

Abstract
Following initiation of chromosomal replication in Escherichia coli, newly initiated origins (oriCs) are prevented from further initiations by a mechanism termed sequestration. During the sequestration period (which lasts about one-third of a cell cycle), the origins remain hemimethylated. The SeqA protein binds hemimethylated oriC in vitro. In vivo, the absence of SeqA causes overinitiation and strongly reduces the duration of hemimethylation. The pattern of immunostained SeqA complexes in vivo suggests that SeqA has a role in organizing hemimethylated DNA at the replication forks. We have examined the effects of overexpressing SeqA under different cellular conditions. Our data demonstrate that excess SeqA significantly increases the time oriC is hemimethylated following initiation of replication. In some cells, sequestration continued for more than one generation and resulted in inhibition of primary initiation. SeqA overproduction also interfered with the segregation of sister nucleoids and caused a delay in cell division. These results suggest that SeqA’s function in regulation of replication initiation is linked to chromosome segregation and possibly cell division.
Keywords: Escherichia coli/initiation/oriC/SeqA/sequestration
Introduction
In Escherichia coli, DNA replication initiates at the fixed chromosomal locus oriC, when the cellular potential to initiate is high (Messer and Weigel, 1996). Decisive for the initiation potential is the activity of the initiator protein, DnaA, the availability of which affects the time of initiation in the cell cycle (Løbner-Olesen et al., 1989; Boye et al., 1996). Binding of DnaA to several recognition sites within oriC (in concert with accessory factors) promotes duplex melting at the AT-rich 13mers (open complex formation) (Bramhill and Kornberg, 1988; Speck and Messer, 2001). Aided by DnaC, the DnaB helicase is loaded onto the separated strands (pre-priming complex), allowing entry of the replication machinery (Kornberg and Baker, 1992).
Initiation occurs once per cell cycle during a short time interval at all origins within a cell (Skarstad et al., 1986). Secondary initiations of the newly replicated origins are avoided by sequestration (Boye et al., 2000). During the sequestration period, the origins are made refractory to reinitiations for up to one-third of a cell cycle (Ogden et al., 1988; Campbell and Kleckner, 1990; Boye, 1991). The sequestered origins are hemimethylated, i.e. adenines of the GATC sites are methylated on only one strand. In general, GATC sites are methylated on both strands by Dam methylase. However, as the replication fork progresses, the newly replicated strands remain unmethylated for ∼1 min (Campbell and Kleckner, 1990; Lu et al., 1994). The prolonged duration of hemimethylation seen at oriC following initiation is presumed to involve binding of the sequestration factor SeqA to oriC, thereby preventing remethylation (Lu et al., 1994; Slater et al., 1995; Boye et al., 2000). Purified SeqA protein is able to inhibit replication initiated from oriC in vitro, both by interfering with the prepriming complex and by affecting DNA topology (Wold et al., 1998; Torheim and Skarstad, 1999; Taghbalout et al., 2000). In vitro, the SeqA protein binds specifically and with high affinity to two segments in oriC containing GATC methylation sites, especially when these sites are hemimethylated (Skarstad et al., 2000). Unmethylated DNA does not bind SeqA (Slater et al., 1995; Skarstad et al., 2000). The in vivo significance of SeqA binding to oriC is not clear, but in both seqA and dam mutant cells the replication pattern is asynchronous and the sequestration period is shortened or absent, respectively (Lu et al., 1994; Skarstad and Løbner-Olesen, 2002). Furthermore, provided there is a persisting high initiation potential, the same origins will, in the absence of SeqA, be refired repeatedly with no detectable time interval (eclipse) between successive initiations (von Freiesleben et al., 2000). In the presence of SeqA, the eclipse is highly influenced by the level of Dam methylase and can be extended or shortened by decreasing or increasing the Dam concentration, respectively (von Freiesleben et al., 2000). In vitro, however, the Dam protein exhibits lower binding affinity for the 13mer region of oriC than SeqA, and is unable to displace SeqA already bound to hemimethylated oriC (Kang et al., 1999).
SeqA protein has been visualized in fixed cells by immunofluorescence microscopy and in living cells as SeqA–green fluorescent protein (GFP) fusions (Hiraga et al., 1998; Onogi et al., 1999). In either case, SeqA forms discrete foci dependent on the presence of Dam. Surprisingly, foci of in situ hybridized oriC are rarely co-localized with SeqA foci during steady-state growth (Hiraga et al., 1998). The number of SeqA foci increases with increasing growth rate, and the appearance of foci shows dependence on DNA replication in the temperature-sensitive dnaA203 and dnaC2 mutants (Brendler et al., 2000; Hiraga et al., 2000). During a synchronous round of replication in the dnaA203 mutant, SeqA foci appear to arise in concert with new replication forks (Hiraga et al., 2000). Hence, SeqA foci are proposed to represent clusters of SeqA bound to hemimethylated DNA behind the ‘replication machines’ at the forks (Brendler et al., 2000; Hiraga et al., 2000). At the forks, SeqA may contribute to organization of the replicated DNA, and this could be an integral part of the process of sister chromosome segregation (Onogi et al., 1999; Brendler et al., 2000; Hiraga et al., 2000). Whether these functions are related to SeqA’s role in regulation of DNA replication initiation is not clear.
Here we demonstrate that an increased level of SeqA protein influences a set of cell cycle-related functions involving oriC sequestration, chromosome segregation and cell division.
