

Abstract
ActVA-Orf6 monooxygenase from Streptomyces coelicolor that catalyses the oxidation of an aromatic intermediate of the actinorhodin biosynthetic pathway is a member of a class of small monooxygenases that carry out oxygenation without the assistance of any of the prosthetic groups, metal ions or cofactors normally associated with activation of molecular oxygen. The overall structure is a ferredoxin-like fold with a novel dimeric assembly, indicating that the widely represented ferredoxin fold may sustain yet another functionality. The resolution (1.3 Å) of the enzyme structure and its complex with substrate and product analogues allows us to visualize the mechanism of binding and activation of the substrate for attack by molecular oxygen, and utilization of two gates for the reaction components including a proton gate and an O2/H2O gate with a putative protein channel. This is the first crystal structure of an enzyme involved in the tailoring of a type II aromatic polyketide and illustrates some of the enzyme–substrate recognition features that may apply to a range of other enzymes involved in modifying a polyketide core structure.
Keywords: actinorhodin/monooxygenase/polyketide/protein structure/Streptomyces coelicolor
Introduction
Natural products such as the polyketide compounds tetracycline, erythromycin, daunorubicin and lovastatin produced by actinomycete bacteria and fungi have enormous therapeutic and commercial significance. Conse quently, considerable research effort has been directed into study of the biosynthetic pathways that lead to these compounds in order to influence the nature and biological activity of the products produced (Hutchinson, 1997). This work has been aided by the cloning, sequence analysis and manipulation of the gene clusters responsible for the biosynthesis of aromatic and macrolide polyketides. Of particular interest have been enzymes that catalyse the assembly and subsequent cyclization of the polyketide chain (Hopwood and Sherman, 1990; Hutchinson and Fujii, 1995; Hopwood, 1997; Hutchinson, 1997). This has allowed many of the events surrounding carbon skeleton formation by condensation of simple building blocks such as a malonyl-CoA or methylmalonyl-CoA to be uncovered, but has left the identity and function of the enzymes that perform the later steps on these pathways relatively unclear. Late-acting, ‘tailoring’ enzymes catalyse a wide range of modifications to polyketide structure, such as hydroxylations, methylations or glycosylations. It is often these subtle changes to the overall molecular shape or charge that give the polyketide molecule its biological activity. A better understanding of the mechanistic and structural parameters of these enzymes may enable engineered biosynthesis (Townsend, 1997) to develop molecules with novel or altered biological activity or a better toxicological profile.
The intense blue colour of the actinorhodin complex produced by Streptomyces coelicolor first attracted researchers into close study of this organism and into developing many of the genetic tools required for genetic manipulation of actinomycete bacteria. These genetic studies have reached their pinnacle with the completion of the entire 8.67 Mb genome sequence of this organism (Bentley et al., 2002). The gene cluster responsible for the biosynthesis of actinorhodin has long been thought of as the paradigm type II polyketide biosynthetic cluster (Malpartida and Hopwood, 1984, 1986). Analysis of the products of genetic mutations and sequence similarities to genes in other biosynthetic pathways allowed enzyme activities encoded by the gene cluster to be assigned tentatively (Hallam et al., 1988; Caballero et al., 1991, 1992; Fernández-Moreno et al., 1991, 1992, 1994). How ever, difficulties in purifying these enzymes, coupled with inadequate knowledge of the pathways and lack of substrates, mean that few of these assignments have been confirmed with biochemical studies of the proteins. While the numbers of natural product gene clusters identified has increased significantly each year, relatively few protein biochemical studies are published. There are still fewer structural studies involving enzymes from polyketide biosynthetic pathways, with the effort focused mainly on the polyketide synthase enzymes or the analogous proteins from fatty acid biosynthesis: the crystal structure of malonyl-CoA acyltransferase (Serre et al., 1995), the solution structure of the actinorhodin ACP (Crump et al., 1997), the crystal structure of plant chalcone synthases (Ferrer et al., 1999; Jez et al., 2000) and the crystal structure of the 6-deoxyerythronolide synthase thioesterase domain (Tsai et al., 2001). The erythromycin cytochrome P450 hydroxylase EryF (Cupp-Vickery and Poulos, 1995) appears to be the only polyketide ‘tailoring’ enzyme structurally characterized to date.
Our work has focused on the enzymes catalysing oxygenations of polyketide biosynthetic pathways, particularly those catalysing the oxidations that introduce hydroxy- or quinone functionality to type II aromatic polyketides. ActVA-Orf6 was characterized as a monooxygenase (Kendrew et al., 1997, 2000) from the actinorhodin pathway that catalyses the oxidation of phenolic compounds to the corresponding quinone. The reaction catalysed in vivo by ActVA-Orf6 (Figure 1) is the conversion of 6-deoxydihydrokalafungin (6-DDHK) to dihydrokalafungin (DHK), intermediates in the biosynthesis of actinorhodin. Here we describe the next stage of our investigation of this enzyme: the high-resolution crystallographic structure at 1.3 Å of the ActVA-Orf6 monooxygenase of S.coelicolor A3 (2) in its native form and in complex with substrate and product analogues. This enzyme is one of a number of homologous monooxygenases (including, for example, TcmH, Shen and Hutchinson, 1993; and ElmH, Rafanan et al., 2001) from Streptomyces species catalysing the oxygenation of their various substrates without the need for prosthetic groups, metal ions or cofactors normally associated with activation of molecular oxygen. This is the first X-ray crystal structure of an enzyme from the actinorhodin biosynthetic pathway and in this perspective is particularly interesting since it allows us to propose a molecular mechanism for an unusual enzymatic reaction and to begin to develop ideas as to how the various tailoring enzymes from these biosynthetic pathways recognize their substrates. This allows us to develop a structural genomic approach to the functional and structural prediction of homologous proteins from other biosynthetic pathways.

Fig. 1. The oxidation of 6-deoxydihydrokalafungin (6-DDHK) to dihydrokalafungin (DHK) catalysed by ActVA-Orf6 monooxygenase.
Results
The overall structure
ActVA-Orf6 monooxygenase crystallizes as a homodimer, as previously suggested (Kendrew et al., 2000) and consistent with previous biochemical studies (Kendrew et al., 1997). The monomers in the asymmetric unit are stabilized by an extensive hydrophobic interface with a buried surface of 3300 Å2. Each monomer belongs to the α–β sandwich group of the α + β fold class and comprises a ferredoxin-like split βαβ-fold (Orengo and Thornton, 1993), with two βαβ motifs forming an antiparallel sheet in which strand B1 lays in between strands B3 and B4 (Figure 2A). A search of the Protein Data Bank carried out with DALI (Holm and Sander, 1993) yields members of the ferredoxin-like fold family and indicates that se quence identity is lower than 15% over ≤90 structurally equivalent amino acids, pointing towards evolutionary convergence, which is not unusual for this type of fold.

