

Abstract
The methylation of lysine residues of histones plays a pivotal role in the regulation of chromatin structure and gene expression. Here, we report two crystal structures of SET7/9, a histone methyltransferase (HMTase) that transfers methyl groups to Lys4 of histone H3, in complex with S-adenosyl-l-methionine (AdoMet) determined at 1.7 and 2.3 Å resolution. The structures reveal an active site consisting of: (i) a binding pocket between the SET domain and a c-SET helix where an AdoMet molecule in an unusual conformation binds; (ii) a narrow substrate-specific channel that only unmethylated lysine residues can access; and (iii) a catalytic tyrosine residue. The methyl group of AdoMet is directed to the narrow channel where a substrate lysine enters from the opposite side. We demonstrate that SET7/9 can transfer two but not three methyl groups to unmodified Lys4 of H3 without substrate dissociation. The unusual features of the SET domain-containing HMTase discriminate between the un- and methylated lysine substrate, and the methylation sites for the histone H3 tail.
Keywords: compact form of AdoMet/9 histone methyltransferase/post-SET helix/SET7/SET domain/substrate-specific channel
Introduction
In eukaryotes, dynamic transition between an extended, transcriptionally-active euchromatin structure and a compact, transcriptionally-silent heterochromatin structure is critical for the regulation of gene expression (van Holde, 1988; Wolffe, 2001). Although it is unclear how the chromatin structure is regulated, a large body of evidence suggests that a variety of post-translational modifications within the basic N-terminal tails of the histones play an important role in alteration of the chromatin structure, and thus the regulation of gene expression (Spencer and Davie, 1999; Strahl and Allis, 2000; Turner, 2000; Zhang and Reinberg, 2001).
Several post-translational modifications of the N-terminal tails of the histones including acetylation, phosphorylation, ubiquitylation and methylation have been reported (van Holde, 1988). The distinct covalent modifications of histone tails are expected to generate a label that can regulate the interaction with chromatin-associated proteins or protein complexes, which in turn control chromatin function (Strahl and Allis, 2000; Jenuwein and Allis, 2001).
Histone methylation occurs on both lysine and arginine residues of histones H3 (Rea et al., 2000; Ma et al., 2001) and H4 (Strahl et al., 2001; Wang et al., 2001b). In histone H3, the lysine methylation is observed on residues 4, 9, 27 and 36, whereas in histone H4, only Lys20 is methylated (van Holde, 1988). Compared with other modifications, the methylation of histone lysines shows some unique features. First, it is an irreversible process that results in a long-term epigenetic label (Byvoet et al., 1972; Duerre and Lee, 1974). This label is required to maintain specific gene expression programmes, and to inherit cell-type identities (Jenuwein, 2001). Furthermore, it allows the distinct chromatin structure of the centromeres to be propagated faithfully during cell division (Karpen and Allshire, 1997). Secondly, the gene regulatory activity mediated by the methylation of histone lysines is determined not only by the location of methylation site(s) but also by the precise methylation status (e.g. mono-, di- or trimethylation; Santos-Rosa et al., 2002).
SUV39H1 protein (Tschiersch et al., 1994) and its homologues were the first identified histone methyltransferases (HMTases) that can specifically methylate Lys9 of histone H3 (H3-K9; Rea et al., 2000). This process is important for the recruitment of the heterochromatin protein 1 (HP1; Bannister et al., 2001; Jacobs et al., 2001; Lachner et al., 2001) and the establishment of heterochromatin (Nakayama et al., 2001; Schotta et al., 2002). In addition to SUV39H1, G9a (Tachibana et al., 2001) and ESET/SETDB1 (Yang et al., 2002; Schultz et al., 2002) have been reported to methylate H3-K9.
The methylation at Lys4 of histone H3 (H3-K4) is another well-studied example of lysine methylation of histones (Wang et al., 2001a; Nishioka et al., 2002). In contrast to H3-K9 methylation that is correlated with transcription repression, H3-K4 methylation is enriched in transcriptionally-active euchromatin (Litt et al., 2001; Boggs et al., 2002; Zegerman et al., 2002). SET7/9 is a recently identified HMTase that methylates H3-K4 (Wang et al., 2001a; Nishioka et al., 2002). The H3-K4 methylation by SET7/9 inhibits the association of the NuRD deacetylase complex with the H3 tail. Furthermore, the methylation on H3-K4 by SET7/9 impairs the SUV39H1-mediated methylation on H3-K9, preventing the placement of a silencing label that is recognized by HP1 (Wang et al., 2001a; Nishioka et al., 2002).
Almost all lysine HMTases contain a highly-conserved SET domain (Jenuwein et al., 1998; Kouzarides et al., 2002). Several lines of evidence suggest that methylation of histone lysine residues is mediated through the SET domain. The pre- and post-SET regions are also required for the activity of the enzyme (Rea et al., 2000). The importance of the SET domain for HMTase activity suggests that the mechanism of methyl transfer to the lysine residue is likely to be conserved among the SET domain-containing family of enzymes (Wang et al., 2001a; Nishioka et al., 2002). The biological importance of the SET domain is underscored by the fact that several SET-containing HMTases are involved in the development of cancer in a positive or negative manner (Peters et al., 2001; Schneider et al., 2002)
Recently, three structures of SET domain-containing methyltransferases (MTases), namely SET7/9 (Wilson et al., 2002), DIM-5 (Zhang et al., 2002) and Rubisco large subunit lysine MTase (Rubisco LSMT; Trievel et al., 2002), have been reported. Each structure has the well-described unique feature of a SET domain. Also, based on biochemical and structural analyses, an active site for each MTase has been proposed. However, in two cases, SET7/9 (Wilson et al., 2002) and Rubisco LSMT (Trievel et al., 2002), significant differences are observed in the cofactor orientation. In addition, the post-SET region that is important for the activity of MTase is well defined only in the Rubisco LSMT structure (Trievel et al., 2002). Thus, further structural characterizations are required to resolve these issues.
To understand the roles of the SET domain and c-SET region in the lysine methyl transfer reaction, we have determined the crystal structure of SET7/9 bound to an S-adenosyl-l-methionine (AdoMet) molecule (Figure 1A). (We refer to the regions immediately prior to and after the SET domain in SET7/9 as n-SET and c-SET, respectively, throughout the text since these regions lack the sequence similarity compared with cysteine-rich pre- and post-SET domains of SUV39H1 and its homologues.) We show that the AdoMet molecule adopts an unusual conformation in the pocket formed between the SET and c-SET domains of SET7/9. We also demonstrate that SET7/9 can transfer two but not three methyl groups to unmethylated H3-K4 without dissociation of a substrate, supporting the importance of a channel formed at the active site for the selection of the lysine substrate.


