

Abstract
The chemical modification of synthetic oligonucleotides has recently been investigated to improve their pharmacological utilization. In addition to chemical alterations of the backbone and of the heterocyclic bases, their conjugation with amphiphylic moieties, such as the polyethylene glycol has been proposed. The large scale production of these molecules as demanded for commercial purposes is hampered by the heterogeneity of the solid-phase processes and by the low reactivity of high-molecular weight PEGs in solution. A new synthetic procedure based on the recently developed liquid-phase method (HELP), has been set up to overcome these limitations.
Keywords: oligonucleotides, polyethylene glycols
Introduction
During the last decade the use of synthetic oligonucleotides as new therapeutic agents has been widely investigated (1). These molecules are able to interact with either specific single-stranded RNA messengers, as antisense (2), or with double-stranded genomic DNA, as antigene (3), thus inhibiting the expression of pathogenic genetic messages. The success of this strategy has increased the demand for a large scaling-up of the oligonucleotide synthesis (4). Solid-phase processes permit a rapid synthesis of even very long sequences, but unfortunately they tend to result in low yields. This limitation is mainly due to diffusion problems inside the resin beads owing to the heterogeneity of the reaction; hence, a large excess of high-cost monomers is required to achieve high yields. Moreover, commercial supports are characterized by a low capacity level and a large amount of support, for a single synthetic cycle, is required to obtain a consistent amount of product. On the other hand, the non-automated solution synthesis requires laborious purification procedures and very skillful operators.
The liquid-phase synthesis was proposed as a method to overcome these limitations (5). In this procedure, the oligonucleotide synthesis is carried out in homogeneous media by linking the growing chain to a soluble supporting polymer. The polymer-bound product is recovered from the reaction mixture by precipitation, thus allowing the rapid elimination of excess reagent and soluble by-products. In a new liquid-phase method called HELP, polyethylene glycol with a molecular weight ranging between 5 and 20 KDa (6) has been employed since it is highly soluble in a large variety of organic solvents and is easily precipitated by the addition of ether as a cosolvent. An automated synthesizer has been also developed (7) to carry out the overall process.The liquid-phase synthesis was proposed as a method to overcome these limitations (5). In this procedure, the oligonucleotide synthesis is carried out in homogeneous media by linking the growing chain to a soluble supporting polymer. The polymer-bound product is recovered from the reaction mixture by precipitation, thus allowing the rapid elimination of excess reagent and soluble by-products. In a new liquid-phase method called HELP, polyethylene glycol with a molecular weight ranging between 5 and 20 KDa (6) has been employed since it is highly soluble in a large variety of organic solvents and is easily precipitated by the addition of ether as a cosolvent. An automated synthesizer has been also developed (7) to carry out the overall process.
The use of synthetic oligonucleotides as new drugs for human therapy is hampered by their fast degradation in vivo caused by endogenous nucleases, and by the difficulty with which these polyanions cross membranes (8). As a consequence, their stability and the ability to achieve high concentrations of oligonucleotides at biological targets is strongly reduced.
To solve these problems, several chemical modifications of these nucleic acid derivatives have been proposed mainly at the level of the sugar-phosphate backbone (9). With this strategy, recognition by degradative enzymes appears reduced and the penetration through cellular barriers seems improved. However, these modified oligonucleotides often present new disadvantages such as low solubility in physiological media, toxicity of their metabolites, and inhibition of the degradative process of duplexes brought about by RNase H.
A possible solution to these problems seems to reside in conjugation with molecules that mask their unfavorable features, such as long-chain alcohols, steroids, cholic acid, peptides, and polymers (10-13). Among polymers, short- and long-chain PEGs have demonstrated some unique behaviors; in fact, these non toxic compounds, when covalently bound to substrates, increase their solubility both in organic and aqueous solutions, decrease their immunogenicity and antigenicity, and extend their in vivo lifetime (14). Owing to these properties, PEG has been used for the preparation of a series of biologically active conjugates (15).
The introduction of high-molecular weight PEG at the 3'- and 5'-end of oligonucleotides has recently been achieved. Modification of the 3'-position is performed by starting from a PEG-modified solid-phase support (16). The PEG chain is introduced at the 5'-end as a post-synthetic modification, or when the oligonucleotide is bound to the solid support, or after its release (17- 18). The amount of modified oligonucleotide produced by these procedures is quite low, owing to the reduced capacity of the solid-phase processes and due to scale-up difficulties. Moreover, in these heterogeneous conditions, high-molecular weight PEGs display a lower reactivity because of the viscosity of their solutions.
In the previously described HELP method, PEG was used as a soluble polymeric support for the large scale synthesis of oligonucleotides of up to 20 monomers. To obtain a therapeutically useful PEG-conjugate, the HELP procedure has been modified by introducing a stable bond between the polymer and the growing chain. PEG was then used as a synthetic helper in the liquid-phase procedure, as well as a biological carrier for the final oligonucleotide, as described in the original article (19).
Materials
Solvents for synthesis
Acetonitrile (AcCN), tert-butyl methylether (MTBE) and 1,2-dichloroethane (DCE) were anhydrous commercial products stored under argon over 4 Å molecular sieves activated at 350°C, and were used with less than 20 ppm of water present, as measured by the Karl-Fisher method. The ethanol (EtOH) was in its absolute form.