Results
Overexpression of SeqA protein in dnaC2 cells extends the duration of the period of hemimethylation at oriC
In order to study the effect of increased intracellular amounts of SeqA on oriC sequestration, an isopropyl-β- d-thiogalactopyranoside (IPTG)-inducible seqA expression plasmid (pMAK7) was transformed into the MG1655 dnaC2 strain. The vector without the seqA gene (pFH2102) served as a control. The dnaC2 strain carries a mutant dnaC allele encoding a thermosensitive DnaC protein that is inactivated reversibly at the non-permissive temperature (38°C). When cultures of dnaC2/pMAK7 or dnaC2/pFH2102 cells growing exponentially at 30°C were shifted to 38°C, initiations ceased but ongoing replication and cell division continued. Thus, after 1 h, most cells ended up containing one or two fully replicated chromosomes (see next paragraph). By subsequently returning the cultures to 30°C, replication was initiated synchronously in all cells. A second round of initiations was prevented by shifting the cultures to 38°C after 8 min at 30°C (Figure 1A). Chromsomal DNA was isolated from the two cell cultures at the indicated times after shifting the temperature to 30°C (Figure 1B). The methylation status of oriC was determined by digestion with restriction enzymes. When the DNA is fully methylated, treatment with XhoI and HphI yields a 492 bp fragment harbouring oriC. Restriction enzyme HphI, whose recognition site (GGTGA) overlaps half a GATC site found within oriC, is methylation sensitive and will not cut the DNA when the site is methylated. Therefore, when oriC is fully methylated, HphI digestion leaves the 492 bp oriC fragment ‘uncut’ (Figure 1B). Unmethylated DNA extracted from dam– cells is fully digested at this site, generating the 234 bp ‘cut’ fragment (Figure 1B, rightmost lane). Following replication, one of the two hemimethylated oriC species generated will be unmethylated at the HphI site and therefore cleaved. Since only one daughter molecule receives an unmethylated HphI site, 50% ‘cut’ DNA (Figure 1B) represents 100% hemimethylated DNA.

Fig. 1. Overexpression of SeqA protein extends the period of hemimethylation at oriC. (A) Schematic representation of the temperature shifts at the indicated times during the experiment. (B) Exponentially growing cultures (30°C) of MG1655 dnaC2 cells harbouring pFH2102 (control) or pMAK7 (excess SeqA) were synchronized with respect to initiation for 1 h at 38°C before shifting the temperature to 30°C. IPTG (0.5 mM) was added to the cultures three mass doublings prior to synchronized initiation. Chromosomal DNA was extracted at the indicated times after returning the cultures to 30°C. The DNA was digested with XhoI and HphI, electrophoresed and hybridized to an oriC probe. The positions of cut and uncut fragments are indicated by arrows. The rightmost lane contains DNA isolated from a dam– strain which reveals the position of the cut fragment. (C) Quantification of the amount of hemimethylated oriC in (B). Squares represent dnaC2/pFH2102 and triangles dnaC2/pMAK7. The percentage of hemimethylated oriC is twice the percentage of the cut fragment in (B) since 50% of the hemimethylated fragments are digested. Time indicates minutes from shift to 30°C after 1 h synchronization at 38°C. (D) Western analyses of samples taken from the cultures at 0 and 20 min after returning the cultures to 30°C. A 1 µg aliquot of total protein of each sample was loaded.
The two cell cultures were induced with 0.5 mM IPTG three doubling times prior to synchronized initiation at 30°C (time 0), and the extent of SeqA overproduction was measured by immunoblotting (Figure 1D). At 0 and 20 min after returning the synchronized cultures to 30°C, the SeqA level was 3–4 times higher in the SeqA-overproducing cells compared with the control cells (Figure 1D).
The sequestration period following initiation is here defined as the period of time oriC is hemimethylated (Campbell and Kleckner, 1990; Lu et al., 1994). The duration of oriC hemimethylation in the control cells was found to be ∼10 min (one-third of the cell cycle under these conditions), calculated from the area under the curve in Figure 1C. In the cells overexpressing SeqA, the duration of hemimethylation was extended at least twice, relative to the control cells (Figure 1C). This result indicates that the level of SeqA present in the cells governs the duration of oriC sequestration. Furthermore, the result supports the idea that there is competition between SeqA inactivation and Dam methylation of oriC following initiation of replication (Kang et al., 1999).
SeqA overexpression delays cell division in dnaC2 cells
By flow cytometric analysis of samples collected from the two dnaC2 cultures studied in Figure 1, we could monitor changes in DNA content distributions following initiation of replication at 30°C. Culture samples were fixed in ethanol and used for both flow cytometry and SeqA immunofluoresence microscopy. After 1 h of incubation at 38°C, the control cells contained mainly one or two chromosomes per cell (Figure 2A). The broad left side of the two chromosome peak indicates that replication runout was not fully completed for all cells. When the control cells were returned to 30°C, replication was initiated synchronously and elongation proceeded at the same rate in all cells, seen as a rightward movement of the one and two chromosome peaks towards the two and four chromosome positions, respectively (Figure 2A–E). The population of one chromosome cells increased rapidly during the first 15 min of the experiment, and the two chromosome peak became sharper (Figure 2A–D). This indicates that cells containing two almost fully replicated chromosomes at the end of 38°C incubation completed previous rounds of replication and continued to divide at 30°C.

Fig. 2. DNA content histograms of dnaC2/pFH2102 (A–E), dnaC2/pMAK7 (F–J) and dnaC2/pSF19 (A, small panel). Samples for flow cytometry of dnaC2/pFH2102 and dnaC2/pMAK7 cells were collected during the temperature shift experiment in Figure 1 at the times indicated. In an identical control experiment, dnaC2/pSF19 cells were harvested for flow cytometry, at the time indicated.
In the culture of cells with excess SeqA, only a small population of cells contained one chromosome after 1 h at 38°C (Figure 2F), while most cells contained two chromosomes. The difference in chromosome content between the control and SeqA-overproducing cells must have arisen during induction of excess SeqA protein since the DNA content distribution of the two strains growing exponentially at 30°C without IPTG was nearly identical (not shown). The result suggests that cell division was inhibited or delayed in the presence of excess SeqA protein. To determine whether the inhibition of cell division was specific for SeqA overexpression and not a general effect of overexpressing any protein, the SeqA2 mutant protein was overexpressed 4- to 5-fold in the dnaC2/pSF19 cells, under experimental conditions identical to those above (pSF19 = pFH2102seqA2). SeqA2 was previously identified as being incapable of sequestering hemimethylated origins (von Freiesleben et al., 1994). Recently, SeqA2 has been found to be unable to bind hemimethylated DNA in vitro or form foci in vivo (Fossum et al., 2003). However, SeqA2 retains the ability to promote degradation of the DnaA204 mutant protein in dnaA204seqA2 cells (Fossum et al., 2003). This means that SeqA2 is not a totally inactive mutant protein, but was considered inactive enough to serve as control in the present experiment. As shown in Figure 2A, the chromosome content distribution in the SeqA2-overexpressing cells (Figure 2A, small panel) was nearly identical to that observed for the control cells, at the end of the 38°C incubation. This indicates that excess SeqA2 had little or no effect on cell division during the replication runout period at non-permissive temperature. Excess SeqA2 was, however, found to affect reinitiation and cell division at later stages (not shown). The reason for this presumably is that higher levels of SeqA2 lead to formation of mixed oligomer complexes that prevent wild-type SeqA activity (Fossum et al., 2003).
