strict. We conclude that helix 1 and helix 2 are the major determinants for dimeric binding among the HMG-box helices.
The chimeric protein consisting of Sox10 dimerization domain and Sox11 HMG domain (CH11α123) was also analyzed for binding to C/C′ in the presence of the isolated Sox10 HMG domain (Fig. 2D). While both proteins bound as monomers, no additional complex was observed indicating that heterodimerization did not occur. Dimerization domain and Sox10-specific HMG domain thus have to be present on the same Sox10 molecule. Intramolecular function of both domains is the simplest interpretation of this result. Alternatively, cooperative binding may require two dimerization domains and two HMG domains interacting with each other in a crosswise manner between two Sox10 molecules.
Cooperative binding of Sox10 is functionally important
The availability of Sox10 mutants with a selective loss of cooperative dimeric binding enabled us to analyze its importance for Sox10 function in vivo. We generated a stable N2A neuroblastoma line which permitted doxycycline-dependent inducible expression of the Sox10 aa1 mutant. Inducibility of protein expression was verified by western blot (Fig. 3A). A similar cell line had been generated before for wild-type Sox10 and used in the study of the P0 promoter (11,12). We have previously shown that the proximal P0 promoter (–435 P0; positions –435 to +48) is activated ∼10-fold after induction of Sox10 (see also below, Fig. 6). This region contains the two high-affinity Sox10-binding sites B and C/C′ as well as other low-affinity sites (11). When either the monomeric site B or the dimeric site C/C′ was mutated (–435 P0mtB and –435 P0mtC, respectively), Sox10-dependent activation was reduced by half. Mutation of both sites was needed to obtain a near complete loss of Sox10 function (11). When the same experiment was carried out with the Sox10 aa1 mutant in the new inducible cell line, the activation pattern of the proximal P0 promoter and its variants differed. In general, activation of the proximal P0 promoter appeared slightly lower. This is, however, difficult to evaluate as it is impossible to compare activation rates obtained in two different stable cell lines. In contrast to previous experiments with wild-type Sox10, mutation of the dimer site C/C′ did not significantly influence the rate of Sox10-dependent transactivation (–435 P0mtC in Fig. 3B). In the case of the Sox10 aa1 mutant, removal of the monomer site B (–435 P0mtB in Fig. 3B) abolished Sox10-dependent activation almost completely arguing that this Sox10 mutant mainly functions through the monomer site. This result proves that dimeric binding is an important aspect of the normal function of Sox10. It furthermore confirms previous results that the C/C′ site is only functional when able to bind Sox10 dimers (11).
Figure 3.

A Sox10 mutant with decreased dimerization capacity fails to mediate Sox10-dependent transcriptional activation of the proximal P0 promoter through site C/C′. (A) Western blot analysis of extracts prepared from an N2A Tet-On cell line generated by stable transfection and capable of inducibly expressing the full-length Sox10 aa1 protein. The cells were kept for 12 h in the absence (–Doxy) or presence (+Doxy) of doxycycline. Expression of the Sox10 aa1 protein was detected using an anti-Sox10 polyclonal antiserum. Wild-type Sox10 protein (Sox10) served as control. Numbers on right indicate size of molecular weight markers in kDa. (B) These Tet-On N2A cells were transfected in quadruplicates with the –435 P0-luciferase reporter (containing positions –435 to +48 of the rat P0 promoter) and various mutants thereof [–435 P0mtC and –435 P0mtB, see also Peirano et al. (11)]. From each transfected quadruplicate, one duplicate was left untreated, while the second one was treated with doxycycline to induce expression of the Sox10 aa1 protein. Luciferase activities in extracts from transfected cells were determined in three independent experiments. Data were normalized as described (11) and are presented as relative activation rates ± SEM with the activation of the wild-type promoter arbitrarily set to 1.
Figure 6.

Variations in bending angle do not impair Sox10-dependent transcriptional activation of the P0 promoter through site C/C′. Tet-On N2A cells capable of inducibly expressing wild-type Sox10 protein (11) were transfected in quadruplicates with the wild-type –435 P0-luciferase reporter (WT) and various mutants thereof (+2, +3, +4, +5) in which the spacing between sites C and C′ in the composite C/C′ site was changed as indicated in Figure 4A. From each transfected quadruplicate, one duplicate was left untreated, while the second one was treated with doxycycline to induce expression of the Sox10 protein. Luciferase activities in extracts from transfected cells were determined in three independent experiments. Data were normalized as described (11) and are presented as relative activation rates ± SEM with the activation of the wild-type promoter arbitrarily set to 1.