Fig. 2. Structural model of ActVA-Orf6 monooxygenase. (A) The B subunit of ActVA-Orf6 monooxygenase viewed from the active site entrance is characterized by a ferredoxin fold, with four β-strands (B1–B4) and four α-helices (A1–A4). The blue strand is the C-terminal domain swap contributed by the A subunit. The mobile loop 34–38 (black arrow), the type II β-turn (red arrow) and the type III β-turn (blue arrow) are highlighted. Residues involved in the enzymatic mechanism are drawn as ball-and-stick. (B) The monomer–monomer interface. The interaction between the two β-sheets (yellow and cyan) is stabilized through a network of inter- and intramolecular hydrogen bonds, which also restrain the N-terminus (residues 2 and 4). Thick and thin sticks represent residues contributed, respectively, by the two subunits. (C) The homodimer of ActVA-Orf6 with nanaomycin D bound to subunit A, in the same orientation as in (F). (D) The homodimer of ActVA-Orf6. The 2-fold axis of symmetry is perpendicular to the plane of the picture (cross). (E) The homodimer of ActVA-Orf6 with nanaomycin D bound to subunit A, in the same orientation as in (G). (F) The bottom of the ActVA-Orf6 active site in the B subunit (yellow van der Waals spheres) shows a narrow tunnel (arrow) gated by Gln37 (purple). Ile110 (orange), belonging to the C-terminal swap of the other monomer, also contributes to the closure of the back of the active site. C- (red) and N- (blue) termini are also shown. (G) The dimer in a different orientation, showing nanaomycin D bound to subunit A (dark grey van der Waals spheres) of ActVA-Orf6 monooxygenase and exposing part of the pyrano-γ-lactone ring to the solvent. This moiety (and C-15 methyl, in particular) obstructs the active site entrance after binding.
The surface of each monomer of ActVA-Orf6 monooxygenase is made up by polar amino acids exposed to the solvent, with a clustering of positive side chains (Arg73, Arg86 and Lys100) around the active site crevice, and negatively charged ones (Glu3, Glu30, Glu38, Glu42, Glu67 and Asp6) on the opposite side of the protein. This is consistent with the requirement for attracting a negatively-charged substrate towards the active site of the enzyme.
A β-turn (Figure 2A, residues: Val43, Pro44, Gly45 and Phe46) positioned between helix A2 and strand B2 can be ascribed to the class of type II β-turns (Lewis et al., 1973). This is not a general feature of ferredoxin-like proteins, which can adopt different conformations for the β-turn in this topological position. Pro44 and Gly45 are strictly conserved among all the homologues of ActVA-Orf6 (Figure 3) and have the highest propensity for these positions in type II β-turns (Hutchinson and Thornton, 1994).

Fig. 3. Multiple sequence alignment of proteins similar to ActVA-Orf6 monooxygenase. The sequences have been selected from the output of a PSI-BLAST search performed using ActVA-Orf6 monooxygenase as query. Not all hits are shown in the alignment, and the alignments are less reliable from residue 75. Above the alignment, the secondary structure of ActVA-Orf6 is shown. The genes are clustered in three groups: a family of monooxygenases with an interface identical to ActVA-Orf6 monooxygenase (orange frame); a second family of putative Streptomyces monooxygenases that probably possess a different interface (green frame); and hypothetical proteins of unassigned function (blue frame). Aligned sequences: protein, organism (gene name and accession No. in SWALL database in parentheses): coelico, ActVA-Orf6 monooxygenase, S.coelicolor (ActVA-Orf6, Q53908); glauces, TcmF1 monooxygenase, S.glaucescens; (TcmH, P39889); olivace, TcmF1 monooxygenase, S.olivaceus (ElmH, Q9L4Y0); violace, C-5 anthrone oxidase, S.violaceus (Jad-Orf7, Q56157); str. C5, oxygenase, unspeciated streptomycete strain C5 (Streptomyces sp. C5; from the Frederick Cancer Research Council) (DauA-OrfE, Q55222); galilae, aklanonic acid anthrone monooxygenase, S.galilaeus (AknX, Q9L552); nogalat, SnoB protein, S.nogalater (SnoB, Q54493); peuceti, anthraquinol monooxygenase, S.peucetius (Dps-Orf8, Q54813); HY coeN, N-terminus of hypothetical 26.4 kDa protein, S.coelicolor (SCI7.27C, Q9X9W3); HY coeC, C-terminus of the same 26.4 kDa protein, S.coelicolor; HY loti, hypothetical protein, Mesorhizobium loti (MLR8211, Q983R7); HY aeru, hypothetical protein, Psuedomonas aeruginosa (PA2274, Q9I1K0). The biochemical function has been assigned experimentally for ActVA-Orf6 monooxygenase from S.coelicolor, TcmH from S.glaucescens, ElmH from S.olivaceus and AknX from S.galilaeus. For all the other proteins, the function has been deduced on the basis of homology with characterized proteins, although other indirect evidence may be available. Full boxes highlight conserved residues: purple, residues essential for tertiary structure (β-turns); red, interface polar residues (quaternary structure); yellow, active site residues; cyan, other identities. Similarities are only reported if they are consistent with structural data: light purple (tertiary structure) and light red (interface). End of the alignment: the first column reports percentage identity calculated for the ‘ferredoxin unit’ lacking helix A4 and the N-terminus (ID%/65, residues 10–74 of ActVA-Orf6 monooxygenase); the second column refers to the ‘minimal structural/functional unit’ (ID%/37, residues 38–74).
A β-turn between strand B3 and helix A3 (Figure 2A, residues: Ser68, Glu69, Gln70 and Ala71) can be defined as a type III β-turn (Crawford et al., 1973; Lewis et al., 1973). The classical hydrogen bond interaction between the carbonyl of the first residue of the turn and the amide proton of the fourth residue is absent in this case. On the contrary, the side chain γ-oxygen and the carbonyl of Ser68 are hydrogen bonded to the amide group of Ala71 and Tyr72, respectively. These two interactions seem to stabilize the backbone of N-terminal residues of helix A3. The multiple alignment (Figure 3) shows that this serine is conserved throughout the homologues of ActVA-Orf6 or, alternatively, can be substituted by an aspartate residue, which is also a hydrogen bond acceptor and thus might play a similar role.
The interface between the two monomers of ActVA-Orf6 is stabilized by hydrophobic interactions contributed by the β-sheets, with additional specific intermolecular contributions provided by Tyr63 and His52 of the two monomers, through tyrosine–tyrosine aromatic stacking and tyrosine– histidine hydrogen bonding (Figure 2B). Interestingly, these two residues are conserved throughout the homologous Tcm F1 monooxygenases from S.glaucescens (Shen and Hutchinson, 1993) and S.olivaceus (Rafanan et al., 2001), anthrone oxidase from S.venezuelae (Yang et al., 1996), and three hypothetical proteins of unassigned function identified in the complete genome sequences of S.coelicolor, Pseudomonas aeruginosa and Mesorhizobium loti (Figure 3). His52 has been implicated previously in catalysis and protein folding by site-directed mutagenesis studies (Kendrew et al., 1997). It now becomes clear that the vastly reduced activity displayed by the refolded mutant was due to disruption of the dimer interface rather than removal of vital catalytic residues. The sequence alignment shows that residues Ser48, Thr50 and Gln65 are also well conserved and appear to form a stabilizing intramolecular net of hydrogen bonds surrounding the interface residue Tyr63, also involving two water molecules (Figure 2B). The difference in gap distribution, degree of homology and conservation of interface residues across the alignment may indicate that there are two families of monooxygenases, with only one stabilized by a dimer interface that is similar to that displayed by ActVA-Orf6 monooxygenase.