Cells with excess SeqA displayed a rightward shift of the chromosome peaks similar to that of the control cells, upon reinitiation of replication at 30°C (Figure 2F–J). However, the increase of the population of one chromosome cells was less pronounced compared with that of the control cells, indicating that cell division continued to be affected at permissive temperature (Figure 2F–J). In cultures grown at a higher growth rate, in richer medium (Luria broth), the difference in increase of the population of one chromosome cells between the control and SeqA-overproducing cells was even more distinct (not shown). Furthermore, the one and two chromosome peaks of cells with excess SeqA broadened with time after initiation (Figure 2I and J). The broadening of the one chromosome peak seems to be due to lack of initiation in some cells (rather than heterogeneous elongation rates), since a subpopulation of one chromosome cells can be distinguished at the 15–20 min time points (Figure 2I and J). The reason for this lack of initiation is possibly due to prolonged sequestration lasting more than one generation. Since the broadening of the one chromosome peak seems to be caused by lack of initiation, it is reasonable to suppose that this is also the case for the two chromosome peak. The two chromosome peak would in this case be composed of cells that either: (i) replicate from both origins; (ii) replicate from one of the two origins; or (iii) do not replicate at all because both origins remain sequestered. In conclusion, these results indicate that SeqA overproduction leads to inhibition of both cell division and primary initiation events of subsequent generations.
SeqA overexpression delays segregation of nucleoids
SeqA has been found to titrate negative supercoils and was proposed to be involved in DNA organization (Torheim and Skarstad, 1999). Also, SeqA is believed to take part in proper segregation of sister chromosomes (Hiraga et al., 1998; Brendler et al., 2000). We found here that excess SeqA inhibited cell division. This effect could be caused by segregation problems. To elucidate whether SeqA has a role in segregation, we therefore investigated how cells with different levels of SeqA manage to separate nucleoids. We made use of the wild-type strain CM735, harbouring pFH2102 (control) or pMAK7 (SeqA overexpression). Exponentially growing cultures were treated for two generations with cephalexin to prevent septum formation and for 15 min with chloramphenicol to condense the nucleoids (von Freiesleben et al., 2000). Overexpression of SeqA was induced by adding IPTG (0, 0.05 or 0.5 mM) to the cell cultures three generations prior to sampling. In CM735/pMAK7 cells cultured in the absence of IPTG, SeqA was 50% overexpressed due to leakage of the promotor, whereas the presence of 0.05 or 0.5 mM IPTG induced a 2- or 5–8-fold SeqA overexpression, respectively (determined by western analysis, not shown).
Light microscopy of Hoechst-stained control cells revealed two major populations of cells, containing four or eight nucleoids. We found that ∼80% of the four nucleoid cells and 70% of the eight nucleoid cells displayed well separated, condensed nucleoids, as shown in Figure 3A. The control cells that did not display four or eight separated nucleoids (20 and 30%, respectively), in most cases contained one or two unseparated nucleoids together with separated ones. Presumably, these cells were not deficient in segregation, but rather represent cells that were in the middle of the segregation process at the time of chloramphenicol addition.
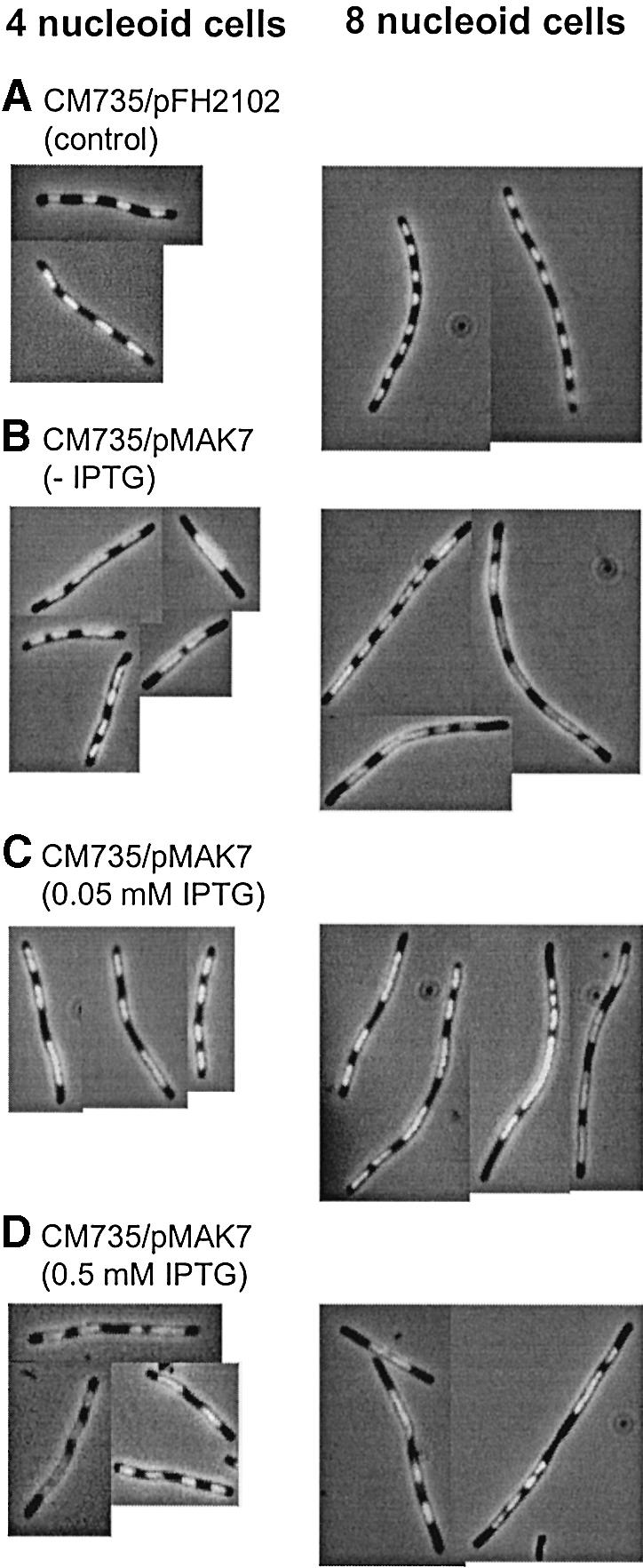
Fig. 3. Hoechst-stained CM735/pFH2102 (A) and CM735/pMAK7 cells (B–D). Cells were grown exponentially in glucose-CAA medium and then incubated with cephalexin (15 µg/ml) for two generations and with chloramphenicol (200 µg/ml) for 15 min. The CM735/pFH2102 (control) culture was incubated with 0.5 mM IPTG, and the CM735/pMAK7 cultures with 0, 0.05 or 0.5 mM IPTG, for three generations prior to sample harvesting.