Fig. 1. (A) Stereo diagram of the 1.7 Å resolution electron density map around the methyl group of AdoMet. The |Fo – Fc| simulated annealing omit map contoured at the 3σ level, with AdoMet and neighbouring residues omitted for map calculation. The refined 1.7 Å structure is superimposed on the map. (B) Sequence alignment of SET7/9, human SUV39H1 and the yeast homologue Clr4. Residues identical between SET7/9, SUV39H1 and Clr4 are highlighted in yellow. The bar graph below the sequence alignment indicates the degree of identity among 11 SET domain-containing HMTases: Hs SET9, Mm Suv39h1, Hs Suv39H1, Dm Su(var)3–9, Hs G9a, Sc SET1, Sp Clr4, Nc dim-5, Hs PR-SET7, Sc SET2, Mm ESET and Hs ESET. Insertions in SUV39H1 and Clr4 sequences are dropped below the alignment with a vertical line for clarity. Loops that form the active site pocket are highlighted in orange, and residues that make up the pocket walls and active site, and those that contact AdoMet are indicated.
Results
Structure determination of SET7/9
In initial crystallization experiments, a thin plate crystal form was obtained from the full-length SET7/9 protein. However, this crystal form did not diffract. To overcome this problem, we carried out a limited proteolytic digestion on full-length protein, and identified two stable polypeptide fragments spanning residues 70–366 (SET7/9L) and residues 109–366 (SET7/9S). Biochemical analyses revealed that both of these fragments retain full HMT activity, suggesting that the missing N-terminal region is not required for substrate binding and catalytic activity (data not shown).
The SET7/9L and SET7/9S proteins were crystallized in well diffracting crystal forms, one in orthorhombic form (1.7 Å resolution) and the other in a tetragonal space group (2.3 Å resolution). Phasing and refinement statistics for these crystals are shown in Table I. Both crystal forms contain one protein–AdoMet complex in the asymmetric unit. The two structures are virtually identical, showing root mean square (r.m.s.) deviations of 0.5 Å for Cα atoms.
Table I. Statistics of data collection and refinement.
| SET 7/9 S | SET 7/9 L | |||
|---|---|---|---|---|
| Data sets | Edge | Peak | Remote | Native |
| Diffraction data | ||||
| Wavelength (Å) | 0.9792 | 0.9791 | 0.9464 | 1.000 |
| Resolution (Å) | 100–1.7 | 100–1.7 | 100–1.7 | 100–2.3 |
| Measured reflections | 300 831 | 297 670 | 308 082 | 140 566 |
| Unique reflections | 29 855 | 29 713 | 30 142 | 20 179 |
| Completeness (%) | 99.7 | 99.7 | 99.9 | 90.1 |
| Average I/σ | 31 | 26 | 24 | 9.43 |
| Rsym (%) | 5.4 | 6.4 | 5.6 | 6.0 |
| Phasing | ||||
| Overall figure of merit (30–1.7 Å) | 0.64 | |||
| Refinement | ||||
| Resolution range (Å) | 30–1.7 | 50–2.3 | ||
| No. of reflections (I/σ>0) | 29 506 | 20165 | ||
| Rwork (%) | 20.0 | 20.4 | ||
| Rfree (%) | 21.7 | 24.3 | ||
| No. of protein/AdoMet atoms | 1835/27 | 2208/27 | ||
| No. of water molecules | 212 | 130 | ||
| R.m.s.d. bond length (Å) | 0.005 | 0.006 | ||
| R.m.s.d. bond angles (°) | 1.25 | 1.25 | ||
Rsym = ΣhΣi|Ih,i – Ih|/ΣhΣiIh,i, where Ih is the mean intensity of the i observations of symmetry-related reflections of h.
R = Σ|Fobs – Fcalc|/ΣFobs, where Fobs = Fp, and Fcalc is the calculated protein structure factor from the atomic model (Rfree was calculated with 5% of the reflections).
R.m.s.d. in bond lengths and angles are the deviations from ideal values, and the r.m.s.d. in B factors is calculated between bonded atoms.
Overall structure
The crystal structure revealed that the SET7/9L consists of three α-helices and 20 β-strands organized in two domains (Figure 1B). The two connected domains are well defined and, upon packing into a whole structure, 2035 Å2 of accessible surface area becomes buried. The N-terminal domain (residues 79–193) is made up entirely of β-strands (Figure 2A and B). The C-terminal domain (residues 194–364) folds into an α/β structure, and most of the highly-conserved residues are concentrated in this region.



Fig. 2. Overall structure of SET7/9–AdoMet. (A) The 2.3 Å SET7/9L structure is shown in a ribbon representation. A bound AdoMet molecule is shown in a ball-and-stick model (oxygen, red; carbon, cyan; nitrogen, blue; and sulfur, green). The n-, SET and c-SET regions in the C-terminal domain are coloured in yellow, red and green, respectively. (B) Topological diagrams of secondary structure elements. Loops forming the active site pocket are in red. The AdoMet-binding site is indicated. The colour scheme is identical to that in (A). (C) Stereo diagram showing an extensive van der Waals contacts network between the SET and c-SET region. The AdoMet is coloured in cyan, and the residues from SET and c-SET domains are in yellow. Other atoms are shown in the same colour as in (A).
The N-terminal domain folds into a β-domain structure with a twisted antiparallel β-sheet and an extra β-strand (Figure 2A and B). The β-sheet is formed with nine β-strands. The first three strands are completely exposed to solvent, whereas strands β4–β7 are packed in a perpendicular manner to the extended part of strand β8, which is bent significantly, and to strand β9. The opposite face of these strands interacts with the C-terminal domain. The N-terminal domain is far from the active site and does not appear to be involved in the enzyme activity or substrate binding. A search for homologous structures using the DALI server revealed two structures, bacterial outer-surface protein A (1OSP; z = 4.9) and porin (3PRN; z = 3.8). The structural similarity of the N-terminal domain to these proteins suggests that this domain may interact with small polymeric molecules or other proteins to mediate the signal to the C-terminal domain.
The C-terminal domain containing a SET domain folds into a novel α/β fold with 10 β-strands and three α-helices (Figure 2A and B). The central feature of this domain includes four motifs, which form the SET domain, and the c-SET helix. The C-terminal domain folds into a tightly-packed structure that contains n-SET, SET and c-SET regions. A search for homologous structures using DALI reveals no proteins with a structural similarity. The first motif in this domain is a β-sheet formed by three twisted β-strands (β13, β14 and β18), and helix α1, which is located next to the β13 strand (Figure 2B). One face of the sheet and helix α1 are packed against the six β-strands (β4–β8 and β10) of the N-terminal sheet. The second motif located at the top of the β-sheet is formed by helix α2 and three antiparallel strands (β15, β16 and β19). The ‘front face’ of this sheet is packed in a perpendicular manner to the β-sheet in the first motif, whereas the ‘back face’ makes extensive interactions with residues from helix α2. The third motif consists of two antiparallel strands (β11 and β12) that are located at the bottom of the first sheet (Figure 2A and B). These strands are perpendicular to the β-strands from both the first and second motifs, and do not make direct contact with them. Strand β20 threads through and underneath strand β17 and loop L4, forming a knot structure, and this fourth motif occupies the space that was formed upon packing of the other three motifs. The c-ET helix α3 is positioned at the opposite end of the N-terminal domain, making extensive interactions with strand β17 and loop L4. This unusual fold of the C-terminal part of the SET domain and c-SET region gives rise to two prominent architectural features; a short and wide pocket and a narrow channel formed in the pocket.