An attractive and clearly visible feature of the dimeric form of ActVA-Orf6 monooxygenase is a swapped strand whereby the C-terminal β-strand (B4, residues 103–113) contributes to the β-sheet of the other monomer by laying antiparallel to strand B2 (Figure 2A). This swap is essential for dimer stability, since its contribution to the buried surface of the interface is ∼2200 Å2 over a total of 3300 Å2. Importantly, the swapped strand takes part in the formation of the bottom of the active site, to which it contributes with Ile110 (Figure 2F). This segment is present in all the homologous sequences that conserve the residues involved in the dimerization contacts (Figure 3), apart from that of C-5 anthrone oxidase from S.violaceus, which is incomplete (Yang et al., 1996) at the C-terminus. Two different ferredoxin-like proteins, the bovine papillomavirus-1 E2 DNA-binding domain (Hegde et al., 1992) and muconolactone isomerase from Pseudomonas putida (Katti et al., 1989), are also dimeric; however, neither of the two displays the β-strand swapping observed in ActVA-Orf6 monooxygenase, and in the E2 DNA-binding domain the 2-fold symmetry axis is orthogonal to that observed in our case.
In summary, examination of the three-dimensional structure suggests that ActVA-Orf6 monooxygenase is a new example of the widely represented ferredoxin-like fold, with a novel dimeric assembly. Furthermore, it is likely that the model will be a template for the tertiary structure of many homologous proteins across a range of microorganisms, and possibly for the quaternary structure of those proteins sharing the same polar residues involved in the dimer interface stabilization.
The active site
ActVA-Orf6 monooxygenase is a small enzyme (113 amino acid residues) that oxidizes a relatively large three-ringed aromatic substrate. Therefore, it is not surprising that all the secondary structure elements are involved in the formation of the active site. This is achieved by disrupting the centre of the helices of the two βαβ elements, resulting in the formation of two arches over a rigid floor contributed by the β-sheets (Figure 2A). The back of the active site cavity is closed by the fifth strand of the sheet and is provided by the other monomer. Within the active site, the accessible surface buried upon binding is ∼200 Å2 and applies to both the protein and the ligand, indicating that there is good shape complementarity. The high resolution and quality of the electron density maps confirm that ActVA-Orf6 monooxygenase, and presumably also the related proteins, does not require a metal ion or a prosthetic group for catalysis, as indicated by the biochemical data. In fact, there is no density that could be attributed to a metal ion, and no cluster of residues with a geometry that would provide a plausible metal coordination site.
The native structure of ActVA-Orf6 monooxygenase (Figure 2A) suggests that four residues may be important for binding and/or catalysis: Tyr51, Asn62 and Trp66 belonging to the β-sheets, and Tyr72 hanging from α-helix A3. The latter residue clearly displays a double conformation on the A subunit, indicating a potential mobility that may be significant in the process of substrate binding or catalysis. Asn62 and Trp66 are conserved across the monooxygenase family, whereas Tyr51 and Tyr72 are less well conserved, perhaps reflecting the differences in the substrates utilized by the homologous enzymes in the family.
In our crystals, the active site of subunit A is less accessible to solvent due to crystallographic packing and appears to be occupied by a polyethylene glycol (PEG) molecule; as we failed to obtain single crystals in the absence of PEG 200, this may be a requirement for crystal growth. To avoid introducing a model bias arising from incorrect positioning of this very mobile molecule, a PEG chain was not fitted to the electron density in the high-resolution model of the A subunit. However, good quality crystals containing a ligand in the A site were obtained by co-crystallization with nanaomycin D, a structural analogue of the natural product, and also with ANSA (1-anilino-8-naphthalenesulfonic acid; data not shown). This was found to bind within the active site of subunit A despite a molecular structure that is significantly different from that of the natural substrate.
The most notable difference between the two monomers is the higher mobility of the region comprising amino acids 34–38 in subunit B (Figure 2A) with respect to subunit A. This difference in mobility is probably induced by crystal packing, but it may also play a functional role or at least points to an intrinsic mobility of the segment since it indicates that it can adopt two different conformations. In particular, in subunit A, Gln37 points towards the centre of the active site, making a hydrogen bond with Tyr51, a residue also implicated in binding or catalysis, whereas in subunit B it adopts a different conformation allowing a water molecule to enter and replace Gln37 in its interaction with the hydroxyl of Tyr51. In the latter conformation, movement of Gln37 opens a channel between the solvent and the bottom of the active site (Figure 2F).
In the absence of the natural substrate, further understanding of the enzymic mechanism was achieved through the binding of substrate and product analogues to the active site. Four structures of ActVA-Orf6 monooxygenase have been solved (Table I), bound to sancycline and acetyldithranol (analogues of the substrate) or to nanaomycin D and the oxidized form of acetyldithranol (analogues of the product) (Figure 4). Nanaomycin D was co-crystallized with ActVA-Orf6 monooxygenase, whereas the other three analogues were diffused into native crystals. The two models of ActVA-Orf6 monooxygenase bound with reduced and oxidized acetyldithranol were obtained using the same preparation of soaked crystals, but freezing after 4 h and 2 weeks, respectively. This protocol was used because preliminary results showed that ActVA-Orf6 monooxygenase appeared (at least to some extent) to catalyse the oxidation of acetyldithranol (data not shown). This fits with other data indicating a broad substrate tolerance for these proteins (Kendrew et al., 1997). In fact, in crystals frozen after 2 weeks, the oxidized form of acetyldithranol can be seen bound at the active site (Figure 5A).
Table I. Summary of crystallographic and refinement statistics of ActVA-Orf6 monooxygenase.
| |
Native |
Sanca |
AcetDit |
AcDOX |
Nana D |
K2PtCl4 |
Mers |
Mers 1 |
|---|---|---|---|---|---|---|---|---|
| Data collection | ||||||||
| Space group: | P212121 | |||||||
| Res. limit (Å) | 1.30 | 1.74 | 1.70 | 1.90 | 2.24 | 2.30 | 2.80 | 2.20 |
| Total refl. (n) | 798 091 | 118 991 | 228 029 | 64 564 | 68 040 | |||
| Unique refl.b (n) | 50 368 | 21 456 | 22 894 | 16 795 | 10 268 | |||
| Completeness (%) | 99.8 | 99.9 | 100.0 | 99.6 | 99.9 | 100.0 | 99.8 | 100.0 |
| (last shell) | 100.0 | 98.6 | 99.8 | 99.4 | 99.0 | |||
| Rmergec | 0.059 | 0.071 | 0.025 | 0.052 | 0.127 | 0.087 | 0.069 | 0.058 |
| (last shell) | 0.136 | 0.329 | 0.098 | 0.235 | 0.591 | |||
| <I>/<σ>d | 36.6 | 21.0 | 99.6 | 19.4 | 12.4 | |||
| (last shell) | 12.2 | 3.8 | 18.3 | 2.9 | 2.1 | |||
| Soak. time (days) |
|
5 |
0.5 |
14 |
|
45 |
7.5 |
21 |
| MIRAS analysis |
|
|
|
|
|
|
|
|
| No. of sites | 4 | 2 | 1 | |||||
| Rcullise | 0.87 | 0.95 | 0.88 | |||||
| Rcullis anoe | 0.95 | 0.93 | 0.80 | |||||
| Phasing powerf |
|
|
|
|
|
0.90 |
0.62 |
0.70 |
| Structure refinement |
|
|
|
|
|
|
|
|
| Low res. (Å) | 20.0 | 15.0 | 20.0 | 20.0 | 20.0 | |||
| High res. (Å) | 1.30 | 1.74 | 1.70 | 1.90 | 2.24 | |||
| Solvent atoms | 258 | 179 | 158 | 121 | 60 | |||
| Work reflections | 47 745 | 20 327 | 21 673 | 15 686 | 9739 | |||
| Free reflections | 2560 | 1098 | 1170 | 834 | 493 | |||
| Rcrystg | 0.142 | 0.190 | 0.206 | 0.202 | 0.214 | |||
| Rfreeg | 0.168 | 0.240 | 0.246 | 0.256 | 0.272 | |||
| R.m.s.d. bonds (Å) | 0.013 | 0.015 | 0.015 | 0.015 | 0.013 | |||
| R.m.s.d. angles (°) | 1.99 | 2.50 | 2.66 | 2.50 | 2.80 |
aDerivatives of ActVA-Orf6: Sanc, sancycline; AcetDit, acetyl dithranol; AcDOX, oxidized acetyl dithranol; Nana D, nanaomycin D; Mers, Mers 1, mersalyl.