Cooperative binding and the architectural function of Sox10
We have shown previously that Sox10 dimers bend C/C′ differently than monomers bend site B (12). Assuming architectural functions for Sox proteins, cooperative binding to C/C′ might be essential for activation of the P0 promoter because of the exact bending angle introduced by the Sox10 dimer. To analyze this question, we changed the spacing between C and C′ from 4 to 6 (+2), 7 (+3), 8 (+4) and 9 (+5) nt (Fig. 4A). C/C′ as well as the spacing mutants were analyzed in electrophoretic mobility shift assays for their ability to bind Sox10 (Fig. 4C). Dimeric binding was virtually indistinguishable for the wild-type and the spacing mutants +2, +3 and +4. In the case of the +5 mutant, which carries 9 nt between C and C′, cooperativity of Sox10 binding is partially lost as indicated by the increased occurrence of monomeric binding and the decreased occurrence of dimeric binding (Fig. 4C). In control experiments with the isolated HMG domain of Sox10, which exclusively binds as a monomer, similar recognition was observed to wild-type C/C′ and all mutant sites [Fig. 4B; see also Peirano and Wegner (12)].
To determine the exact bending angle imposed by Sox10 dimers, the spacing mutants were analyzed in circular permutation assays (Fig. 5A). Contributions to the apparent bending angle will stem from the Sox10 molecules bending C and C′ as well as from the overall topology of the DNA-bound dimer. The wild-type C/C′ site yielded an overall apparent bending angle of 103.5°, in close agreement with the value previously determined for C/C′ in the context of a slightly longer fragment (12). Increasing the spacing from 4 to 6 nt led to an increase in the apparent bending angle by 5.6 to 109.1° (Fig. 5B and C). Additional insertion of 1 or 2 nt further increased the bending angle to 118.4 and 122.0°, respectively. Thus, the bending angle could be increased by 18.5° without loss of cooperativity, arguing for a remarkable ability of the dimer to compensate for overall topological changes. Comparable to the +4 mutant, insertion of an additional 5 nt led to an increase of the bending angle by 18.1° (Fig. 5C). This time, however, we observed a concomitant reduction in cooperative binding (Figs 4C and 5C), indicating that there is no strict correlation between magnitude of the apparent bending angle and cooperativity.
If binding of Sox10 to C/C′ involves an architectural function, one would expect this dramatic change in the apparent bending angle to have severe consequences on the ability of Sox10 to activate the P0 promoter. We introduced the spacing mutations into the context of the proximal P0 promoter (–435 P0) and analyzed the resulting P0 promoter mutants for their ability to be activated in a Sox10-dependent manner (Fig. 6). On average, we obtained an ∼10-fold activation of the wild-type proximal P0 promoter by Sox10. This level was set to 1 and all other activation rates were expressed relative to the one obtained with the wild-type promoter. Surprisingly, insertion of an additional 2–4 nt did not significantly change the ability of the promoter to be activated by Sox10. Sox10-dependent activation of the proximal P0 promoter was drastically reduced only upon addition of 5 nt between C and C′. As the bending angle in the +5 mutant closely resembles the one in the +4 mutant, this reduction in Sox10 responsiveness is unlikely to be caused by the increased bending angle but rather by the reduction of cooperative binding observed in this mutant. Thus, C/C′-bound Sox10 dimers can accommodate large changes in bending, but little changes in cooperativity.
DISCUSSION
The recent years have seen the identification of many different Sox proteins in diverse species (1,2). These Sox proteins can be classified in 10 subgroups (A–J). With the exception of class D proteins Sox5, Sox6 and Sox13, Sox proteins bind to DNA as monomers. Class D proteins efficiently dimerize in solution due to the presence of a leucine zipper, and their strong preference for dimeric DNA-binding sites over monomeric sites directly follows from this (18).
We are primarily interested in class E proteins Sox8, Sox9 and Sox10. In particular, we have studied Sox10 function and shown it to be important for the development of neural crest cells and glial cells. In the absence of Sox10, certain neural crest-derived cell types such as melanocytes, enteric neural crest and peripheral glia are completely missing (6–8). Additionally, oligodendrocytes, the myelinating glia of the central nervous system, fail to terminally differentiate and to form myelin (9). Heterozygous loss of Sox10 is already accompanied by defects, which in affected human patients become visible as a combination of Hirschsprung disease (aganglionosis of the distal colon), Waardenburg syndrome (partial depigmentation and melanocyte-dependent hearing deficits) and myelinopathies in peripheral and central nervous systems (19–21). Several genes that code for myelin proteins are among the identified Sox10 target genes (9–11), arguing that one of the functions of Sox10 in glial cells may be to regulate myelin genes and the process of myelination. One of these myelin genes is the gene for the myelin protein zero (P0). Its promoter is activated directly by Sox10 and contains multiple binding sites for this transcription factor (11).