The role of the c-SET domain
The c-SET region comprising residues 345–366 is critical for the function of SET7/9, and it has been shown that removal of this region completely abolishes the HMTase activity of SET7/9 (Wilson et al., 2002). Our structure shows that a part of this region (residues 344–349) forms a flexible loop, whereas residues 350–360 form an amphipathic helix. In this helix α3, the interacting residues, including Pro350, Trp352, Tyr353, Leu357 and Phe360, form an extensive network of van der Waals contacts with His297 in strand β17 and Phe299 in loop L4 (Figure 2C). This network is also connected to Tyr335 and Tyr337 in loop L5. The c-SET helix and a part of the L5 loop are not present in the recently reported crystal structure of SET7/9 (Wilson et al., 2002). As we describe later, in the presence of this helix, a significant portion of the cofactor-binding site is protected compared with that in the absence of the helix, and the AdoMet molecule can gain access from limited directions.
The adenine base of AdoMet is tightly packed between Trp352 of the c-SET helix and loop L1/strand β17, and mediates the interaction between them (Figure 2C; see the section entitled ‘The SET7/9–AdoMet interaction’). The L5 loop that connects the c-SET helix and SET domain is flexible, and the c-SET region may regulate access of the AdoMet molecule to the binding site by altering the relative position of this helix.
AdoMet-binding pocket
A pocket with a width of 9.5 Å × 13 Å and a depth of 8.5 Å is formed by the interaction of loops and strands from the SET domain and the c-SET helix in the C-terminal domain (Figure 3A and B). The bottom of the pocket is lined with strand β17 that contains the R/HxxNHS motif, the most conserved sequence found in members of the SET domain-containing protein family, and with residues 335–337 of loop L5 in the c-SET region. One side of the wall is formed by loop L1 between strands β11 and β12. The opposite side of the wall is covered with residues 263–266 in loop L2. Two other ends of the wall are lined with residues 293–295 in loop L3 and the c-SET helix α3.
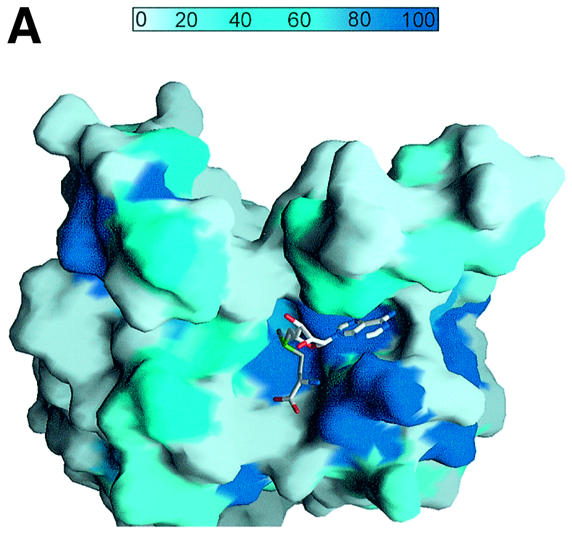



Fig. 3. The AdoMet-binding pocket. (A) Surface representation of the AdoMet-binding pocket. The surface is coloured according to the residue identity as in Figure 1B, highlighting the conservation of residues that make up the AdoMet-binding site. The AdoMet molecule is shown in a bond representation. The orientation of the figure is similar to that in (B). (B) The AdoMet-binding pocket in SET7/9S. Key residues discussed in the text are shown in a ball-and-stick model. The AdoMet molecule is shown with same colour scheme as in (A), except that carbon atoms are shown in cyan. (C) Stereo diagram showing the conformations of AdoMet molecules. Three AdoMet molecules, extended (red), intermediate (blue) and compact (yellow) forms, are superimposed. The extended AdoMet molecule was drawn using AdoMet bound to isoflavone-O-methyltransferase [Protein data bank (PDB) identifiaction code: 1FPX], and the intermediate form was from AdoMet bound to methionine repressor protein (Metj; PDB identification code: 1CMA). (D) A schematic drawing showing direct interactions between SET7/9 and AdoMet. Hydrogen bonds are shown by dashed lines. An arc next to the residue name indicates that the amino acid is involved in a van der Waals interaction with AdoMet. The residues coloured in blue indicate side chain interactions, while those in red are involved in backbone interactions.
An AdoMet with a compact conformation binds to this pocket (Figure 3A–C). The methyl group of the methionine moiety and N6 of the adenine base moiety are directed towards the binding pocket, whereas the hydroxyl group of adenosine ribose points outwards to the solvent.
The conformation of AdoMet is identical in both crystal forms, indicating that this unusual conformation is not the result of different crystal packing. In the SET7/9–AdoMet structure, the adenine base is in an anti-conformation with its ring parallel to the β-strand plane at the bottom of the pocket. The adenine ribose has 2′-exo sugar puckering (Figure 3A–C). We have compared the conformation of AdoMet in SET7/9 with those of 20 other AdoMet molecules that bound to different proteins. Although there are slight differences in each AdoMet conformation, they can be grouped into two classes. Together with the compact form of AdoMet in SET7/9, we can divide these conformations into extended, intermediate and compact forms (Figure 3C). The intermediate form of AdoMet is found in methionine repressor protein (Metj; Somers and Phillips, 1992) and adenine-N6-DNA-methyltransferase (Zubieta et al., 2001), whereas the extended form is observed in the rest of the MTase structures. When these molecules are superimposed by placing their centres on the sugar rings, the compact conformation of an AdoMet molecule in SET7/9 shows some unique features (Figure 3C): (i) the adenine base of the compact form is perpendicular to that of the extended or intermediate form; and (ii) the methionine moiety of AdoMet in SET7/9 is rotated significantly relative to the adenine ribose moiety around both the C4–C′5 and the C′5–Sδ bonds. This rotation results in a highly compact form of AdoMet, and the side chain axis of the methionine moiety is directed perpendicular to the sugar plane, whereas it is parallel to the sugar ring plane in an extended or intermediate form.
The distinctive compact conformation of AdoMet plays crucial roles in the structure and function of SET domain-containing HMTases. First, it allows the binding of AdoMet into the pocket, which is not possible in the extended or intermediate conformation. Secondly, in this conformation, a sulfonium methyl group (Cε) in the methionine moiety can be located close to a number of residues that form a channel where a substrate lysine can enter (see below). The methyl group would be positioned ‘incorrectly’ in an extended or intermediate conformation, and the substrate could not be reached.