bUnique reflections with I/σ>1, where I is the measured intensity and σ is the error of the intensity.
cRmerge = Σhkl Σ|Ij(hkl) – <I(hkl)>|/ΣhklΣj<I(hkl)>, with Ij(hkl) representing the intensity of measurement j and <I(hkl)> the mean of measurements for the reflection hkl.
d<I >/<σ> = ratio between the mean intensity and the mean error of the intensity.
eRcullis = ΣE/ΣD, where E is the residual lack of closure error and D is the isomorphous difference and the anomalous difference for Rcullis and Rcullis ano, respectively.
fPhasing power = <FH>/E, where <FH> is the root mean square heavy-atom structure factor.
gRcryst = Σhkl||Fcalc(hkl)| – |Fobs(hkl)||/Σhkl|Fobs(hkl)|, where Fcalc and Fobs are the calculated and measured structure factors, respectively. Rfree is calculated in the same way on a set of structure factors (5% of the total) that have not been used for model refinement.

Fig. 4. Structure of the four ligands bound to ActVA-Orf6 crystals.

Fig. 5. Active site analysis. (A) 2Fo – Fc electron density map contoured at 1.0σ cut-off around oxidized acetyl dithranol and active site residues in the B subunit. (B) 2Fo – Fc electron density map contoured at 1.0σ cut-off around nanaomycin D in the A site. (C and D) Superposition in two different orientations of active sites from the five models of ActVA-Orf6 and significant hydrogen bond distances. Pink, native active site B; dark red, nanaomycin D in active site A (A = 2.9, B = 2.6, C = 2.4, D = 2.4, E = 2.8, F >13 Å); cyan, sancycline bound to the B site (A = 3.1, B = 3.1, F = 4.1 Å); green, acetyl dithranol bound to the B site (A = 3.3, B = 3.4, F = 3.5 Å); yellow, oxidized acetyl dithranol in the B site (A = 3.1, B = 3.4, F = 2.7 Å). The water molecule is only seen when nanaomycin D is bound to active site A. The different conformations of Gln37 in the two sites are clearly visible. The side chain oxygen of Gln37 is hydrogen-bonded to Tyr51 in the A site (dark red) and replaced by a water molecule (not shown) in the B site of all the other structures. Arg86 is forced out of the active site in the A subunit (dark red) by crystallographic packing. In the B subunit, it moves towards Tyr72 after diffusion of the ligands (green and yellow). A complete model of the dimer including nanaomycin D and side chains as in (C) and (D) is presented in the Supplementary data available at The EMBO Journal Online.
The four analogues used share many of the structural features of 6-DDHK and polycyclic aromatic polyketide intermediates that undergo the oxygenation catalysed by this family of proteins. The hydroxyl groups at positions C-11 and C-13 form hydrogen bonds with Trp66 and Tyr72, which act as hydrogen bond donor and acceptor, respectively. These bonds are crucial to fix the position of the substrate in the active site (Figure 5C and D). Only in the case of nanaomycin D are these hydrogen bonds <3.0 Å, allowing a water molecule to bind on the opposite side of the rings, hydrogen bonded to Tyr51, Asn62 and the C-6 keto group of the product analogue (Figure 5B, C and D). By superimposing the A and B subunits of the five models, we have observed that the positions of Asn62 and Tyr51 are relatively fixed, whereas Trp66 and Tyr72 undergo a displacement that is related to the motion of helix A3 (Figure 5C and D).
It appears that the active sites of the two monomers are not equally available to the different substrate/product analogues. In fact, substrate analogues such as sancycline and acetyldithranol bind only to subunit B, whereas nanaomycin D, which in some ways is more similar to the product, was found to bind only to subunit A in both soaking and co-crystallization experiments. The structural basis for the apparent discrimination between substrate-like and product-like molecules appears to be related to the different conformations of Gln37 in subunit A and B induced by crystal packing; this residue is close to the carbon atom C-6 that undergoes oxidation. Oxidized acetyldithranol, bound to subunit B, derives from the substrate acetyldithranol, which has not escaped after catalysis. Comparing the native and analogue-bound models of ActVA-Orf6 monooxygenase, we observed that Arg86 (a residue well conserved among the homologous sequences; see Figure 3), like Gln37, can assume different conformations. This residue is placed at the entrance of the active site but on the opposite side to Tyr72 (Figures 2A and 5). In subunit B of the native structure, it has a double conformation, either pointing towards the active site ∼5 Å from Tyr72 or extending far from the binding cavity. On the other hand, in subunit A, it points outside the active site, due to an electrostatic interaction with Glu84 from a symmetry-related B subunit (Figure 5B, C and D). The conformation of Arg86 is also variable in the models of the complexes of ActVA-Orf6 with the various analogues (Figure 5C and D). In the sancycline derivative, Arg86 of subunit B retains a similar conformation to the native enzyme, whereas after binding of acetyldithranol this residue is in a fixed position (confirmed by a strong electron density) with the guanidine group hydrogen bonded (3.4 Å) to Tyr72. This distance is even shorter (2.7 Å) in the complex with oxidized acetyldithranol. This all suggests that the different ligands and possibly the size/charge of the ligands have an effect on the position of this invariant residue.
