


Abstract
The enzyme endonuclease V nicks uracil-containing DNA at the second or third phosphodiester bond 3′ to uracil sites. I applied the enzyme to random fragmentation of DNA to revise the complex DNA shuffling protocol. The merit of using endonuclease V is that cleavage occurs at random sites and the length of the fragments can easily be adjusted by varying the concentration of dUTP in the polymerase chain reaction. Unlike the conventional method using DNase I, no partial digestion or gel separation of fragments is required. Therefore, labor is dramatically reduced and reproducibility ensured. I applied this method to recombine two truncated green fluorescent protein (GFP) genes and demonstrated successful DNA shuffling by the appearance of the fluorescent full-length GFP genes.
INTRODUCTION
DNA recombination in vitro as an evolution strategy was first reported by Stemmer in 1994 (1). Stemmer’s method, which he called DNA shuffling, uses enzymatic digestion (most commonly with DNase I) of parent genes to generate a pool of random DNA fragments. These fragments can be assembled by iterative cycles of denaturation, annealing and extension with thermostable DNA polymerase—known as the polymerase chain reaction (PCR). This reaction generates a mixture of products in length and combination, from which full-length genes are amplified by PCR with flanking primers. Although the method has already proven to be extremely useful in directed evolution (1,2), the experimental procedure requires careful monitoring of each reaction step and, hence, is extremely labor intensive and time consuming. In particular, DNA fragmentation is most problematic since the reaction has to be carefully controlled in order to obtain fragments of appropriate length. In addition, the length of the fragments obtained by DNase I digestion varies greatly with minor changes in conditions, including the amount of nuclease, the source (supplier) or lot of nuclease, the reaction temperature and the purity of DNA substrates. This makes the digestion experiment non-reproducible. Furthermore, it is known that DNase I digests double-stranded DNA preferentially at sites adjacent to pyrimidine nucleotides (3). Therefore, using fragments generated by DNase I digestion may induce a sequence bias into the recombination (4).
To overcome these drawbacks, I attempted to develop an alternative random digestion method whereby complex libraries can be obtained through a simple and reproducible approach. In this study, I used endonuclease V, which is known to nick uracil-containing DNA (and other damaged DNA having inosine, abasic sites, and so on) at the second or third phosphodiester bond 3′ to uracil sites (5,6). Although the cleavage sites are always two or three bases downstream of a thymidine (substituted by uracil) site, this method is expected to produce much fewer hot and cold spots that decrease random representation of fragments which occur with DNase I cleavage (3,4). Using the DNA fragments generated by endonuclease V digestion, successful DNA shuffling was achieved with shuffling efficiency equivalent to DNase I.
MATERIALS AND METHODS
Reagents
Endonuclease V was purchased from Trevigen (Gaithersburg, MD), Taq2000 DNA polymerase was from Stratagene (La Jolla, CA), deoxynucleotides were from Amersham (Piscataway, NJ). Plasmid pGFPuv was obtained from Clontech (Palo Alto, CA). Competent Escherichia coli JM109 cells were purchased from Toyobo (Osaka, Japan). A plasmid, pTGuv1, contained the green fluorescent protein (GFP) gene (2) under control of a tac promoter.
Preparation of uracil-containing recombination templates
Uracil-containing recombination templates were prepared by PCR in the presence of dUTP (7). The PCR mixture contained 10 mM Tris–HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM each of dATP, dGTP and dCTP, and 0.2 mM of a dTTP/dUTP mixture, 25 pmol each of primers, 50 ng of each template plasmid, and 1.25 U of Taq2000 DNA polymerase in a total volume of 50 µl. The concentration of dTTP/dUTP was varied (0.2 mM/0 mM, 0.15 mM/0.05 mM, 0.1 mM/0.1 mM, 0.05 mM/0.15 mM or 0 mM/0.2 mM) to find a condition which gives fragments of appropriate length. The PCR mixture was heated at 95°C for 1 min, followed by 25 cycles of incubation at 94°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min. Amplified DNA (∼1.5 µg) was separated by agarose gel electrophoresis, purified in a QIAquick spin column (Qiagen, Tokyo, Japan), and dissolved in 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonate (HEPES)–KOH pH 7.4, 50 mM NaCl, 0.5 mM MnCl2.
Endonuclease V digestion of the recombination templates
To the solution, 1 U of endonuclease V was added and incubated at 37°C for 12 h, followed by heating at 95°C for 10 min. A portion of the products (3 µl) was used for agarose gel (1.6%) electrophoresis to check the fragment lengths. The rest of the sample was purified in a QIAquick spin column, eluted in 30 µl of water, and used for the subsequent assembly reaction.
Assembly reaction
The assembly reaction of DNA fragments was carried out in 10 mM Tris–HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM each of dNTP, 5 µl each of DNA fragments, and 1 U of Taq2000 DNA polymerase in a total volume of 20 µl. Reaction mixtures were heated at 95°C for 1 min, followed by 30 cycles of incubation at 94°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final incubation at 72°C for 5 min.