Molecular determinants for cooperative binding are located both outside and within the HMG domain of Sox10
Class E Sox proteins are unusual among Sox proteins in that they bind efficiently both as monomers and as dimers to DNA (11). Both monomeric and dimeric binding sites have been identified in several target gene promoters including myelin basic protein, connexin-32 and P0 (9–11). This binding behavior appears to be important for the biological function of this group of Sox proteins. Within the P0 promoter, site B is a monomer site and site C/C′ is a dimer site. C/C′ will loose its function after mutation of the low-affinity C′ half-site, arguing that binding of a monomer to one half-site is not sufficient (12).
We have previously shown that dimeric binding cannot be obtained with the isolated HMG domain. A 40 amino acid region immediately preceding the HMG domain is additionally required (12). This region is conserved only between class E Sox proteins explaining the restriction of this type of dimeric binding. Previous analyses have failed to give any indication for a direct role of this domain in DNA binding as there is no detectable difference in affinity or kinetics of binding to monomer sites in the absence or presence of this domain. Here, we have analyzed the requirements of dimeric binding in greater detail. We show that some of the conserved amino acids within this 40 amino acid domain, in particular cysteine-71 to arginine-73 and to a lesser extent tyrosine-83 to tryptophane-85, are indeed essential for dimeric binding. Several other conserved residues, however, are dispensable, arguing that the conserved region could have other functions besides dimerization.
Interestingly, the effect of mutations within this conserved domain can be modulated by changes within the HMG domain. A single alanine-to-valine substitution at position 117 within the first helix of the HMG domain decreases the ability of the Sox10 aa2 mutant for dimeric binding dramatically. When modeled onto the HMG-domain of Sry, alanine-117 is not in direct contact with DNA and does not seem to be important for the overall structure of the HMG domain. This functional interaction of amino acids more than 30 residues apart in the primary sequence is indicative of a cooperation between the HMG domain and the preceding conserved region in dimeric binding. Our results are most compatible with an intramolecular, rather than an intermolecular, interaction between both domains.
If both domains cooperate there might be some degree of specificity in this interaction. This seems to be the case, as the HMG domain of Sox11 fails to functionally substitute for the HMG domain of Sox10 in a chimeric molecule consisting of N-terminal Sox10 sequences and Sox11 HMG domain. Corroborating specificity in the interaction between the two domains, alanine-117 within the first helix of the HMG domain of Sox10 is not generally conserved among Sox proteins, but present in all class E members (5).
All HMG domains are known to have a similar three-dimensional structure with three α-helices and adjacent regions assembled into a twisted L shape (17,22). Helix 1 and helix 2 form the short arm of the L, helix 3 and the N-terminal strand the long arm. We took the chimera consisting of N-terminal Sox10 sequences and the HMG domain of Sox11, and tried to reconstitute dimeric binding ability by exchanging increasingly larger regions of Sox11 against the corresponding ones of Sox10. Precaution was taken to leave the α-helices intact. These experiments point to an important role of α-helix 1 in dimeric binding, the same helix that also contains alanine-117. Replacement of α-helix 1, however, reconstitutes dimeric binding only at a suboptimal level. For full recovery of dimeric binding, α-helix 2 (and thus the complete short arm of the L) must also be Sox10-derived. In contrast, the exact amino acid composition of α-helix 3 does not appear to be crucial for dimeric binding. It has previously been shown for the HMG domain of Sox5 that its association with DNA proceeds with a significant decrease of conformational entropy (23). This reduction in conformational mobility upon association with DNA might be needed in Sox10 for an efficient interaction of the HMG domain with the preceding region which in turn might be a prerequisite for DNA-dependent dimerization.