The carboxyl group of the substrate extends from the active site pocket and is exposed to solvent (Figure 2G). Thus, this portion of the molecule does not appear to play a significant role in substrate recognition and is consistent with the ability of this enzyme to oxidize a range of substrates including larger substrates, such as TcmF1, which contain additional aromatic rings, and may also go some way to help explain the apparent differences in timing of the monooxygenation step (i.e. before or after final ring cyclization), which occurs across the different biosynthetic pathways of aromatic polyketides. This consideration may be generalized to this class of monooxygenases: experimental evidence shows that TcmF1 monooxygenase, aklanonic acid anthrone monooxygenase and ActVA-Orf6 (Shen and Hutchinson, 1993; Fujii and Ebizuka, 1997; Kendrew et al., 1997) can accept substrates that differ in size or chemical character from the natural ones on the side that would remain external from the active site, since they are not fully buried in the protein by a ‘lid closure’ mechanism. On the other hand, ActVA-Orf6 can catalyse the oxidation of TcmF1, a substrate that possesses a hydroxy functionality in the first ring, which is not part of the natural substrate, so it is clear that this group is not a primary determinant of specificity. However, we could not identify an obvious candidate residue that potentially might bind this hydroxyl group in our modelling studies with TcmH. As far as the specific interactions that determine substrate binding in other proteins are concerned, the main determinant seems to be the strictly conserved Trp66 and Tyr72, which in other monooxygenases might be substituted by other residues in the C-terminal region of the sequence, where homology is too low to identify topologically equivalent positions reliably. The overall shape and hydrophobic character of the active site are clearly important requirements for substrate binding in this class of enzymes.
Discussion
The discovery that the reaction catalysed by some monooxygenases involved in the biosynthesis of aromatic polyketide antibiotics does not require metals or other cofactors was unexpected and originally led to the proposal of a protein radical mechanism (Shen and Hutchinson, 1993; Fujii and Ebizuka, 1997). According to this hypothesis, the enzyme can extract a proton and an electron from the substrate to yield a phenolic radical form of the polyketide substrate, which is activated for the subsequent reaction with molecular oxygen. This leaves a radical species on the enzyme. The conserved tryptophan (position 66 in ActVA-Orf6) has been considered the most likely candidate to be involved in the radical mechanism and in dioxygen activation with the formation of the peroxide (a protonated superoxide anion) that could then attack the substrate. However, examination of the high-resolution three-dimensional structure of ActVA-Orf6 monooxygenase shows that this is unlikely, as Trp66 is buried deep in the active site and located on the opposite side with respect to the reactive carbon C-6 of the substrate. This residue is always >7.5 Å away from this carbon and not accessible to O2 upon ligand binding. Further evidence for the role of Trp66 comes from studies (Chung et al., 2002) in which site-directed mutants of this tryptophan in the homologous protein AknX are active but possess an increased Km for the substrate emodinanthrone. Furthermore, the few characterized enzymatic systems involved in radical reactions contain prosthetic groups, metal ions or at least modified amino acids such as dihydroxylated tyrosines or tryptophan tryptophylquinones, features (Pedersen and Finazzi-Agrò, 1993) clearly not visible in the structure of ActVA-Orf6. It has been proposed that His52 might act as a general base to catalyse the dehydration of a putative peroxy intermediate formed by a radical or other reaction mechanism (Kendrew et al., 1997). Again the structure shows that His52 lies away from the active site and is involved in stabilizing the dimer interface. This shows the value of comprehensive structural studies and clearly illustrates the difficulties involved in predicting enzymatic reaction mechanism using site-directed mutagenesis.
In an effort to shed further light on the enzymatic mechanism of ActVA-Orf6 monooxygenase, we have turned to some of the models explaining autoxidation of anthrones such as dithranol or similar antipsoriatic drugs (Muller and Gurster, 1993; Hayden et al., 1994; Muller, 1996). In these systems, a carbanionic form coupled to deprotonation is stabilized by their aromatic systems as well as by intramolecular hydrogen bonds between adjacent hydroxyl and keto groups positioned on the same side of the molecule. A subsequent oxidation, coupled for instance to reduction of O2 to O2·– (superoxide anion), leads to the formation of the radical form, stabilized in the same manner as the carbanion. This radical reacts with molecular oxygen or the superoxide anion radical to form a peroxy intermediate. In systems in which the superoxide anion cannot be detected, there is evidence for anthronyl-peroxyl radical formation instead (Hayden et al., 1994). While carbanionic anthrones react with molecular oxygen to give the anthraquinone form (directly analogous to the products of polyketide monooxygenases), the radical intermediates can also lead to the formation of many oxidatively coupled products, as well as leading to reactive radical species of anthrones and oxygen, some of which can be very toxic (Hayden et al., 1994).
Using the autoxidation of anthrones as a potential model, it is possible to build a reaction mechanism utilizing the ActVA-Orf6 structural characteristics. The invariant residue Trp66 seems to be essential for specific recognition of the C-11 hydroxyl (Figure 5), which is not only characteristic of 6-DDHK but common to all the substrates of this class of enzymes, and may be a necessary functional group for catalysis as only anthrones containing an equivalent group undergo autoxidation (Hayden et al., 1994). This hydrogen bond between the C-11 hydroxyl and Trp66 helps to orientate the substrate in the active site, with the indole NH acting as a hydrogen donor, enhances the formation of a network of intramolecular hydrogen bonds on the substrate (Figure 6A) and promotes the deprotonation of a hydroxyl group: the first step of the catalysis (Figure 6B). This leads to the formation of a C-6 carbanion (Figure 6C) as one of the favoured possible resonant forms, resembling the primary species of anthrone anions during autoxidation in solution. Depro tonation of the substrate probably involves Arg86 and Tyr72. The latter residue plays an essential role in this mechanism because, via its hydrogen bond with Arg86, it represents a likely route for the extracted proton to the solvent.

Fig. 6. Putative catalytic mechanism of ActVA-Orf6 monooxygenase. Once 6-DDHK (shown here with alternative tautomeric form) enters the active site, stabilization of intramolecular hydrogen bonds (A) is associated with H+ transfer to Tyr72 (B) to yield the deprotonated form. C-6 carbanion can react with molecular oxygen (C). This leads to a peroxy intermediate (D), stabilized by hydrogen bonds with Asn62 and Tyr51. Protonation followed by dehydration is then necessary to complete the reaction.
Whereas in 6-DDHK the methyl group at C-15 prevents solvent exposure of C-13 hydroxyl after binding into the active site, the natural substrates of homologous monooxygenases often lack this methyl, but instead have either extra aromatic rings, or hydroxyl or keto substituents. These may fulfil the role played by the C-13 group in 6-DDHK, and extend the intramolecular hydrogen bond network generated on the substrate. Unlike in ActVA-Orf6, deprotonation of these groups might then be carried out directly by the solvent or Arg86, or by other residues taking the role of Tyr72 but placed elsewhere in the primary sequence. This may explain why, unlike Arg86, Tyr72 is not conserved throughout the family.
The binding of substrate via Trp66 and subsequent formation of the C-6 carbanion appears to be the vital step, allowing the correct chemistry for reaction with oxygen. Once the carbanion at C-6 is formed, attack of molecular oxygen may occur, using the same kind of chemistry as proposed for autoxidation of anthrones in solution. Though forbidden on the basis of spin conservation, this process closely resembles the direct reaction between singlet reduced flavins and triplet O2 occurring both in solution and in many flavoenzymes (Massey, 1994), such as cyclohexanone monooxygenase (Ryerson et al., 1982), 2-hydroxybiphenyl 3-monooxygenase (Suske et al., 1999) and the FMN-dependent bacterial luciferase (Xin et al., 1991; Huang and Tu, 1997). For flavoenzymes, it is assumed that the reduced flavin (a carbanion on C-4a) may directly reduce molecular oxygen through the formation of two intermediate species, a semiquinone-superoxide (anion) caged radical and, after spin inversion, a C(4a)-hydroperoxide anion (Massey, 1994). More recently, the same mechanism has been proposed for the activation of molecular oxygen towards an intramolecular oxygenation in the case of 1H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase (Fischer and Fetzner, 2000).