In the CM735/pMAK7 cells, nucleoid segregation was significantly disturbed at all three levels of SeqA overexpression (Figure 3B–D). The appearance of aberrant segregation in the cells of the three groups was similar (Figure 3B–D). Already at 50% SeqA overexpression, the proportion of four and eight nucleoid cells displaying well separated nucleoids was decreased considerably to 45 and 20%, respectively (Figure 3B). When SeqA was overexpressed 2- or 5–8-fold, the proportion of cells with eight separated nucleoids was ∼15%, whereas the proportion of cells with four separated nucleoids remained at ∼45% (Figure 3C and D). To rule out the possibility that SeqA overproduction simply mediated non-specific effects on nucleoid segregation, DnaB protein was overproduced 2- to 3-fold in a wild-type strain. No effects on segregation could be detected (not shown).
In conclusion, successful segregation of sister nucleoids was highly sensitive to small increases in SeqA levels. The result indicates that excess SeqA delays or inhibits the nucleoid segregation process. This could in turn delay the next cell division, which was what we observed in Figure 2.
Excess SeqA adds to existing SeqA complexes and prevents segregation of SeqA foci in dnaC2 cells
SeqA focus formation following initiation of replication was visualized by immunofluorescence microscopy in samples collected during the temperature shift experiment in Figures 1 and 2. SeqA foci were visible even after 1 h incubation at non-permissive temperature (Figure 4A and D ). The corresponding flow cytometric data of the control cells show that at the end of the 38°C incubation most cells contained one or two apparently complete chromosomes. A smaller population of cells had not finished replication and contained between one and two chromosomes (Figure 2A). Some of the SeqA foci seen in these cells can therefore be explained by ongoing DNA replication. However, considering the flow cytometry data, most cells should have completed replication, suggesting that some fluorescent SeqA foci represent non-functional complexes or SeqA accumulations left behind after previous replication. An indication of disappearing foci might be the variation of focus intensity in individual cells, seen in Figure 4A.

Fig. 4. Immunofluorescent SeqA foci in dnaC2/pFH2102 (A–C) and dnaC2/pMAK7 (D–F) cells. Cells were from the same samples as those analysed by flow cytometry in Figure 2 (0, 10 and 20 min).
Since the control cells after 1 h at 38°C (Figure 4A) in general have a maximum of two active replication forks, the number of foci per cell fits best with an assumption of one SeqA focus per fork. In Figure 4A, smaller cells with one or two closely situated foci most probably represent cells containing one chromosome. Shortly after replication was reinitiated in such cells, two closely located foci were observed, now probably representing the two new forks (Figure 4B). Larger cells with two foci spaced further apart (Figure 4A) must represent cells with two chromosomes that have been separated. When these cells reinitiate replication, two pairs of closely situated SeqA foci appear, preferentially at each end of the cell (Figure 4B). Sometimes one or two additional foci were found in between the two pairs, indicating finishing replication from earlier rounds or non-functional complexes (Figure 4B). Large cells containing more than two foci were also found in Figure 4A. Most of them, however, fall into the categories described above following initiation of replication, even though rare and extremely filamentous cells with aberrant focus patterns were seen. When replication had been allowed to proceed for 20 min, the SeqA foci in individual cells had moved further apart (Figure 4C). Some of the larger cells still contained additional foci in between the two pairs (Figure 4C). Considering the appearance of the flow cytometry histogram (Figure 2E), these foci are now less likely to be explained by ongoing replication from earlier rounds since the ‘former two chromosome’ peak was sharp, indicating completion of earlier rounds.
In cells with excess SeqA, foci appeared larger (Figure 4D–F). The extra SeqA molecules seemed to add to existing complexes rather than building more complexes or being freely dissociated in the cell (Figure 4D). When replication was reinitiated in these cells, the foci became more distinct but, compared with the control cells, the larger SeqA foci were more clustered and cells with pairs of two separated foci were rarely observed (Figure 4E). At 20 min after initiation of replication, pairs of foci were observed, but seemed to be separated to a lesser extent compared with the pairs in the control cells (compare Figure 4F with B and C). This indicates that either the segregation of nucleoids or the segregation of foci is delayed by the excess SeqA.
The number of SeqA foci in wild-type cells equals about half the number of forks
The above experiments indicate that one SeqA focus is formed per replication fork. However, the non-physiological initiation event in temperature shift experiments using temperature-sensitive mutants might not be entirely representative of what occurs in wild-type cells during initiation of replication. Therefore, we also performed SeqA immunofluorescence microscopy in exponentially growing wild-type cells. Samples for SeqA immunofluorescence staining and flow cytometry were taken simultaneously. Growing in glucose-CAA medium at 30°C, MG1655 cells replicated mainly from four and eight origins (70 and 30% of the population, respectively), determined from DNA content histograms of rifampicin-treated cells (not shown). This means that initiation of replication occurs at four origins in the later part of the cell cycle. A dividing cell therefore contains eight new forks and four old forks, while a newborn cell contains four new forks and two old forks. The number of SeqA foci seen in individual wild-type cells in Figure 5A indicates that there are too few foci to ascribe one focus to each proceeding fork. Cells have either two, three, two pairs of two, or two clusters of 3–4 foci per cell, co-localized with the nucleoids (Figure 5A and B). Under the same (permissive) growth conditions, similar results were obtained for the dnaC2 strain (not shown). Thus, the data suggest that the number of SeqA foci in wild-type cells equals about half the number of forks. There may be two explanations for why there are fewer foci than replication forks: (i) there exist higher forms of coordination between pairs of replication forks during normal growth than what is seen in the dnaC2 mutant during temperature shift experiments; or (ii) the fine details of SeqA complexes in exponentially growing cells are not resolved.