Cooperative binding is important for Sox10 function, but its mode of action can not be reduced to differential DNA bending
Our identification of Sox10 mutants deficient in dimeric binding has also allowed us to test the importance of dimeric binding for target gene activation. Here we show that Sox10 mutants deficient in dimeric binding are no longer able to activate a target gene promoter through a functionally important dimeric site while still working through an equally important monomeric site. This nicely complements the previous finding, that conversion of a dimeric into a monomeric site in the P0 promoter did not rescue the activity of this site (12). It clearly argues that Sox10 and possibly other class E Sox proteins might act on their target gene promoters through two fundamentally different mechanisms that can now be separately addressed. One possibility could be that monomeric and dimeric binding both mediate differential architectural function. Sox proteins like other HMG domain proteins bind to the minor groove of DNA and concomitantly introduce strong bends (reviewed in 2). We have previously shown that upon binding to monomeric sites, Sox10 introduces a bend of 75–80° into DNA (12) in good agreement with predictions from NMR studies on the SRY–DNA complex (3). However, on the C/C′ dimeric site from the P0 promoter, the apparent overall bending angle proved to be much larger. Thus, monomeric and dimeric binding could be used at different positions of a promoter (or enhancer) to force it into the optimal conformation required for enhanceosome formation and function. Here, we have tested this model for the P0 promoter.
By changing the spacing between the C and C′ half-sites, we were able to dramatically alter the overall apparent bending angle between 103 and 122°. These changes should also lead to dramatic overall changes in the conformation of an enhanceosome formed on this promoter. Assuming a critical role of bending in the function of Sox10 dimers, these conformational alterations should in turn lead to significant changes in Sox10-dependent promoter activation. At least for the P0 promoter, this was not the case. Despite extreme variations in the bending angle, promoter activation remained fairly constant. Thus, Sox10 does not appear to solely function on C/C′ in the P0 promoter through bending, although it has to be taken into account that the behavior of a transfected promoter on a plasmid may not fully resemble behavior in its natural chromatin environment. If Sox10 function is architectural, it should therefore be by mechanisms other than just DNA bending.
Whereas activation of the P0 promoter did not correlate with changes in bending angle at the dimeric site, it correlated well with the cooperativity of binding. Insertion of 4 or 5 bp between the C and C′ half-sites led to similar changes in bending angle. Insertion of 5 bp, however, selectively diminished cooperativity, and was the only promoter mutation resulting in strongly reduced Sox10-dependent activation. This importance of cooperative binding of two Sox10 molecules could for instance be explained by a differential ability of dimer versus monomer to interact with transcription factors bound to adjacent sites in the promoter or with select components of the transcriptional apparatus and chromatin remodeling activities.
A previous study (24) used a Sox2 mutant with reduced bending capacity as well as less bendable versions of a Sox2-binding site to show that spatially precise bending is an essential element for Sox2-dependent promoter activation. We would like to point out that the two studies do not contradict each other. While Scaffidi and Bianchi (24) used a monomeric site and a class B Sox protein, we performed our analysis on a dimeric site and a class E member. Thus, it is possible that architectural function is restricted to monomeric sites with dimeric sites having other functions. It must also be taken into account that both studies used specific sites in defined promoter contexts. At present it is difficult to generalize beyond the specific promoters analyzed. Nevertheless, our study clearly proves the importance of dimeric binding as well as the fact that Sox protein function cannot be reduced to DNA bending.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by a grant from the Deutsche Forschungsgemeinschaft to M.W. (We1326/7-2).
REFERENCES
- 1.Bowles J., Schepers,G. and Koopman,P. (2000) Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol., 227, 239–255. [DOI] [PubMed] [Google Scholar]
- 2.Wegner M. (1999) From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res., 27, 1409–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werner M.H., Huth,J.R., Gronenborn,A.M. and Clore,G.M. (1995) Molecular basis of human 46X,Y sex reversal revealed from the three-dimensional solution strucutre of the human SRY–DNA complex. Cell, 81, 705–714. [DOI] [PubMed] [Google Scholar]