The SET7/9–AdoMet interaction
The recently reported structure of SET7/9–AdoMet (Wilson et al., 2002) differs significantly from our structure with regard to the orientation of AdoMet in the active site. These differences may be explained by the lack of a c-SET helix in the published structure or by the limited resolution at which the orientation of AdoMet cannot be determined accurately. The orientation of cofactor in Rubisco LSMT (Trievel et al., 2002) is similar to that in our structure. The interactions observed between SET7/9 and the AdoMet molecule in our structure can be grouped according to adenine base, adenosine ribose and methionine moieties of AdoMet (Figure 3B and D).
The adenine base is buried between the SET and c-SET domains through van der Waals contacts and hydrogen bonds (Figure 3A and B). The indole ring of Trp352 from the c-SET helix packs against the face of the adenine base, and Ala226, Ile223 and the aliphatic part of Asn296 make van der Waals contacts with the other face of the base. Nitrogen N6 forms a hydrogen bond with the Oε1 and Oε2 atoms of Glu356, and N6 and N7 interact with the backbone carbonyl group of His297.
No significant interactions are observed between the ribose ring and SET7/9 because the sugar moiety of AdoMet is oriented in a manner such that its hydroxyl group points towards the solvent (Figure 3B). The only detectable intermolecular interaction is a hydrogen bond between O4′ of the ribose ring and Nδ2 of Asn296. However, O4′ of the ribose ring makes an intramolecular hydrogen bond with the amide group of the methionine moiety, and this interaction may contribute to the compact conformation of the AdoMet molecule.
The carboxylate group of the methionine moiety interacts with the side chain of Lys294 and the backbone amide of Glu228 through a salt bridge and a hydrogen bond, respectively (Figure 3B and D). The amino group forms hydrogen bonds with the backbone carbonyl groups of Ala226 and Glu228 and the side chain of Asn296. The sulfur atom of AdoMet makes contact with Nδ1 of Asn265. Interestingly, the methyl group is located in a polar environment formed by hydroxyl groups of Tyr245 (4.4 Å), Tyr305 (5.5 Å) and Tyr335 (3.5 Å), and carbonyl oxygen atoms of Gly264 (3.8 Å), His293 (3.0 Å) and Ala295 (4.2 Å). This polar environment presumably helps the access of the Nε of a substrate lysine. Previous mutational analysis suggested that two conserved residues, His293 and His297, are important for the HMTase activity of this enzyme family (Wang et al., 2001a; Nishioka et al., 2002). Although these histidine residues are located in the AdoMet-binding pocket, they are far from this methyl group (6.5 Å for His293 and 9.5 Å for His297). The side chain of His297 is located near the adenine base moiety (4.1 Å from N6 of the base), whereas the side chain of His293 is at a distance of 6.5 Å from the methionine moiety. The sulfur group is in between these.
The substrate-specific channel
The methyl group of AdoMet points to a narrow channel that is seen within the binding pocket (Figures 3A and 4A). This channel, with an opening diameter of 6 Å and a depth of 6 Å, is formed by the side chains of Tyr245, Leu267, Tyr305, Tyr335 and Tyr337, and the backbone of residues 264–267 (Figure 4A and B). Most of the residues lining the channel are highly conserved among SET domain-containing HMTases, indicating that the channel is a key feature of this protein family (Figure 4A and B). The inner surface inside the channel where the methyl group is located is highly polar, which is due to the presence of several hydroxyl and carbonyl groups.
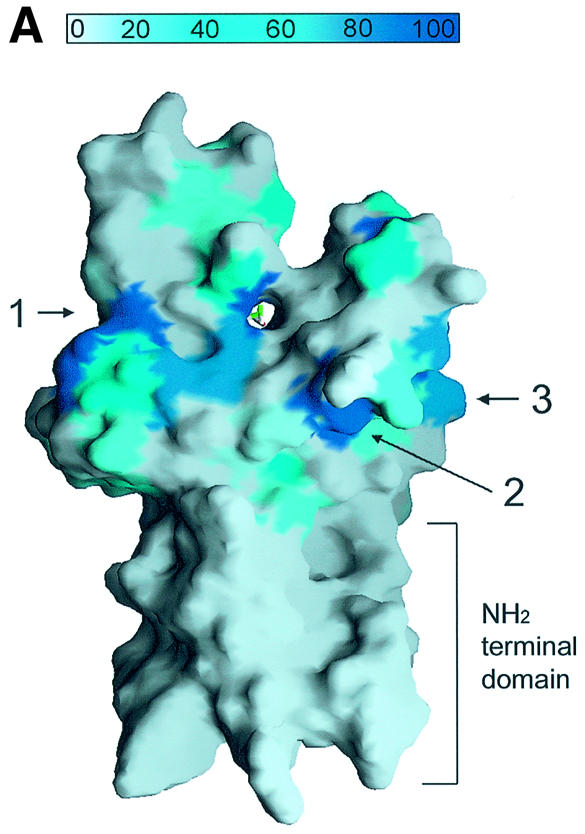
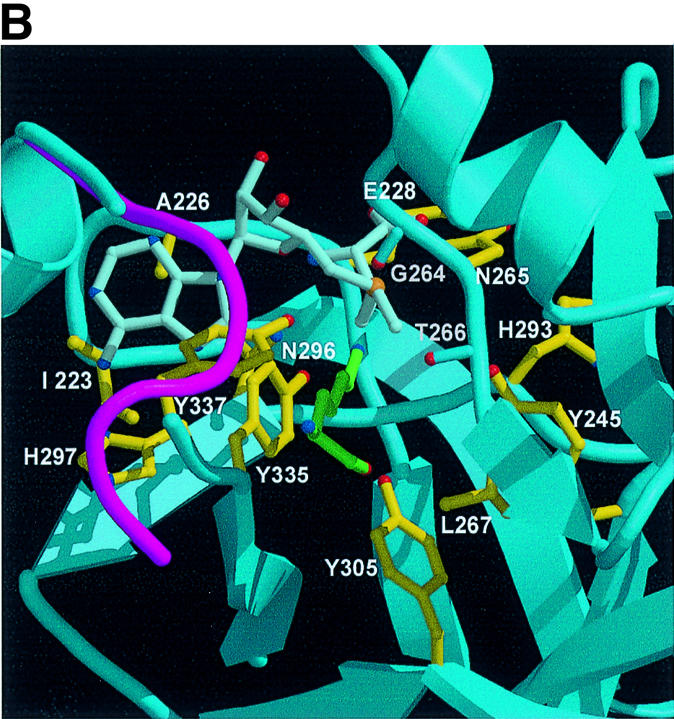

Fig. 4. A channel at the active site pocket. (A) A surface representation showing the location of the putative lysine-binding channel and a conserved shallow groove for the substrate-binding site (indicated by an arrow). See the text for sites 1, 2 and 3. The colouring scheme is identical to that in Figure 3A. AdoMet is shown in a bond model. The N-terminal domain is not coloured since it is not homologous to those of other SET-containing HMTases (Figure 1B). (B) A diagram showing the substrate-specific channel in SET7/9S. Key residues discussed in the text are shown in a ball-and-stick model. The AdoMet molecule is shown with the same colour scheme as in Figure 3B. (C) SET7/9 methylates unmethylated H3 peptide but cannot add methyl group(s) to already methylated peptide. An N-terminal peptide with amino acids 1–8 of unmodified, mono- or dimethylated H3-K4 was used for the assay of SET7/9 HMTase activity. The full-length SET7/9 was used for the assay. (D) Methylation specificity of SET7/9. Histone H3 (Roche) was methylated by SET7/9. The reaction products were resolved by SDS–PAGE, blotted to nitrocellulose and probed with either a H3-K4 mono- or dimethyl antibody, or a H3-K4 trimethyl antibody as indicated. The H3-K4, K9 trimethyl antibody also gave the same result as that of the H3-K4 trimethyl antibody (data not shown).