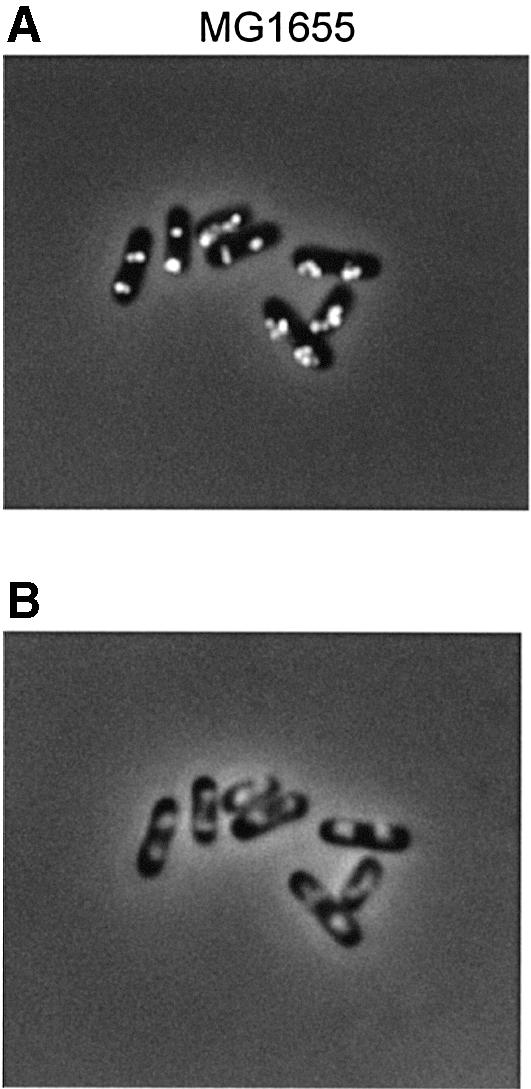
Fig. 5. Immunofluorescent SeqA foci (A) and Hoechst-stained nucleoids (B) in the same cells. Wild-type (MG1655) cells were grown exponentially in glucose-CAA medium to OD450 = 0.15. Samples were fixed in ethanol and subjected to immunofluorescence microscopy.
Discussion
The sequestration period
We show here that dnaC2 cells overexpressing SeqA protein display an extended period of oriC hemimethylation following initiation of chromosomal replication. It was found previously that the oriC hemimethylation period was shorter when seqA was deleted or Dam methylase was overproduced (Campbell and Kleckner, 1990; Lu et al., 1994), suggesting competition between Dam and SeqA for the same substrate (Kang et al., 1999). In both these cases, the newly replicated origins apparently were released from sequestration soon after initiation, while the initiation potential was still high. Secondary initiations on newly fired origins therefore occurred, causing an asynchronous replication phenotype (Boye and Lobner-Olesen, 1990; Lu et al., 1994). Dam-deleted cells, which lack sequestration altogether, presumably initiate chromosomal replication asynchronously for the same reason (Boye and Lobner-Olesen, 1990). Our data strongly suggests that when SeqA is overexpressed, remethylation of oriC is delayed and thus the sequestration period is prolonged. This result supports the idea that there is competition between inactivation by SeqA and methylation by Dam at newly initiated origins. Hence, origin sequestration is driven in opposite directions by the activities of Dam methylase and SeqA.
Recently, von Freiesleben et al. (2000) reported that excess SeqA does not extend the length of the eclipse, i.e. the minimal time between successive initiations on the same origin. Newly formed origins in the temperature-sensitive dnaA46 mutant were found to be inert to reinitiations for 25–30 min, irrespective of the presence of excess SeqA or not. This is somewhat surprising considering our study of sequestration in dnaC2 cells. However, the eclipse and sequestration periods are measured in different ways and obtained in different mutants, and thus the results are not directly comparable. In agreement with our data, SeqA overexpression was found to inhibit cell division and consequently mediate increased cell mass (von Freiesleben et al., 2000).
The sequestration process and downstream events
It is reasonable to assume that the SeqA-mediated sequestration of oriC and the binding of SeqA to other hemimethylated areas occur mechanistically in the same way. Although fully methylated oriC and SeqA foci are not co-localized (Hiraga et al., 1998), hemimethylated oriC may well be situated within foci. Since the binding of SeqA to GATC-containing regions is highly cooperative (Slater et al., 1995; Brendler and Austin, 1999), areas with many GATC sites, like oriC, might stay bound by SeqA complexes for longer periods than areas with few GATC sites. Thus, the cooperative interaction between SeqA molecules bound at the many GATC sites in oriC could explain the prolonged duration of hemimethylation seen at newly fired origins. Likewise, the large number of GATC sites in the dnaA gene would explain the prolonged state of hemimethylation here also.
We found that overproduction of the SeqA protein further increased the duration of oriC hemimethylation (Figure 1) and also caused SeqA foci to become larger (Figure 4). Taken together, these two observations may indicate that at higher SeqA concentrations, larger regions of hemimethylated DNA in general are held within larger SeqA structures, and that SeqA is displaced more slowly before giving access to Dam methylase.
The subsequent cell cycle events, sister chromosome segregation and cell division, were also inhibited or delayed due to SeqA overproduction (Figures 2 and 3). Following synchronized initiation of replication in dnaC2 cells, SeqA foci were found to separate and move apart (Figure 4). This movement of SeqA foci, which was prevented or delayed in SeqA-overproducing cells, may reflect either replisome movement or ongoing segregation of sister nucleoids. The building of SeqA complexes at GATC-containing sites might directly contribute to organization of newly replicated hemimethylated DNA into higher order structures. These structures could be important for the segregation process. Hence, the aberrant nucleoid distribution seen in SeqA-overproducing cells in Figure 3B–D could be explained by the presence of large SeqA complexes interfering with the assembly or function of a segregation apparatus. This would in turn delay downstream events including cell division. The results thus suggest that SeqA-mediated sequestration of hemimethylated regions is closely linked to nucleoid segregation and cell division. An alternative explanation for why SeqA overexpression affects nucleoid segregation and cell division should, however, also be considered. SeqA protein previously was found to be a transcriptional activator (Slominska et al., 2001). It could thus be that excess SeqA influences transcription of genes involved in nucleoid segregation and cell division.