We believe that oxygen may be activated in a similar way by the substrate itself in the active site of ActVA-Orf6 monooxygenase, leading to the formation of a peroxy intermediate stabilized by a hydrogen bond with Asn62 and Tyr51 (Figure 6D). Geometrical inspection of a putative reaction intermediate shows that these two residues are in a favourable position for this task (Figure 7). This is confirmed by the presence of a water molecule in the active site of ActVA-Orf6 monooxygenase bound to nanaomycin D (Figure 5B), which perhaps mimics the second oxygen atom of a peroxy intermediate. The exact role of Tyr51, although clearly in the active site, is uncertain, because, unlike Asn62, it is not conserved. However, as we have observed that all other monooxygenases have a strictly conserved asparagine (topologically equivalent to Val15 in ActVA-Orf6), it may be possible that this asparagine could flank Asn62, to take the role of Tyr51 (though on the opposite side with respect to the position of Tyr51). This possibility is also suggested by homology models of TcmF1 monooxygenases from S.glaucescens and S.olivaceus (data not shown), which we have obtained using automatic servers for protein modelling (Guex et al., 1999). The final step in the catalytic cycle would be protonation and dehydration of the peroxy intermediate, with consequent formation of a water molecule.
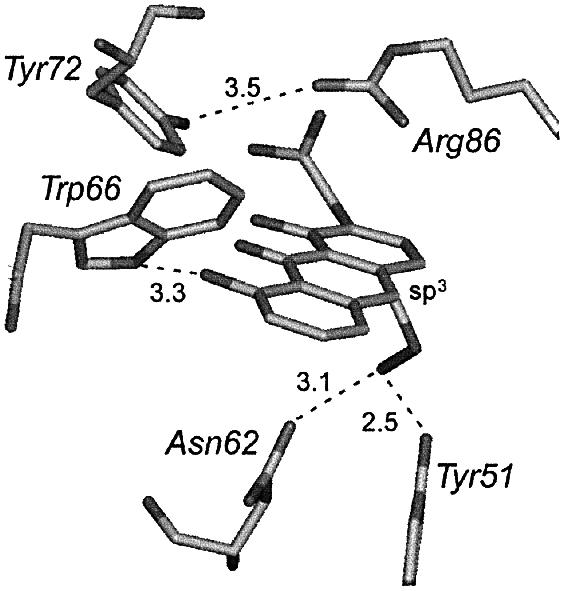
Fig. 7. Hypothetical peroxy intermediate built on the atomic coordinates of acetyl dithranol in the active site B of ActVA-Orf6 monooxygenase. The model assumes sp3 geometry for the reactive carbon C-6. Asn62 and Tyr51 are in favourable stereochemistry to stabilize such an intermediate by hydrogen bonding.
The model for the binding of the substrate to the active site of ActVA-Orf6 monooxygenase accounts for the preferential oxidation of C-6, rather than other potentially reactive carbons. This is because all the potentially reactive atoms except this one are in direct contact with the protein matrix through van der Waals or polar interactions. To make them accessible to attack by molecular oxygen, a significant displacement of the side chains of the rigid enzyme scaffold would be required. Hence, the active site cavity acts as a shield, leaving only the reactive C-6 carbanion (and the carboxylate group) exposed to the solvent bulk. On the other hand, the mobile loop 34–38 and particularly Gln37 appear to represent a gate, opening a narrow tunnel (Figure 2F) for diffusion of O2 from the bulk towards the carbanion at C-6, allowing oxygen addition, whereas the main crevice of the active site is obstructed by the pyrano-acetyl moiety of the substrate (Figure 2G). This Gln37-gated tunnel may also allow the diffusion of H2O out into the bulk, as the final catalytic step.
In conclusion, we have solved the structure of the first member of a class of small monooxygenases carrying out oxygenation without the assistance of cofactors. This indicates that yet another functionality may be compatible and sustained by the ferredoxin fold, adopted by convergent evolution to carry out a wide range of tasks. Furthermore, we propose a catalytic mechanism for ActVA-Orf6 monooxygenase, one of the tailoring enzymes involved in the biosynthesis of the aromatic polyketide actinorhodin, based on the high resolution crystallographic structure of the native enzyme and its complex with several substrate/product analogues. The mechanism that we are proposing will be tested by site-directed mutagenesis using the natural substrate, which is not yet available. This scheme allows us to visualize the mechanism of binding and activation of the 6-DDHK substrate for attack by molecular oxygen, and utilization of two gates for the reaction components, with a H+ gate represented by Tyr72/Arg86 and, on the opposite side of the bound substrate, an O2/H2O gate consisting of a putative protein channel and involving Gln37. This is the first crystal structure of an enzyme involved in the tailoring of a type II aromatic polyketide and has illustrated some of the enzyme–substrate recognition features that may apply to the range of other enzymes involved in modifying the polyketide core structure.
Materials and methods
Crystallization and data collection
Recombinant ActVA-Orf6 monooxygenase was expressed and purified from Escherichia coli containing the gene ActVA-Orf6 from S.coelicolor under the T7 promoter, following the methods previously described (Kendrew et al., 1997). The 98% pure protein has been used extensively for growing crystals following a determined procedure (Kendrew et al., 2000). The optimal crystallization conditions were determined to be 1.5 M ammonium sulfate, 100 mM HEPES/Tris–HCl pH 7.0/8.0, 10–15% PEG 200, 293 K. We attempted to obtain better crystals using both hanging- and sitting-drop vapour diffusion techniques and, although addition of KBr or KI markedly improved crystal growth, we could not detect halogens co-crystallized in the final structures. Micro- and macroseeding techniques have been used to grow large single crystals (up to 0.5 × 0.5 × 1 mm). Co-crystallization of ActVA-Orf6 monooxygenase and nanaomycin D under the same conditions produced smaller isomorphous crystals that diffracted to 2.2 Å. However, diffusing this compound into previously grown native crystals yielded poorly diffracting crystals. Except for nanaomycin D, all the derivatives of ActVA-Orf6 were obtained by soaking native crystals previously grown or transferred to a sitting drop in 20 µl of mother liquor, which contained 2–10 mM of each ligand or heavy metal salt. Soaking times are reported in Table I. Acetyl dithranol has been synthesized according to Tanzer et al. (1988).
All data were collected at cryogenic temperature (100–120 K) at the synchrotrons ELETTRA (Trieste, Italy) and ESRF (Grenoble, France), equipped with a MAR 345 image plate and a MAR CCD detector, respectively. Data were indexed with DENZO and scaled and reduced with SCALEPACK (Otwinoski and Minor, 1996). Heavy metal derivative data were scaled with SCALA and FHSCALE in the CCP4 package (CCP4, 1994). The space group was orthorhombic P21212 1, with unit cell dimensions a = 47.03 Å, b = 60.17 Å, c = 71.38 Å for the native crystals.