Amplification of full-length genes
Full-length genes were amplified using a set of flanking primers. The PCR mixture contained 10 mM Tris–HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM each of dNTP, 25 pmol each of flanking primers, 0.5 µl of assembly product, and 1.25 U of Taq2000 DNA polymerase in a total volume of 50 µl. Reaction mixtures were heated at 95°C for 1 min, followed by 25 cycles of incubation at 94°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final incubation at 72°C for 5 min.
Library production and screening
Amplified fragments were purified and cloned back into the expression plasmid by ligation. Competent E.coli JM109 cells were transformed with the ligation mixture and grown overnight at 37°C on LB agar plates containing 100 µg/ml ampicillin and 0.1 mM isopropyl-β-d-thiogalactopyranoside. The total number of colonies was counted under white light and the number of fluorescent colonies was counted under UV light (∼365 nm).
RESULTS AND DISCUSSION
In order to overcome the technical problems of DNase I digestion and to achieve high shuffling efficiency, Kikuchi et al. (8) used frequent-cutter restriction enzymes. Their method is effective at shuffling and is simple and reproducible. However, it requires DNA sequence information a priori in order to select appropriate restriction enzymes and hence it is not versatile. In addition, the cleavage sites and the length of the obtained fragments are not random. Therefore, the resultant library will severely lack complexity. To overcome these drawbacks, I attempted to develop an alternative random digestion method whereby complex libraries can be obtained through a simple and reproducible approach.
In this study, I used endonuclease V, which is known to nick uracil-containing DNA (and other damaged DNA having inosine, abasic sites and so on) at the second or third phosphodiester bond 3′ to uracil sites (5,6). Although the endonuclease V specifically recognizes uracil, the PCR technique allows uracil to distribute at random over the sequence. In addition, the cleavage site is not adjacent to the uracil site but at the second or third 3′ bond to the uracil site. For these reasons, digestion sites are considered to be random, as with DNase I. Another determinant for efficient shuffling is the length of fragments. With endonuclease V, the length can be controlled by adjusting the number of uracil residues in the DNA simply by varying the concentration of dUTP in the PCR. All these features unique to endonuclease V are suitable for random DNA fragmentation, compared with the conventional method using DNase I.
Based on this idea, I tested the validity of endonuclease V-mediated fragmentation in DNA shuffling. For recombination templates, two truncated GFP genes were prepared in which the full-length GFP sequence (∼750 bp) was truncated at amino acids Leu60 and Val120, respectively, by replacing the amino acid codons with a stop codon (TAA). Recovery of fluorescence requires recombination between two variants (GFPuv60* and GFPuv120*, respectively) to restore a full-length GFP gene, and this system allows a quick assay for DNA shuffling (Fig. 1) (9).
Figure 1.

Recombination assay system. Two GFP genes having stop codons (GFPuv60* and GFPuv120*) were used for recombination templates. If recombination occurs between the two stop codons and the resultant gene does not contain stop codons, the gene recovers the fluorescence. The fluorescent gene is shown as white bars and non-fluorescent variants having stop codon(s) are shown as shaded bars.
In the experiment, the ratio of dTTP to dUTP was first varied in the PCR preparation of full-length recombination templates. After treatment with endonuclease V, a portion of the products (1/10) was separated by agarose gel electrophoresis (Fig. 2). As predicted, no digestion occurred for the sample prepared in the absence of dUTP (Fig. 2, lane 1), and the average length of fragments decreased as the ratio of dTTP to dUTP decreased. The full-length gene was visible for the samples prepared in 0.2 mM dTTP/0 mM dUTP, 0.15 mM dTTP/0.05 mM dUTP and 0.1 mM dTTP/0.1 mM dUTP (Fig. 2, lanes 1–3), suggesting that these conditions were not suitable for DNA shuffling. DNA fragments prepared in 0.05 mM dTTP/0.15 mM dUTP gave 100–300-bp fragments (Fig. 2, lane 4) and the condition was considered to be appropriate for DNA shuffling. If dTTP was completely substituted by dUTP, the resultant fragments were hardly visible, probably because the fragments became too short (Fig. 2, lane 5). This condition was also considered to be unsuitable for DNA shuffling.
Figure 2.
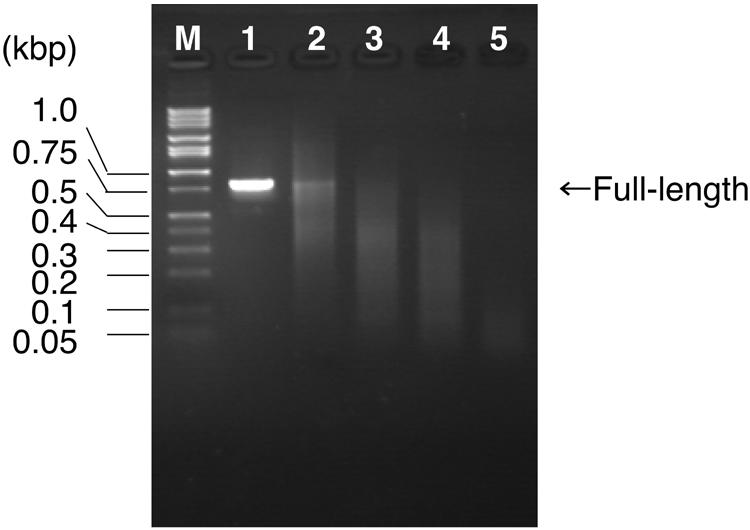
Monitoring the endonuclease V digestion by agarose gel (1.6%) electrophoresis. M, DNA size standard (1 kb DNA ladder; Takara, Tokyo, Japan); lane 1, 0.2 mM dTTP/0 mM dUTP; lane 2, 0.15 mM dTTP/ 0.05 mM dUTP; lane 3, 0.1 mM dTTP/0.1 mM dUTP; lane 4, 0.05 mM dTTP/0.15 mM dUTP; lane 5, 0 mM dTTP/0.2 mM dUTP. See text for the other components of the reaction mixture.