The Nε atom of histone lysines can be mono-, di- or trimethylated, and substrates with differently methylated states may bind to HMTase. However, the narrow diameter and the electrostatic properties of a channel formed at the active site may prevent the access of a methylated lysine substrate. To address whether the functional role of the channel is to discriminate between unmethylated and methylated lysine substrates, we tested whether the enzyme can methylate a mono- or a dimethylated lysine substrate. Figure 4C shows that SET7/9 can transfer a methyl group(s) to unmethylated K4 of H3 but not to either mono- or dimethylated K4 of H3. Wilson et al. (2002) have also observed that SET7/9 cannot add a methyl group to a monomethylated peptide.
However, since multiple methyl groups can be transferred to the lysine side chain without dissociation of a substrate peptide, we have explored this issue further by employing tested antibodies that specifically recognize mono-, di- and trimethylated K4 of H3. Although these antibodies are highly specific for each methylated state of the peptide as judged by enzyme-linked immunosorbent assay (ELISA; data not shown) and peptide competition experiments (Santos-Rosa et al., 2002), in order to remove completely any possible non-specific interaction between each antibody and the product(s), we have carried out a peptide competition assay. In this assay, mono- and trimethylated peptides were added to the dimethylated K4 antibody prior to detecting the reaction product(s). Similar peptide competition methods were also employed for the mono- or trimethylated K4 antibody assay. As shown in Figure 4D, the dimethylated K4 antibody can recognize the recombinant H3 after it has been methylated by SET7/9, whereas the trimethylated antibody fails to do so. Figure 4D also shows that the dimethylated peptide specifically competed with the reaction product against the dimethylated K4 antibody, further supporting the specificity of the dimethylated K4-H3 antibody and the dimethylated reaction product formed by SET7/9. The binding of dimethylated K4 antibody to the reaction product of H3 by SET7/9 has been observed consistently by another group (B.Hamilton and J.Bone, Upstate Biotechnology, unpublished data). These results suggest that SET7/9 can transfer two methyl groups but not three methyl groups to the K4 of H3, and multiple rounds of methylation occur on the Nε atom of a lysine without being released from the enzyme when unmodified substrate is bound to the active site.
The histone-binding site
The surface representation in Figure 4Aa shows that a conserved shallow groove is formed around the channel that is extended continuously to one side of the structure at site 1. This shallow groove is formed with highly-conserved residues from strand β18 and helix α3 near the channel, and strand β20 at the side of the structure (Figure 4A). Since a lysine substrate enters the channel, another part of the N-terminal tail must be in the vicinity of this region. We propose that this shallow groove provides the binding site for a part of the histone tail. Alternatively, another highly-conserved region is located near the channel (site 2 in Figure 4A) comprising strands β16 and β19, and extends to site 3 where helix α2 is located. Mutation on an exposed residue, Asp270 (strand β16) or Glu254 (helix α2), to alanine reduced the binding of a substrate peptide to SET7/9 (Wilson et al., 2002), suggesting the possibility that a substrate binds to this region.
The structure–function correlations
Guided by the structural information, we carried out mutagenesis on eight residues, His293, His297, Lys294, Asp276, Trp352, Tyr305, Tyr245 and Tyr335, at or near the active site of SET7/9 (Figure 5A). These residues were changed individually to alanines, and the HMTase activities of the mutants were measured. Interestingly, most of the mutants exhibited a significantly decreased HMTase activity (Figure 5B).


Fig. 5. (A) Stereo diagram showing the active site region in SET7/9S. Eight residues discussed in the text along with interacting residues are shown in a ball-and-stick model. (B) HMTase activities of the SET7/9 point mutants. Reactions contained wild-type or mutant SET7/9, 1 µM AdoMet and 1 µg of H3 from calf thymus. Assays were performed in triplicate.
Lys294 and Trp352 interact directly with the carboxylate group and adenine base of AdoMet, respectively. The decreased HMTase activity observed for the mutants reflects the importance of these interactions. Mutation of Lys294 or Trp352 is also likely to disrupt the local structure of the pocket. The Nε of atom of Lys294 forms a salt bridge with Oε1 of Glu228 in loop L1, which forms a wall of the binding pocket. The indole ring of Trp352 makes van der Waals contacts with the side chain rings of His297, Tyr335, Tyr337 and Tyr353 (Figure 5A).
The side chain of Asp279 in strand β16 forms a salt bridge with the conserved Arg258 residue in loop L2 that is located on top of the channel. The decrease in enzyme activity seen for the Asp276 mutant may be explained by the disruption of this salt bridge that could perturb the local channel structure.
The side chain rings of Tyr245, Tyr305 and Tyr335 are important components that form the wall of the channel in the binding pocket. All the hydroxyl groups of these residues are directed to the methyl group of the AdoMet molecule, and a water molecule (3.4 Å from the AdoMet methyl group) is hydrogen bonded to Tyr305 in the centre of a channel. It is likely that the role of these residues is to position the lysine substrate correctly, which is crucial for catalysis. Furthermore, they could deprotonate the Nε of the lysine substrate to generate a nucleophile intermediate for the methylation reaction.
His293 and His297 are far from the catalytic site and are unlikely to participate directly in the methylation reaction. These residues may therefore play an important role in the maintenance of the local structure around the active site. His293 is surrounded by Tyr245, Val277 and Tyr287. His297 is surrounded by Phe299, Glu356, Trp352, Tyr353 and Tyr335. However, rotations of the imidazole ring of His293 and His297 around the χ1 axis of the side chain would position their Nε atoms within a distance 3 Å from the hydroxyl group of Tyr245 and Tyr335, respectively, indicating the histidine residues may assist the tyrosine residues with their catalytic function.