- 4.Giese K., Cox,J. and Grosschedl,R. (1992) The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell, 69, 185–196. [DOI] [PubMed] [Google Scholar]
- 5.Kuhlbrodt K., Herbarth,B., Sock,E., Hermans-Borgmeyer,I. and Wegner,M. (1998) Sox10, a novel transcriptional modulator in glial cells. J. Neurosci., 18, 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbarth B., Pingault,V., Bondurand,N., Kuhlbrodt,K., Hermans-Borgmeyer,I., Puliti,A., Lemort,N., Goossens,M. and Wegner,M. (1998) Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc. Natl Acad. Sci. USA, 95, 5161–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Southard-Smith E.M., Kos,L. and Pavan,W.J. (1998) Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nature Genet., 18, 60–64. [DOI] [PubMed] [Google Scholar]
- 8.Britsch S., Goerich,D.E., Riethmacher,D., Peirano,R.I., Rossner,M., Nave,K.A., Birchmeier,C. and Wegner,M. (2001) The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev., 15, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stolt C.C., Rehberg,S., Ader,M., Lommes,P., Riethmacher,D., Schachner,M., Bartsch,U. and Wegner,M. (2002) Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev., 16, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bondurand N., Girard,M., Pingault,V., Lemort,N., Dubourg,O. and Goossens,M. (2001) Human Connexin 32, a gap junction protein altered in the X-linked form of Charcot-Marie-Tooth disease, is directly regulated by the transcription factor SOX10. Hum. Mol. Genet., 10, 2783–2795. [DOI] [PubMed] [Google Scholar]
- 11.Peirano R.I., Goerich,D.E., Riethmacher,D. and Wegner,M. (2000) Protein zero expression is regulated by the glial transcription factor Sox10. Mol. Cell. Biol., 20, 3198–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peirano R.I. and Wegner,M. (2000) The glial transcription factor Sox10 binds to DNA both as monomer and dimer with different functional consequences. Nucleic Acids Res., 28, 3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhlbrodt K., Schmidt,C., Sock,E., Pingault,V., Bondurand,N., Goossens,M. and Wegner,M. (1998) Functional analysis of Sox10 Mutations found in human Waardenburg–Hirschsprung patients. J. Biol. Chem., 273, 23033–23038. [DOI] [PubMed] [Google Scholar]
- 14.Kuhlbrodt K., Herbarth,B., Sock,E., Enderich,J., Hermans-Borgmeyer,I. and Wegner,M. (1998) Cooperative function of POU proteins and Sox proteins in glial cells. J. Biol. Chem., 273, 16050–16057. [DOI] [PubMed] [Google Scholar]
- 15.Kim J., Zwieb,C., Wu,C. and Adhya,S. (1989) Bending of DNA by gene-regulatory proteins: construction and use of a DNA bending vector. Gene, 85, 15–23. [DOI] [PubMed] [Google Scholar]
- 16.Sock E., Renner,K., Feist,D., Leger,H. and Wegner,M. (1996) Functional comparison of PML-type and archetype strains of JC virus. J. Virol., 70, 1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Houte L.P.A., Chuprina,V.P., van der Wetering,M., Boelens,R., Kaptein,R. and Clevers,H. (1995) Solution structure of the sequence-specific HMG box of the lymphocyte transcriptional activator Sox-4. J. Biol. Chem., 270, 30516–30524. [DOI] [PubMed] [Google Scholar]
- 18.Lefebvre V., Li,P. and de Crombrugghe,B. (1998) A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J., 17, 5718–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue K., Tanabe,Y. and Lupski,J.R. (1999) Myelin deficiencies in both the central and peripheral nervous system associated with a SOX10 mutation. Ann. Neurol., 46, 313–318. [DOI] [PubMed] [Google Scholar]
- 20.Pingault V., Bondurand,N., Kuhlbrodt,K., Goerich,D.E., Prehu,M.-O., Puliti,A., Herbarth,B., Hermans-Borgmeyer,I., Legius,E., Matthijs,G., Amiel,J., Lyonnet,S., Ceccherini,I., Romeo,G., Smith,J.C., Read,A.P., Wegner,M. and Goossens,M. (1998) Sox10 mutations in patients with Waardenburg–Hirschsprung disease. Nature Genet., 18, 171–173. [DOI] [PubMed] [Google Scholar]
- 21.Touraine R.L., Attie-Bitach,T., Manceau,E., Korsch,E., Sarda,P., Pingault,V., Encha-Razavi,F., Pelet,A., Auge,J., Nivelon-Chevallier,A., Holschneider,A.M., Munnes,M., Doerfler,W., Goossens,M., Munnich,A., Vekemans,M. and Lyonnet,S. (2000) Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. Am. J. Hum. Genet., 66, 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Werner M.H. and Burley,S.K. (1997) Architectural transcription factors: proteins that remodel DNA. Cell, 88, 733–736. [DOI] [PubMed] [Google Scholar]
- 23.Privalov P.L., Jelesarov,I., Read,C.M., Dragan,A.I. and Crane-Robinson,C. (1999) The energetics of HMG box interactions with DNA: thermodynamics of the DNA binding of the HMG box from mouse sox-5. J. Mol. Biol., 294, 997–1013. [DOI] [PubMed] [Google Scholar]
- 24.Scaffidi P. and Bianchi,M.E. (2001) Spatially precise DNA bending is an essential activity of the sox2 transcription factor. J. Biol. Chem., 276, 47296–47302. [DOI] [PubMed] [Google Scholar]