Structure determination and refinement
ActVA-Orf6 monooxygenase crystal structure was determined by MIRAS (multiple isomorphous replacement and anomalous scattering) using three heavy metal derivatives (see Table I). The heavy atom positions for the derivatives were determined in the Patterson maps using FFT/PEAKMAX (CCP4, 1994) and SHELXS (Sheldrick et al., 1993) and confirmed by the difference cross-Fourier technique. The heavy atom positions were refined together with MLPHARE (Otwinowski, 1991) to produce a first set of phases. A density modification procedure based on solvent flattening and histogram matching algorithms was then applied using the program DM (Cowtan, 1994). Although the initial maps were not readily interpretable, they allowed production of the native structure by running the automated building procedure in the program Arp/Warp (Perrakis et al., 1999). The model was then refined at 1.30 Å resolution using REFMAC (Murshudov et al., 1997) from the CCP4 package. Every step of the refinement was verified and improved by inspection of the electron density maps, correction of the side chain geometry and addition of solvent molecules using QUANTA (Molecular Structure Inc.). Using the atom coordinates of the native structure for phasing, four isomorphous structures were subsequently determined and refined in the same way, with the enzyme in complex with analogues of substrate and product bound to the active site (Table I). Each model of the protein contains a different number of water molecules depending on the resolution limits of the respective data collection (Table I). All of the 113 residues of the polypeptide chain are visible in the electron density map obtained for each individual model refined, except for Met1, which is processed during expression in E.coli.
Accessible surfaces have been calculated using AREAIMOL (CCP4, 1994), and buried surfaces as differences between them. Accessible volumes have been calculated using Insight II (Dayringer et al., 1986) and CASTp (Liang et al., 1998). Putative three-dimensional structures of homologues of ActVA-Orf6 monooxygenase have been obtained using the automatic modeller program SWISS-MODEL (Guex et al., 1999). The coordinates and structure factors of native and complexed ActVA-Orf6 have been deposited in the Protein Data Bank, with accession codes 1LQ9, 1N5Q, 1N5S, 1N5T and 1N5V.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Professors S.Ômura and D.Hopwood for the kind gift of nanaomycin D, and to Hovione Ltd (Portugal) for the generous gift of sancycline. Data were collected at the synchrotrons ELETTRA (Trieste, Italy) and ESRF (Grenoble, France). We thank Ed Mitchell (ESRF, Grenoble) for expert support at beamline ID-14, and V.Lamzin (EMBL, Hamburg) for invaluable guidance in the application of Arp/Warp for structure solution. We are also grateful to Professors M.Brunori and A.Tramontano for critical reading of this manuscript. This work was partially supported by Istituto Pasteur-Fondazione Cenci Bolognetti (to S.G.K.), MIUR of Italy (PRIN ‘Bioenergetica: aspetti genetici, biochimici e fisiopatologici’ to F.M.) and by the Italian CNR-Progetto Finalizzato Biotecnologie.
References
- Bentley S.D. et al. (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature, 417, 141–147. [DOI] [PubMed] [Google Scholar]
- Caballero J.L., Martinez,E., Malpartida,F. and Hopwood,D.A. (1991) Organisation and functions of the actVA region of the actinorhodin biosynthetic gene cluster of Streptomyces coelicolor. Mol. Gen. Genet., 230, 401–412. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Chung J., Fujii,I., Harada,S., Sankawa,U. and Ebizuka,Y. (2002) Expression, purification, and characterization of AknX oxygenase, which is involved in aklavinone biosynthesis in Streptomyces galilaeus. J. Bacteriol., 184, 6115–6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. (1994) DM: an automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newslett. Protein Crystallogr., 31, 34–38. [Google Scholar]
- Crawford J.L., Lipscomb,W.N. and Schellman,C.G. (1973) The reverse turn as a polypeptide conformation in globular proteins. Proc. Natl Acad. Sci. USA, 70, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump M.P., Crosby,J., Dempsey,C.E., Parkinson,J.A., Murray,M., Hopwood,D.A. and Simpson,T.J. (1997) Solution structure of the actinorhodin polyketide synthase acyl carrier protein from Streptomyces coelicolor. Biochemistry, 36, 6000–6008. [DOI] [PubMed] [Google Scholar]
- Cupp-Vickery J.R. and Poulos,T.L. (1995) Structure of cytochrome P450eryF involved in erythromycin biosynthesis. Nat. Struct. Biol., 2, 144–153. [DOI] [PubMed] [Google Scholar]
- Dayringer H.E., Tramontano,A., Sprang,S.R. and Fletterick,R.J. (1986) Interactive program for visualization and modelling of proteins, nucleic acids and small molecules. J. Mol. Graphics, 4, 82–87. [Google Scholar]
- Fernández-Moreno M.A., Caballero,J.L., Hopwood,D.A. and Malpartida,F. (1991) The act cluster contains regulatory and antibiotic export genes, direct targets for translational control by the bldA tRNA gene of Streptomyces. Cell, 66, 769–780. [DOI] [PubMed] [Google Scholar]
- Fernández-Moreno M.A., Martinez,E., Boto,L., Hopwood,D.A. and Malpartida,F. (1992) Nucleotide sequence and deduced functions of a set of cotranscribed genes of Streptomyces coelicolor A3(2) including the polyketide synthase for the antibiotic actinorhodin. J. Biol. Chem., 267, 19278–19290. [PubMed] [Google Scholar]
- Fernández-Moreno M.A., Martinez,E., Caballero,J.L., Ichinose,K., Hopwood,D.A. and Malpartida,F. (1994) DNA sequence and functions of the actVI region of the actinorhodin biosynthetic gene cluster of Streptomyces coelicolor A3(2). J. Biol. Chem., 269, 24854–24863. [PubMed] [Google Scholar]
- Ferrer J.L.,Jez,J.M., Bowman,M.E., Dixon,R.A. and Noel,J.P. (1999) Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol., 6, 775–784. [DOI] [PubMed] [Google Scholar]
- Fischer F. and Fetzner,S. (2000) Site-directed mutagenesis of potential catalytic residues in 1H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase, and hypothesis on the catalytic mechanism of 2,4-dioxygenolytic ring cleavage. FEMS Microbiol. Lett., 190, 21–27. [DOI] [PubMed] [Google Scholar]
- Fujii I. and Ebizuka,Y. (1997) Anthracycline biosynthesis in Streptomyces galilaeus. Chem. Rev., 97, 2511–2523. [DOI] [PubMed] [Google Scholar]
- Guex N., Diemand,A. and Peitsch,M.C. (1999) Protein modelling for all. Trends Biochem. Sci., 24, 364–367. [DOI] [PubMed] [Google Scholar]
- Hallam S.E., Malpartida,F. and Hopwood,D.A. (1988) Nucleotide sequence, transcription and deduced function of a gene involved in polyketide antibiotic synthesis in Streptomyces coelicolor. Gene, 74, 305–320. [DOI] [PubMed] [Google Scholar]
- Hayden P.J., Free,K.E. and Chignell,C.F. (1994) Structure–activity relationships for the formation of secondary radicals and inhibition of keratinocyte proliferation by 9-anthrones. Mol. Pharmacol., 46, 186–198. [PubMed] [Google Scholar]