Discussion
The evolutionarily conserved SET domain is found in a large and rapidly increasing number of proteins. At present, >350 proteins are known to contain SET domains (Schultz et al., 1998). It is unknown, however, whether all these proteins possess HMTase activity. One important requirement for a SET-containing protein to possess HMTase activity is the presence of specific pre (n)- and post (c)-SET regions (Rea et al., 2000). In contrast to the conserved SET domain, the n- and c-SET regions vary significantly among the members of this enzyme family, and a particular arrangement of the n-SET and c-SET regions appears to be important in HMTase function.
The role of a SET domain and a c-SET helix
Our structure shows that the n-SET region and the SET domain are tightly packed together in the C-terminal domain. The n-SET region is therefore likely to be involved in the folding of the SET domain and contribute to its stability. The SET domain and c-SET region have more direct roles in the HMTase activity of SET7/9 because an active site is located within two domains. In particular, the c-SET helix is essential to protect the binding site from one side. Thus, in the presence of a c-SET helix, the access of an AdoMet molecule is more restricted than in the absence of this helix. The region immediately adjacent to this helix is a flexible loop. The movement of the c-SET helix relative to the SET domain may regulate the access of the AdoMet molecule to the active site. Secondly, the more important role of a c-SET region may be to stabilize the structure of a substrate-specific channel and the positioning of the catalytic Tyr335 (see below). Thus, disruption of the local structure of the interface between the SET domain and the c-SET helix may alter the conformation of the substrate-specific channel where a lysine substrate can enter, and/or may perturb the position of Tyr335, which would affect the catalytic activity. Consistent with this explanation, a deletion mutant in which this c-SET helix is removed is inactivated completely while retaining cofactor- or substrate-binding affinity similar to that of wild-type SET7/9 (Wilson et al., 2002). In addition, two point mutants, Trp352Ala and Tyr353Ala, which would disturb the local structure of the interface between the SET domain and the c-SET helix, did not exhibit HMTase activity (Wilson et al., 2002). It should be noted that the c-SET region is not defined in the structure of apoSET7/9 (Wilson et al., 2002). Thus, it is possible that the structure of the c-SET helix may be induced from the unfolded state upon binding of AdoMet to SET7/9.
Two key features in the active site of SET7/9
Our structure shows two important features of the active site of SET7/9. First, instead of an extended conformation that is observed in most other MTases, the AdoMet molecule in SET7/9 adopts a compact conformation that results in it fitting tightly into the binding pocket. This conformation is important to position the methyl group of AdoMet towards the substrate-specific channel where a lysine binds on the opposite side. Accordingly, a major role of the SET domain and the c-SET helix in SET7/9 may be to force the AdoMet molecule into a compact conformation.
The second feature is a narrow substrate-specific channel lined with several highly-conserved residues that is formed within the active site. It is well established that lysine residues of histones exist in a mono-, di- and trimethylated form. Recent results demonstrated that the precise methylation state of lysine residues defines the gene expression status (Santos-Rosa et al., 2002).
Our structural and biochemical data show that SET7/9 can transfer methyl groups only to the unmethylated K4 of H3, suggesting that the narrow substrate-specific channel controls the access of the already methylated substrates to the active site. Our data also clearly demonstrate that the bound substrate lysine is subjected to two rounds of methylation without having to be released from SET7/9. Since little is known about the processivity of the methyl transfer reaction, it would be interesting to see if these multiple rounds of the methyl transfer mechanism are a general feature in other MTases.
Several residues forming the substrate-specific channel are not conserved. This variability of amino acid residues may alter the diameter of a channel in other SET domain-containing HMTases. Since the monomethylated peptide substrate that may barely fit into the channel cannot gain access to SET7/9, factors other than the size of the channel may be involved in discriminating between substrates. It is possible that the surface electrostatic property of the channel is an additional factor. Taken together, although it is unknown where the substrate binds to the C-terminal domain, the selectivity of the methylation site provided by the SET and c-SET domain, and the specificity of the substrate methylation state (unmethylated versus methylated) provided by the channel represent two key features for the regulation of gene expression by lysine methylation of histones.
The recently reported crystal structure of the large subunit of the Rubisco methyltransferase where a c-SET region is well defined contains a similar channel in the binding pocket (Trievel et al., 2002), and an HEPES ion binds to this channel mimicking the lysine binding. Our results in conjunction with this observation suggest that the substrate-specific channel is likely to be an important feature in members of the SET domain-containing HMTase enzyme family.
Methyl transfer mechanism of SET7/9
Based on our structure in combination with mutational analysis, we propose the following mechanism for the AdoMet-mediated methyl transfer reaction catalysed by SET domain-containing HMTases. It has been proposed previously that the lysine methyl transfer process is an SN2 reaction in which the substrate lysine, a methyl group and the leaving thioester group form a linear arrangement (Coward, 1977). In this arrangement, the unprotonated lysine residue makes a nucleophilic attack on the methyl group of AdoMet.
When lysine is positioned within the channel in such a way that the methyl group of AdoMet is near the Nε atom, the two OH groups of Tyr245 and Tyr335 are located within a distance of 3.5–4 Å. Tyr335 is absolutely conserved among this family of enzymes, and could help to neutralize the Nε atom of the lysine substrate. However, the pKa of the hydroxyl group of tyrosine is ∼10, which is rather high to deprotonate the Nε atom of the lysine substrate. The closely located His297 may serve as a proton acceptor of the hydroxyl group of Tyr335. A simple rotation around the χ1 angle of the conserved His297 would locate the Nε of His297 within 2.5 Å of the hydroxyl group of Tyr335. This assumption is supported by the significantly decreased HMTase activity of a His297Ala mutant (Figure 5B). Alternatively, we cannot exclude the possibility that the hydroxyl group of Tyr245 deprotonates the substrate lysine. As in the Tyr335–His297 proton relay system, a rotation of the side chain of His293 would locate the Nε group of His293 within a distance of 2.5 Å from the hydroxyl group of Tyr245. In contrast to His297, His293 is not well conserved, however, and is substituted by arginine in several enzymes of the family.
In summary, our structure combined with mutational analysis provides new insights into the catalytic mechanism of SET domain-containing HMTases. The key findings of our study are that: (i) the AdoMet molecule adopts a compact conformation within the active site pocket; (ii) SET7/9 can transfer methyl groups only to the unmethylated substrate lysine; (iii) two but not three methyl groups are added to the lysine without dissociation of a substrate; and (iv) the neutralization of lysine substrate is likely to be achieved by tyrosine residue(s) with or without the assistance of a histidine residue. These findings together with the site specificity for methylation provided by the SET domain and the c-SET helix provide an explanation for the basis of the HMTase activity of SET7/9.
The high-resolution structure reported here may also serve as a basis for the design of small molecules that mimic the distinctive conformation of the AdoMet molecule to block the channel of the binding pocket. These molecules could be used as drugs to modulate the gene expression mediated by SET domain-containing HMTases.