Although SeqA foci in synchronized dnaC2 cells were found to move apart, it is not clear whether foci in wild-type cells must move. It may be that a wild-type SeqA focus stays in the same location during an entire round of replication and disappears thereafter. The very rapid movement of SeqA foci (Ohsumi et al., 2001; Sunako et al., 2001) seen in some experiments alternatively could represent the disappearance of old foci and appearance of new foci. It is also not clear what parts of the chromosome are contained within a SeqA focus. Though it is reasonable to assume that these foci represent organized regions of newly replicated DNA, it is unclear how many forks, and which forks, are included in each focus. The results presented here show that a SeqA focus in wild-type cells (or dnaC2 cells growing at 30°C) contains at least two forks (Figure 5). However, when replication is restarted in dnaC2 cells following synchronization at 38°C, each focus seems to contain only one fork (Figure 4). The reason for this difference is not known. The number of forks per foci found in wild-type cells indicates that an organization of replisomes into replication factories, containing at least two replication forks, as reported for Bacillus subtilis (Lemon and Grossman, 1998), is likely to also be true for E.coli wild-type cells. Synchronized dnaC2 cells seem to have lost this level of organization and apparently replicate with one replisome per fork. These results therefore suggest that such a higher order organization of replisomes is dependent on a proper coupling of cell cycle events with cell growth.
Materials and methods
Bacterial strains, plasmids and media
All strains used were of E.coli K-12. The dnaC2 allele has been transduced from the PC2 strain (Carl, 1970) into the wild-type MG1655 (F–, λ–, rph-1) strain which provides a clearer experimental system than PC2 (Withers and Bernander, 1998). The MG1655 dnaC2 (F–, λ–, rph-1, thr::Tn10 dnaC2) strain was transformed with the IPTG-inducible expression plasmids pMAK7 (ori-pBR322, placPA1-04/03-seqA, bla, lacI) or pSF19 (ori-pBR322, placPA1-04/03-seqA2, bla, lacI) or the vector without seqA genes, pFH2102 (ori-pBR322, placPA1-04/03, bla, lacI) (von Freiesleben et al., 2000). The CM735 (metE46, trp-3, his-4, thi-1, galK2, lacY1 or lacZ4, mtl-1, ara-9, tsx-3, ton-1, rps-8 or rps-9, supE44 λ–) (Hansen and von Meyenburg, 1979; Torheim et al., 2000) wild-type strain transformed with pFH2102 or pMAK7 was used for studies of nucleoid segregation in the presence of excess SeqA. The MC1061 [araD139, Δ(ara leu)7697, lacX74, galU, galK, hsr, strA] wild-type strain carrying pINGK vector (control) or the corresponding DnaB expression plasmid, pINB (Allen and Kornberg, 1991; Skarstad and Wold, 1995), was utilized for DnaB overexpression. Growth was in AB minimal medium (Clark and Maaløe, 1967) supplemented with 1 µg/ml thiamine, 0.2% glucose, 0.5% casamino acids (glucose-CAA medium). To cultures of MG1655 derivatives 100 µg/ml uridine and 10 µg/ml threonine were also added, whereas 50 µg/ml tryptophan were added to cultures of CM735 derivatives. Mass growth was monitored by measuring optical density at 450 nm (OD450).
Temperature shift experiments
Overnight cultures of dnaC2/pFH2102, dnaC2/pMAK7 and dnaC2/pSF19 were diluted 1/1000 in glucose-CAA medium, grown exponentially to OD450 ∼0.07 at 30°C (permissive temperature) before shifting to 38°C (non-permissive temperature) for 1 h. Replication was initiated subsequently in the dnaC2/pFH2102 and dnaC2/pMAK7 cells by returning the cultures to 30°C by adding appropriate amounts of 4°C medium. After 8 min at 30°C, the cultures were shifted to 38°C to prevent initiation of further rounds of replication. All cell cultures were induced by addition of 0.5 mM IPTG shortly before shifting to 38°C (∼3 mass doublings prior to reinitiation of replication at 30°C).
Isolation of chromosomal DNA
Samples of 50 ml of cell culture taken during the temperature shift experiment were incubated for 10 min at about –10°C (ice-water bath, saturated with NaCl) to stop cellular reactions. Cells were pelleted by centrifugation at 4°C, resuspended in TE and lysed by adding SDS buffer (1.4% SDS, 4 mM EDTA). The mixture was incubated at 65°C for 5 min. NaCl (0.2 M) and a 0.7 vol. of isopropanol were then added for precipitation of DNA. The resulting DNA pellet was resuspended in TE, RNase (100 µg/ml) was added and the solution was incubated for 1 h at 37°C. The DNA solution was then treated with proteinase K (50 µg/ml) overnight at 37°C. The remaining proteins were precipitated from the solution by adding a one-third volume of protein precipitation solution (Promega), and the DNA was subsequently precipitated from the supernatant by adding a 0.7 vol. of isopropanol. The resulting DNA pellet was washed in 70% ethanol and resuspended in TE.
Digestion of chromosomal DNA with restriction enzymes and Southern blotting
Restriction enzyme digestions to identify hemimethylated DNA were performed by incubating 1 µg of chromosomal DNA and 3 U of HphI and XhoI restriction enzymes (New England Biolabs) for 1.5 h at 37°C (Campbell and Kleckner, 1990). The restriction digestions were run on 1.7% agarose gels. The DNA in the gel was denatured and neutralized with 0.37% HCl for 20 min, denaturing buffer (0.5 M NaOH, 1.5 M NaCl) for 40 min and neutralization buffer (0.5 M Tris–HCl pH 7.2, 3 M NaCl) for 1 h. The gel was washed twice with dH2O between each treatment. The DNA was then transferred overnight by capillary action to a nylon membrane (Hybond N+ Membrane, Amersham) in 10× SSC. The DNA was fixed to the membrane by baking at 80°C for 1 h. The membrane was then pre-hybridized for 2 h at 60°C in a pre-hybridization buffer (6× SSC, 5× Denhardt, 0.5% SDS, 100 µg/ml sonicated salmon sperm DNA). The probe used for hybridization was a purified PCR fragment (225 bp) of the oriC region spanning bp –41 to 184. The oriC probe (25 ng) was labelled with [α-32P]dCTP (50 µCi, 3000 Ci/mmol) using Random Primer Labelling of DNA (Promega). The labelled probe was added to the hybridization buffer (6× SSC, 5× Denhardt, 0.5% SDS, 100 µg/ml sonicated salomon sperm DNA, 20 mM EDTA, [α-32P]dCTP-labelled oriC probe) and the membrane was hybridized overnight at 60°C. The membrane was washed as follows: wash I (2× SSC, 0.5% SDS) for 5 min, wash II (2× SSC, 0.1% SDS) for 15 min, and wash III (0.1× SSC, 0.5% SDS) three times for 30 min. The membrane was scanned on a Storm 840 (Molecular Dynamics) and quantified using IMAGEQUANT software (Molecular Dynamics).