- Hegde R.S., Grossman,S.R., Laimins,L.A. and Sigler,P.B. (1992) Crystal structure at 1.7 Å of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature, 359, 505–512. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Hopwood D.A. and Sherman,D.H. (1990) Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu. Rev. Genet., 24, 37–66. [DOI] [PubMed] [Google Scholar]
- Hopwood D.A. (1997) Genetic contributions to understanding polyketide synthases. Chem. Rev., 97, 2465–2498. [DOI] [PubMed] [Google Scholar]
- Huang S. and Tu,S.-C. (1997) Identification and characterization of a catalytic base in bacterial luciferase by chemical rescue of a dark mutant. Biochemistry, 36, 14609–14615. [DOI] [PubMed] [Google Scholar]
- Hutchinson C.R. (1997) Biosynthetic studies of daunorubicin and tetracenomycin C. Chem. Rev., 97, 2525–2536. [DOI] [PubMed] [Google Scholar]
- Hutchinson C.R. and Fujii,I. (1995) Polyketide synthase gene manipulation: a structure–function approach in engineering novel antibiotics. Annu. Rev. Microbiol., 49, 201–238. [DOI] [PubMed] [Google Scholar]
- Hutchinson E.G. and Thornton,J.M. (1994) A revised set of potentials for β-turn formation in proteins. Protein Sci., 3, 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jez J.M., Austin,M.B., Ferrer,J., Bowmann,M.E., Schroeder,J. and Noel,J.P. (2000) Structural control of polyketide formation in plant-specific polyketide synthases. Chem. Biol., 7, 919–930. [DOI] [PubMed] [Google Scholar]
- Katti S.K., Katz,B.A. and Wyckoff,H.W. (1989) Crystal structure of muconolactone isomerase at 3.3 Å resolution. J. Mol. Biol., 205, 557–571. [DOI] [PubMed] [Google Scholar]
- Kendrew S.G., Hopwood,D.A. and Marsh,E.N.G. (1997) Identification of a monooxygenase from Streptomyces coelicolor A3(2) involved in the biosynthesis of actinorhodin: purification and characterization of the recombinant enzyme. J. Bacteriol., 179, 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrew S.G., Federici,L., Savino,C., Miele,A., Marsh,E.N.G. and Vallone,B. (2000) Crystallization and preliminary X-ray diffraction studies of a monooxygenase from Streptomyces coelicolor A3(2) involved in the biosynthesis of the polyketide actinorhodin. Acta Crystallogr. D, 56, 481–483. [DOI] [PubMed] [Google Scholar]
- Lewis P.N., Momany,F.A and Sheraga,H.A. (1973) Chain reversals in proteins. Biochim. Biophys. Acta, 303, 211–229. [DOI] [PubMed] [Google Scholar]
- Liang J., Edelsbrunner,H. and Woodward,C. (1998) Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci., 7, 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpartida F. and Hopwood,D.A. (1984) Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature, 309, 462–464. [DOI] [PubMed] [Google Scholar]
- Malpartida F. and Hopwood,D.A. (1986) Physical and genetic characterisation of the gene cluster for the antibiotic actinorhodin in Streptomyces coelicolor A3(2). Mol. Gen. Genet., 205, 66–73. [DOI] [PubMed] [Google Scholar]
- Massey V. (1994) Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem., 269, 22459–22462. [PubMed] [Google Scholar]
- Muller K. (1996) Antipsoriatic anthrones: aspects of oxygen radical formation, challenges and prospects. Gen. Pharmacol., 27, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Muller K. and Gurster,D. (1993) Hydroxyl radical damage to DNA sugar and model membranes induced by anthralin (dithranol). Biochem. Pharmacol., 46, 1695–1704. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum likelihood method. Acta Crystallogr. D., 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Orengo C.A. and Thornton,J.M. (1993) α plus β folds revisited: some favoured motifs. Structure, 1, 105–120. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. (1991) Maximum likelihood refinement of heavy atom parameters. In Wolf,W., Evans,P.R. and Leslie,A.G.W. (eds), Isomorphous Replacement and Anomalous Scattering. Science and Engineering Research Council, Warrington, UK, pp. 80–86. [Google Scholar]
- Otwinowski Z. and Minor,W. (1996) Processing X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pedersen J.Z. and Finazzi-Agrò,A. (1993) Protein-radical enzymes. FEBS Lett., 325, 53–58. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris,R. and Lamzin,V.S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Rafanan E.R., Le,L., Zhao,L., Decker,H. and Shen B. (2001) Cloning, sequencing, and heterologous expression of the elmGHIJ genes involved in the biosynthesis of the polyketide antibiotic elloramycin from Streptomyces olivaceus Tu2353. J. Nat. Prod., 64, 444–449. [DOI] [PubMed] [Google Scholar]
- Ryerson C.C., Ballou,D.P. and Walsh,C. (1982) Mechanistic studies on cyclohexanone oxygenase. Biochemistry, 21, 2644–2655. [DOI] [PubMed] [Google Scholar]
- Serre L., Verbree,E.C., Dauter,Z., Stuitje,A.R. and Derewenda,Z.S. (1995) The Escherichia coli malonyl-CoA:acyl carrier protein transacylase at 1.5-Å resolution. Crystal structure of a fatty acid synthase component. J. Biol. Chem., 270, 12961–12964. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M., Dauter,Z., Wilson,K.S., Hope,H. and Sieker,C. (1993) The application of direct methods and Patterson interpretation to high resolution native protein data. Acta Crystallogr. D, 49, 18–23. [DOI] [PubMed] [Google Scholar]
- Shen B. and Hutchinson,C.R. (1993) Tetracenomycin F1 mono oxygenase: oxidation of a naphtacenone to a naphtacenequinone in the biosynthesis of tetracenomycin C in Streptomyces glaucescens. Biochemistry, 32, 6656–6663. [DOI] [PubMed] [Google Scholar]
- Suske W.A., van Berkel,W.J.H. and Kohler,H.-P.E. (1999) Catalytic mechanism of 2-hydroxybiphenyl 3-monooxygenase, a flavoprotein from Pseudomonas azaleica HBP1. J. Biol. Chem., 274, 33355–33365. [DOI] [PubMed] [Google Scholar]
- Tanzer H., Seidel,M. and Wiegrebe,W. (1988) Hydrophilic derivatives of dithranol. Arch. Pharmacol., 321, 447–449. [Google Scholar]
- Townsend C.A. (1997) Structural studies of natural product biosynthetic proteins. Chem. Biol., 4, 721–730. [DOI] [PubMed] [Google Scholar]
- Tsai S.C., Miercke,L.J., Krucinski,J., Gokhale,R., Chen,J.C., Foster,P.G., Cane,D.E., Khosla,C. and Stroud,R.M. (2001) Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: versatility from a unique substrate channel. Proc. Natl Acad. Sci. USA, 98, 14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin X., Xi,L. and Tu,S.-C. (1991) Functional consequences of site-directed mutation of conserved histidyl residues of the bacterial luciferase α subunit. Biochemistry, 30, 11255–11262. [DOI] [PubMed] [Google Scholar]
- Yang K., Han,L., Ayer,S.W. and Vining,L.C. (1996) Accumulation of the angucycline antibiotic rabelomycin after disruption of an oxygenase gene in the jadomycin B biosynthetic gene cluster of Streptomyces venezuelae. Microbiology, 142, 123–132. [DOI] [PubMed] [Google Scholar]