While this manuscript was under review, two structures for SET domain-containing HMTases (Jacobs et al., 2002; Min et al., 2002) have been reported. Neither structure contains the c-SET region.
Materials and methods
Protein expression and purification
SET7/9L (residues 70–366) or SET7/9S (residues 109–366) was inserted into PET15b vector and expressed in Escherichia coli BL21 as a His-tagged form. The protein was isolated initially using a nickel column. After thrombin digestion, the protein was purified further by anion exchange (Mono-Q) and gel filtration (Superdex 200) chromatography. Each protein was concentrated by ultrafiltration to 20 mg/ml in 50 mM Tris–HCl pH 7.5, 200 mM NaCl, 5 mM DTT, and stored –70°C.
Crystallization
Concentrated SET7/9 protein was mixed with AdoMet at a final concentration of 5 mM AdoMet, and the mixture was incubated on ice for 30 min to allow complex formation. SET7/9–AdoMet complex crystals were grown by the hanging drop vapour diffusion method. The orthorhombic crystals were grown at 4°C in conditions containing 100 mM Tris pH 8.5 and 30% PEG 6K. Se-Met-substituted orthorhombic crystals were grown under the same conditions. The crystals belong to the space group P21212 with cell dimensions a = 102.2 Å, b = 38.7 Å, c = 66.6 Å. X-ray diffraction data were collected at –170°C from a crystal flash-frozen in crystallization buffer containing 30% glycerol using the 19ID2 beamline in Advanced Photon Source (APS). The tetragonal crystals were grown at 18°C in a condition containing 100 mM Tris pH 8.5, 1.2 M ammonium sulfate, 0.5 M lithium sulfate and 5 mM DTT. The tetragonal crystal belongs to the space group P43212 with cell dimensions a = 93.45 Å, b = 93.45 Å and c = 110.47 Å. The diffraction data were collected at –170°C from a crystal flash-frozen in crystallization buffer containing 30% glycerol using the B6 beamline in Pohang Accelerator Laboratory (PAL). Data were processed with the programs DENZO and SCALEPACK (Otwinowski and Minor, 1997).
Structure determination
The orthorhombic structure was solved first using the multiple amorphous diffraction (MAD) method. A Se-Met derivative was used for MAD phasing. The SOLVE program (Terwilliger and Berendzen, 1999) identified six Se positions and provides the initial phasing with three wavelength (λ1 = 0.9792 Å, λ2 = 0.9791 Å and λ3 = 0.9464 Å) Se MAD data at 1.7 Å resolution. Solvent flattening using RESOLVE (Terwilliger and Berendzen, 1999) was used to improve the accuracy of phases. The graphic programs CHAIN (Sack, 1988) and O (Jones et al., 1991) were used for model building, and CNS (Brünger et al., 1998) was used for refinement. The model was refined against a 1.7 Å Se–Met derivative data set collected at 0.9792 Å (λ1) wavelength. During the refinement, the Rfree value was monitored by using 5% of the data. The final model has 87% of the protein main chain (φ/ψ) angles in the most favoured region and none in the disallowed region of the Ramachandran plot, calculated using PROCHECK (Laskowski et al., 1993).
The tetragonal structure was solved by molecular replacement using the Amore program suite (CCP4, 1994) and diffraction data in the 15.0–4.0 Å resolution range. The initial R-factor for the molecular replacement solution was 42% for all the data ≤3.0 Å resolution. The model was refined to 2.3 Å resolution (5% of the data were set aside and used to calculate Rfree), and 85% of the main chain φ/ψ angles are in the most favoured region, and none in the disallowed region of the Ramachandran plot.
In both structures, most of the regions are clearly defined. However, the N-terminal part (residues 109–115 in SET7/9S and residues 70–78 in SET7/9L), the C-terminal part (residues 364–366) and the loop region connecting strand β20 and helix α3 (residues 338–348 in SET7/9S and residues 338–339 in SET7/9L) are disordered and not modelled.
HMTase activity assay and western blot
Site-directed mutagenesis was performed with either a PCR-based method or the QuickChange site-directed mutagenesis kit (Stratagene). The mutation in the SET7/9 sequence was confirmed by DNA sequence analysis.
SET9 protein (1 µg) was incubated in 20 mM Tris–HCl pH 8.0, 50 mM NaCl, 10% glycerol, 1 mM phenylmethylsulfonylfluoride (PMSF) with l µl of 3H-labelled AdoMet (Amersham) and 1 µg of H3 from calf thymus (Roche) or 1 µg of histone peptide. Following incubation at 30°C for 30 min, the reaction product was spotted on Wattman p81 paper, washed three times for 10 min each with 50 mM NaHCO3 pH 9.2 and briefly with acetone, dried completely and measured by scintillation counting.
For western blot, the reaction was carried out for 180 min. The reaction product was resolved by 15% SDS–PAGE, blotted to a nitrocellulose membrane and probed with the histone3 K4 mono- (Abcam; ab8895) or dimethyl antibody (Upstate Biotech, 07-030), K4, K9 trimethyl (Abcam; ab1322) or K4 trimethyl antibody (Abcam; ab8580). In the antibody assay, peptide competitors were used to remove any non-specific interaction between each antibody and the product(s).
Acknowledgments
Acknowledgements
We thanks to B.Hamilton and J.Bone (Upstate Biotechnology) for sharing unpublished data, members of PAL for their help with data collection, and R.Kammere for critical reading of the manuscript. This work was supported by funds from the Korean Ministry of Science and Technology (National Creative Research Initiative programme, Frontier 2000 programme), the Ministry of Education (programme BK21) and the Korean Academy of Science and Technology. The coordinates have been deposited in the RCSB database (PDB ID of 1N6A and 1N6C).
References
- Bannister A.J., Zegerman,P., Partridge,J.F., Miska,E.A., Thomas,J.O., Allshire,R.C. and Kouzarides,T. (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature, 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Boggs B.A., Cheung,P., Heard,E., Spector,D.L., Chinault,A.C. and Allis,C.D. (2002) Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosomes. Nat. Genet., 30, 73–76. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Byvoet P., Shepherd,G.R., Hardin,J.M. and Noland,B.J. (1972) The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys., 148, 558–567. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Coward J.K. (1977) Chemical mechanisms of methyl transfer reactions: comparison of methylases with nonenzymic ‘model reactions’. In Salvatore,F., Borek,E., Zappia,V., Williams-Ashman,H.G. and Schlenk,F. (eds), The Biochemistry of Adenosylmethionine. Columbia University Press, New York, NY, pp. 127–144.