In order to survey the appropriate preparation of the fragments for DNA shuffling, the fragments were purified and subjected to subsequent assembly and amplification reactions. At this stage, no products were produced from the fragments prepared in the absence of dTTP (complete substitution to dUTP), indicating the unsuitability for DNA shuffling. The rest of the preparations gave a full-size gene. The gene was then cloned and mutant libraries were created. As for the result of screening for fluorescence, libraries created by using the fragments prepared in dTTP/dUTP of 0.2 mM/0 mM, 0.15 mM/0.05 mM and 0.1 mM/0.1 mM gave virtually no fluorescent colonies (<0.1%), indicating the contamination of the full-length gene was problematic. In contrast, DNA fragments prepared in 0.05 mM dTTP/0.15 mM dUTP gave fluorescent colonies at a high frequency; 10 out of 100 transformants were fluorescent. This fraction of fluorescent colonies (10%) was smaller than the theoretical prediction of 25% (10), but was close to the value obtained by the DNase I method, which was done in nearly the same assay system (10). Five fluorescent colonies were picked and their DNA sequences checked. In all the colonies, the sequence of the GFP gene was identical to that of the parent GFP gene.
In my case, fragments of appropriate length were obtained in the 0.05 mM dTTP/0.15 mM dUTP condition. However, this does not mean that the condition is immediately applicable to any other genes. This is because base composition varies from sequence to sequence. In our case, the G+C content of the GFP gene was 41%, slightly AT-rich. If the G+C content is high, as is often seen in some bacteria such as Streptomyces and Thermus, it will be necessary to increase the ratio of dUTP to dTTP accordingly. Besides the base composition, the incorporation of uracil also depends upon the PCR conditions (e.g. DNA polymerase, number of cycles, amount of template) (7). For these reasons, it is always necessary to optimize the PCR conditions to get fragments of appropriate length.
In conclusion, random DNA fragmentation by endonuclease V is a handy and reproducible method and can be used instead of fragmentation by DNase I, which is technically problematic, without sacrificing shuffling efficiency.
REFERENCES
- 1.Stemmer W.P. (1994) Rapid evolution of a protein in vitro by DNA shuffling. Nature, 370, 389–391. [DOI] [PubMed] [Google Scholar]
- 2.Crameri A., Whitehorn,E.A., Tate,E. and Stemmer,W.P. (1996) Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat. Biotechnol., 14, 315–319. [DOI] [PubMed] [Google Scholar]
- 3.Sambrook J. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 4.Joern J.M., Meinhold,P. and Arnold,F.H. (2002) Analysis of shuffled gene libraries. J. Mol. Biol., 316, 643–656. [DOI] [PubMed] [Google Scholar]
- 5.Huang J., Lu,J., Barany,F. and Cao,W. (2001) Multiple cleavage activities of endonuclease V from Thermotoga maritima: recognition and strand nicking mechanism. Biochemistry, 40, 8738–8748. [DOI] [PubMed] [Google Scholar]
- 6.Yao M. and Kow,Y.W. (1997) Further characterization of Escherichia coli endonuclease V. Mechanism of recognition for deoxyinosine, deoxyuridine and base mismatches in DNA. J. Biol. Chem., 272, 30774–30779. [DOI] [PubMed] [Google Scholar]
- 7.Slupphaug G., Alseth,I., Eftedal,I., Volden,G. and Krokan,H.E. (1993) Low incorporation of dUMP by some thermostable DNA polymerases may limit their use in PCR amplifications. Anal. Biochem., 211, 164–169. [DOI] [PubMed] [Google Scholar]
- 8.Kikuchi M., Ohnishi,K. and Harayama,S. (1999) Novel family shuffling methods for the in vitro evolution of enzymes. Gene, 236, 159–167. [DOI] [PubMed] [Google Scholar]
- 9.Volkov A.A., Shao,Z. and Arnold,F.H. (2000) Recombination and chimeragenesis by in vitro heteroduplex formation and in vivo repair. Nucleic Acids Res., 27, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volkov A.A. and Arnold,F.H. (2000) Methods for in vitro DNA recombination and random chimeragenesis. Methods Enzymol., 328, 447–456. [DOI] [PubMed] [Google Scholar]