SeqA western blotting
Samples of 3 ml of cell culture were harvested during the temperature shift experiment (OD450 ∼0.20–0.25) and in the nucleoid segregation experiment (OD450 ∼0.28). Sample preparation and procedures were as previously described, using rabbit antiserum raised against SeqA protein or DnaB protein (Torheim et al., 2000).
Flow cytometry
Aliquots of 1 ml of exponentially growing cell cultures were collected at OD450 = 0.15. Volumes of flow samples collected at other ODs, e.g. during the temperature shift experiments, were adjusted to contain the same amount of cells. For origin content determination, rifampicin (300 µg/ml) and cephalexin (10 µg/ml) were added to samples and incubation continued for 3 h at 30°C to complete ongoing rounds of replication. The samples were washed in filtered TE (10 mM Tris–HCl pH 7.5, 1 mM EDTA) and fixed in 70% ethanol-TE buffer. Procedures for flow cytometry were as previously described (Torheim et al., 2000).
Nucleoid segregation
Overnight cultures of CM735 carrying pFH2102 or pMAK7 were diluted 1/1000 in glucose-CAA medium and grown exponentially to OD450 ∼0.035 at 37°C. The cultures were then grown in the presence of IPTG (0, 0.05 or 0.5 mM) for three generations, cephalexin (15 µg/ml) for two generations and chloramphenicol (200 µg/ml) for 15 min, prior to sampling. The MC1061 strain carrying pINGK or pINB was cultured as above, apart from that DnaB expression was induced by adding 0.2% arabinose instead of IPTG, one generation prior to sampling to obtain 3- to 4-fold overexpression. Samples of 1 ml were harvested for microscopy. Cells were pelleted by centrifugation and fixed in 70% ethanol-TE buffer. Samples of 3 ml were also harvested for western analysis. The fractions of cell populations displaying similar nucleoid phenotypes were calculated after counting >50 cells of each phenotype.
Immunofluorescence microscopy
The procedure was carried out as described (Hiraga et al., 1998), with the following modifications: ethanol-fixed cell samples were used instead of 80% methanol-suspended cells. The polyclonal first step antibody against SeqA was pre-treated with cell extract from strain CM735ΔseqA. The Cy3-labelled second step antibody (goat anti-rabbit IgG, Amersham) was not pre-treated with an E.coli cell extract. The immunostained sample on the glass slide was stained with Hoechst buffer (1.5 µg/ml Hoechst 33258, 40% glycerol in phosphate-buffered saline pH 7.5) instead of a 4′,6-diamidino-2-phenylindole (DAPI)-containing buffer.
Visualization of immunostained cells was performed using a Zeiss Axioplan2 phase-contrast/fluorescence microscope equipped with a 63× objective and a BP546/12 excitation filter. DNA was visualized with a BP365/12 excitation filter. Pictures were taken using a MicroMax CCD camera (Princeton Instruments Inc.) that was connected to a computerized image analysis system (Axiovision2 Multichannel, Zeiss). The optical images, which showed cell outlines, were merged with the fluorescent images, that showed fluorescent SeqA proteins or DNA.
Acknowledgments
Acknowledgements
We are grateful to Helen C.Withers for kindly providing the MG1655 dnaC2 strain. We thank Kirsti Solberg of the Institute for Cancer Research Flow Cytometry Core Facility and Anne Wahl for excellent technical assistance, and Erik Boye, Anders Løbner-Olesen and Flemming Hansen for critical reading of the manuscript. This work was supported by the Norwegian Research Council.
References
- Allen G.-C. Jr and Kornberg,A. (1991) Fine balance in the regulation of DnaB helicase by DnaC protein in replication in Escherichia coli. J. Biol. Chem., 266, 22096–22101. [PubMed] [Google Scholar]
- Boye E. (1991) The hemimethylated replication origin of Escherichia coli can be initiated in vitro. J. Bacteriol., 173, 4537–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye E. and Lobner-Olesen,A. (1990) The role of dam methyltransferase in the control of DNA replication in E.coli. Cell, 62, 981–989. [DOI] [PubMed] [Google Scholar]
- Boye E., Stokke,T., Kleckner,N. and Skarstad,K. (1996) Coordinating DNA replication initiation with cell growth: differential roles for DnaA and SeqA proteins. Proc. Natl Acad. Sci. USA, 93, 12206–12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye E., Lobner-Olesen,A. and Skarstad,K. (2000) Limiting DNA replication to once and only once. EMBO Rep., 1, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramhill D. and Kornberg,A. (1988) Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E.coli chromosome. Cell, 52, 743–755. [DOI] [PubMed] [Google Scholar]
- Brendler T. and Austin,S. (1999) Binding of SeqA protein to DNA requires interaction between two or more complexes bound to separate hemimethylated GATC sequences. EMBO J., 18, 2304–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendler T., Sawitzke,J., Sergueev,K. and Austin,S. (2000) A case for sliding SeqA tracts at anchored replication forks during Escherichia coli chromosome replication and segregation. EMBO J., 19, 6249–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.L. and Kleckner,N. (1990) E.coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell, 62, 967–979. [DOI] [PubMed] [Google Scholar]
- Carl P.L. (1970) Escherichia coli mutants with temperature-sensitive synthesis of DNA. Mol. Gen. Genet., 109, 107–122. [DOI] [PubMed] [Google Scholar]
- Clark D.J. and Maaløe,O. (1967) DNA replication and the division cycle in Escherichia coli. J. Mol. Biol., 23, 99–112. [Google Scholar]
- Fossum S., Søreide,S. and Skarstad,K. (2003) Lack of SeqA focus formation, specific DNA binding and proper protein multimerization in the Escherichia coli sequestration mutant seqA2. Mol. Microbiol., in press. [DOI] [PubMed] [Google Scholar]
- Hansen F.G. and von Meyenburg,K. (1979) Characterization of the dnaA, gyrB and other genes in the dnaA region of the Escherichia coli chromosome on specialized transducing phages λ tna. Mol. Gen. Genet., 175, 135–144. [DOI] [PubMed] [Google Scholar]
- Hiraga S., Ichinose,C., Niki,H. and Yamazoe,M. (1998) Cell cycle-dependent duplication and bidirectional migration of SeqA-associated DNA–protein complexes in E.coli. Mol. Cell, 1, 381–387. [DOI] [PubMed] [Google Scholar]
- Hiraga S., Ichinose,C., Onogi,T., Niki,H. and Yamazoe,M. (2000) Bidirectional migration of SeqA-bound hemimethylated DNA clusters and pairing of oriC copies in Escherichia coli. Genes Cells, 5, 327–341. [DOI] [PubMed] [Google Scholar]
- Kang S., Lee,H., Han,J.S. and Hwang,D.S. (1999) Interaction of SeqA and Dam methylase on the hemimethylated origin of Escherichia coli chromosomal DNA replication. J. Biol. Chem., 274, 11463–11468. [DOI] [PubMed] [Google Scholar]
- Kornberg A. and Baker,T.A. (1992) DNA Replication, 2nd edn. Freeman, New York, NY.