- Duerre J.A. and Lee,C.T. (1974) In vivo methylation and turnover of rat brain histones. J. Neurochem., 23, 541–547. [DOI] [PubMed] [Google Scholar]
- Jacobs S.A., Taverna,S.D., Zhang,Y., Briggs,S.D., Li,J., Eissenberg,J.C., Allis,C.D. and Khorasanizadeh,S. (2001) Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J., 20, 5232–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S.A., Harp,J.M., Devarakonda,S., Kim,Y., Rastinejad,F. and Khorasanizadeh,S. (2002) The active site of the SET domain is constructed on a knot. Nat. Struct. Biol., 9, 833–838. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. (2001) Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol., 11, 266–273. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis,C.D. (2001) Translating the histone code. Science, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Jenuwein T., Laible,G., Dorn,R. and Reuter,G. (1998) SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell. Mol. Life Sci., 54, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Karpen G. and Allshire,R.C. (1997) The case for epigenetic effects on centromere identity and function. Trends Genet., 13, 489–496. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2002) Histone methylation in transcriptional control. Curr. Opin. Genet. Dev., 12, 198–209. [DOI] [PubMed] [Google Scholar]
- Lachner M., O’Carroll,D., Rea,S., Mechtler,K. and Jenuwein,T. (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature, 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.A. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Litt M.D., Simpson,M., Gaszner,M., Allis,C.D. and Felsenfeld,G. (2001) Correlation between histone lysine methylation and developmental changes at the chicken β-globin locus. Science, 293, 2453–2455. [DOI] [PubMed] [Google Scholar]
- Ma H. et al. (2001) Hormone-dependent, CARM1-directed, arginine-specific methylation of histone H3 on a steroid-regulated promoter. Curr. Biol., 11, 1981–1985. [DOI] [PubMed] [Google Scholar]
- Min J., Zhang,X., Cheng,X., Grewal,S.I. and Xu,R.M. (2002) Structure of the SET domain histone lysine methyltransferase Clr4. Nat. Struct. Biol., 9, 828–832. [DOI] [PubMed] [Google Scholar]
- Nakayama J., Rice,J.C., Strahl,B.D., Allis,C.D. and Grewal,S.I. (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science, 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Nishioka K., Chuikov,S., Sarma,K., Erdjument-Bromage,H., Allis,C.D., Tempst,P. and Reinberg,D. (2002) Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev., 16, 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Peters A.H. et al. (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell, 107, 323–337. [DOI] [PubMed] [Google Scholar]
- Rea S. et al. (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature, 406, 593–599. [DOI] [PubMed] [Google Scholar]
- Sack J.S. (1988) CHAIN: a crystallographic modeling program. J. Mol. Graphics, 6, 224–225. [Google Scholar]
- Santos-Rosa H., Schneider,R., Bannister,A.J., Sherriff,J., Bernstein,B.E., Emre,N.C., Schreiber,S.L., Mellor,J. and Kouzarides,T. (2002) Active genes are tri-methylated at K4 of histone H3. Nature, 419, 407–411. [DOI] [PubMed] [Google Scholar]
- Schneider R., Bannister,A.J. and Kouzarides,T. (2002) Unsafe SETs; histone lysine methyltransferases and cancer. Trends Biochem. Sci., 27, 396–402. [DOI] [PubMed] [Google Scholar]
- Schotta G., Ebert,A., Krauss,V., Fischer,A., Hoffmann,J., Rea,S., Jenuwein,T., Dorn,R. and Reuter,G. (2002) Central role of Drosophila Su(var)3–9 in histone H3-K9 methylation and heterochromatin-dependent gene silencing. EMBO J., 21, 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Ayyananthan,K., Negorev,D., Maul,G.G. and Rauscher,F.J. (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by krab zinc-finger proteins. Genes Dev., 16, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J., Milpetz,F., Bork,P. and Ponting,C.P. (1998) SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl Acad Sci USA, 95, 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers W.S. and Phillips,S.E. (1992) Crystal structure of the met repressor–operator complex at 2.8 Å resolution reveals DNA recognition by β-strands. Nature, 359, 387–393. [DOI] [PubMed] [Google Scholar]
- Spencer V.A. and Davie,J.R. (1999) Role of covalent modifications of histones in regulating gene expression. Gene, 240, 1–12. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. and Allis,C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. et al. (2001) Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol., 11, 996–1000. [DOI] [PubMed] [Google Scholar]
- Tachibana M., Sugimoto,K., Fukushima,T. and Shinkai,Y. (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem., 276, 25309–25317. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr., 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trievel R.C, Beach,B.M., Dirk,L.M.A., Houtz,R.L. and Hurley,J.H. (2002) Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell, 111, 91–103. [DOI] [PubMed] [Google Scholar]
- Tschiersch B., Hofmann,A., Krauss,V., Dorn,R., Korge,G. and Reuter,G. (1994) The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3–9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J., 13, 3822–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B.M. (2000) Histone acetylation and an epigenetic code. BioEssays, 22, 836–845. [DOI] [PubMed] [Google Scholar]
- vanHolde K.E. (1988) Histone modifications. In Rich,A. (ed.), Chromatin, Springer Series in Molecular Biology. Springer Press, New York, pp. 111–148.
- Wang H., Cao,R., Xia,L., Erdjument-Bromage,H., Borchers,C., Tempst,P. and Zhang,Y. (2001a) Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell, 8, 1207–1217. [DOI] [PubMed] [Google Scholar]
- Wang H. et al. (2001b) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science, 293, 853–857. [DOI] [PubMed] [Google Scholar]
- Wilson J.R., Jing,C., Walker,P.A., Martin,S.R., Howell,S.A., Blackburn,G.M., Gamblin,S.J. and Xiao,B. (2002) Crystal structure and functional analysis of the histone methyltransferase SET7/9. Cell, 111, 105–115. [DOI] [PubMed] [Google Scholar]
- Wolffe A.P. (2001) Transcriptional regulation in the context of chromatin structure. Essays Biochem., 37, 45–57. [DOI] [PubMed] [Google Scholar]
- Yang L., Xia,L., Wu,D.Y., Wang,H., Chansky,H.A., Schubach,W.H., Hickstein,D.D. and Zhang,Y. (2002) Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene, 21, 148–152. [DOI] [PubMed] [Google Scholar]
- Zhang X., Tamaru,H., Khan,S.I., Horton,J.R., Keefe,L.J., Selker,E.U. and Cheng,X. (2002) Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell, 111, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. and Reinberg,D. (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev., 15, 2343–2360. [DOI] [PubMed] [Google Scholar]
- Zegerman P., Canas,B., Pappin,D. and Kouzarides,T. (2002) Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J. Biol. Chem., 277, 11621–11624. [DOI] [PubMed] [Google Scholar]
- Zubieta C., He,X.-Z., Dixon,R.A., Noel,J.P. (2001) Structures of two natural product methyltransferases reveal the basis for substrate specificity in plant O-methyltransferases. Nat. Struct. Biol., 8, 271–279. [DOI] [PubMed] [Google Scholar]