- Lemon K.P. and Grossman,A.D. (1998) Localization of bacterial DNA polymerase: evidence for a factory model of replication. Science, 282, 1516–1519. [DOI] [PubMed] [Google Scholar]
- Løbner-Olesen A., Skarstad,K., Hansen,F.G., von Meyenburg,K. and Boye,E. (1989) The DnaA protein determines the initiation mass of Escherichia coli K-12. Cell, 57, 881–889. [DOI] [PubMed] [Google Scholar]
- Lu M., Campbell,J.L., Boye,E. and Kleckner,N. (1994) SeqA: a negative modulator of replication initiation in E.coli. Cell, 77, 413–426. [DOI] [PubMed] [Google Scholar]
- Messer W. and Weigel,C. (1996) Initiation of chromosome replication. In Neidhart,F.C. et al. (eds), Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd edn. ASM Press, Washington, DC, pp. 1579–1601.
- Ogden G.B., Pratt,M.J. and Schaechter,M. (1988) The replicative origin of the E.coli chromosome binds to cell membranes only when hemimethylated. Cell, 54, 127–135. [DOI] [PubMed] [Google Scholar]
- Ohsumi K., Yamazoe,M. and Hiraga,S. (2001) Different localization of SeqA-bound nascent DNA clusters and MukF–MukE–MukB complex in Escherichia coli cells. Mol. Microbiol., 40, 835–845. [DOI] [PubMed] [Google Scholar]
- Onogi T., Niki,H., Yamazoe,M. and Hiraga,S. (1999) The assembly and migration of SeqA–Gfp fusion in living cells of Escherichia coli. Mol. Microbiol., 31, 1775–1782. [DOI] [PubMed] [Google Scholar]
- Skarstad K. and Løber-Olesen,A. (2002) Stable coexistence of separate replicons in Escherichia coli is dependent on once-per-cell-cycle initiation. EMBO J., 22, 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K. and Wold,S. (1995) The speed of the Escherichia coli fork in vivo depends on the DnaB:DnaC ratio. Mol. Microbiol., 17, 825–831. [DOI] [PubMed] [Google Scholar]
- Skarstad K., Boye,E. and Steen,H.B. (1986) Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J., 5, 1711–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K., Lueder,G., Lurz,R., Speck,C. and Messer,W. (2000) The Escherichia coli SeqA protein binds specifically and co-operatively to two sites in hemimethylated and fully methylated oriC. Mol. Microbiol., 36, 1319–1326. [DOI] [PubMed] [Google Scholar]
- Slater S., Wold,S., Lu,M., Boye,E., Skarstad,K. and Kleckner,N. (1995) E.coli SeqA protein binds oriC in two different methyl-modulated reactions appropriate to its roles in DNA replication initiation and origin sequestration. Cell, 82, 927–936. [DOI] [PubMed] [Google Scholar]
- Slominska M., Wegrzyn,A., Konopa,G., Skarstad,K. and Wegrzyn,G. (2001) SeqA, the Escherichia coli origin sequestration protein, is also a specific transcription factor. Mol. Microbiol., 40, 1371–1379. [DOI] [PubMed] [Google Scholar]
- Speck C. and Messer,W. (2001) Mechanism of origin unwinding: sequential binding of DnaA to double- and single-stranded DNA. EMBO J., 20, 1469–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunako Y., Onogi,T. and Hiraga,S. (2001) Sister chromosome cohesion of Escherichia coli. Mol. Microbiol., 42, 1233–1241. [DOI] [PubMed] [Google Scholar]
- Taghbalout A., Landoulsi,A., Kern,R., Yamazoe,M., Hiraga,S., Holland,B., Kohiyama,M. and Malki,A. (2000) Competition between the replication initiator DnaA and the sequestration factor SeqA for binding to the hemimethylated chromosomal origin of E.coli in vitro. Genes Cells, 5, 873–884. [DOI] [PubMed] [Google Scholar]
- Torheim N.K. and Skarstad,K. (1999) Escherichia coli SeqA protein affects DNA topology and inhibits open complex formation at oriC. EMBO J., 18, 4882–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torheim N.K., Boye,E., Lobner-Olesen,A., Stokke,T. and Skarstad,K. (2000) The Escherichia coli SeqA protein destabilizes mutant DnaA204 protein. Mol. Microbiol., 37, 629–638. [DOI] [PubMed] [Google Scholar]
- von Freiesleben U., Rasmussen,K.V. and Schaechter,M. (1994) SeqA limits DnaA activity in replication from oriC in Escherichia coli. Mol. Microbiol., 14, 763–772. [DOI] [PubMed] [Google Scholar]
- von Freiesleben U., Krekling,M.A., Hansen,F.G. and Lobner-Olesen,A. (2000) The eclipse period of Escherichia coli. EMBO J., 19, 6240–6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers H.L. and Bernander,R. (1998) Characterization of dnaC2 and dnaC28 mutants by flow cytometry. J. Bacteriol., 180, 1624–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold S., Boye,E., Slater,S., Kleckner,N. and Skarstad,K. (1998) Effects of purified SeqA protein on oriC-dependent DNA replication in vitro. EMBO J., 17, 4158–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]