HPLC solvents and buffers
Acetonitrile was of HPLC grade. Water was double-distilled and filtered through a Millipore GS 0.22 μm sterile filter. Triethylamine (TEA) was distilled just before use. Glacial acetic acid and concentrated ammonia solution were commercial products at high level of purity.
0.1 M TEAAc (pH 7.0): 1.0 M stock solution was made up by adding to double-distilled water (100 ml), freshly distilled TEA (27.8 ml) and acetic acid (11.4 ml). The solution was then diluted to 200 ml and the pH adjusted to 7.0 by addition of either TEA or acetic acid as required. Stock solution was stored at 4-7°C. Stock solution was diluted to 0.1 M and the pH adjusted to 7.0. 0.1 M buffer was then filtered through a 0.22 μm sterile filter and degassed by sonication (30 min.) before use.
0.1 M TEAAc (pH 7.0): AcCN = 20:80 (v/v): AcCN (800 ml) was added to a recently filtered 0.1 M TEAAc (pH 7.0) (200 ml) and the mixture degassed by sonication (30 min.).
0.5 M NaCl (pH 12.0):</u> 1.0 M stock solution was made up by adding to double-distilled water (1.0 l) NaCl (58.44 g, 1 mole); the pH was adjusted to 12 with 5N NaOH. Stock solution was stored at 4-7°C. Stock solution was diluted to 0.5 M and the pH adjusted to 12. This 0.5 M solution was filtered through a Millipore GS 0.22 μm sterile filter and degassed by sonication (30 min.) prior to use.
0.05 M NaCl (pH 12.0): 0.5 M NaCl was diluted to 0.05 M and the pH adjusted to 12. The solution was then degassed by sonication (30 min.) before use.
Chemicals
Polyethylene glycol monomethylether (MPEG) was a commercial product from Shearwater Polymers, Inc., Huntsville AL (USA), with a very narrow MW distribution value, and was stored in a dessicator over KOH pellets. 1H-tetrazole was stored at room temperature in the presence of a dessicator, while phosphoramidites were stored at -20°C, in a dessicator. These products were from Pharmacia Biotech Italia, Cologno Monzese, Milano (Italy). Trichloroacetic acid (TCA), 2,6-lutidine, N-methylimidazole (NMI) were used as such; acetic anhydride was distilled over anhydrous sodium acetate; tert-butyl hydroperoxide (TBHP) was a commercial 80% solution in di-tert-butylperoxide. All these chemicals as well as all anhydrous organic solvents were obtained from Fluka Chimica or Aldrich Chimica, Milano (Italy).
Glassware and plasticware
The glassware used in each step of the oligonucleotide synthesis was first dried in an oven (110°C) for a few hours and then stored in a dessicator over KOH pellets. All glass apparatus was furnished with CaCl2 tubes to avoid moisture absorption. Polypropylene syringes and rubber septa were single-used.
Preparation of reagent solutions
0.5 M 1H-tetrazole in AcCN: 1H-tetrazole (2 g), in a bottle sealed with a rubber cap, was dissolved in dry AcCN (57.1 ml), injected with a syringe. The solution was stored under argon, at room temperature.
6% TCA in DCE (w/v): Detritylation solution was prepared before use by dissolving TCA (6 g) in dry DCE (100 ml). Unused solution was stored in a dark bottle at 4-7°C.
0.1 M DMT-dN-phosphoramidite in AcCN: Phosphoramidite solution was prepared immediately before use; DMT-dN-phosphoramidite (1 g), in a dark bottle sealed with a rubber cap, was dissolved in the right volume of dry AcCN, injected with a syringe. The solution was stored under argon at -20°C.
Thin-layer chromatography (TLC)
TLC was performed on precoated silica gel sheets using the following elution systems: Eluent 1: acetone:ethyl acetate:water = 10:5:1 (v/v/v), eluent 2: n-propanol:30% ammonia:water = 65:35:10 (v/v/v). Plates were observed using a UV light source and developed by spraying 60% perchloric acid:ethanol = 3:2 (v/v) to reveal DMT-bearing compounds (dark orange spots). Moreover, after the acidic treatment, the plates were heated at 100°C for 10 min. to achieve the carbonization of the sugar moiety of the oligonucleotide (dark spots). MPEG-bearing compounds were also detected by exposure to iodine vapor (brown spots).
Methods
1. Functionalization Of MPEG
Synthesis of 5'-DMT-dN1 (first nucleoside) -3'-βCE-phosphite-MPEG (Coupling)
MPEG 5,000 (1 g, 0.2 mmole), placed in a 250 ml three-necked round-bottom flask provided with a three-way stopcock, sealed with rubber caps, was coevaporated with dry AcCN (3 x 10 ml) and carefully dried under vacuum. Argon was flushed through the flask for 4-5 minutes. To the PEG residue, wetted with 1.0 ml of dry AcCN, 0.1 M 5'-DMT-dN1 -3'-(βCE)(N,N-iPr2 )-phosphoramidite in dry AcCN solution (5.0 ml, 0.5 mmole, 2.5 equivalents) and 0.5 M 1H-tetrazole in dry AcCN solution (4.0 ml, 2 mmole, 10 equivalents) were then simultaneously added by syringes through the rubber septum. The solution was stirred, under argon atmosphere, at room temperature, for 5 min. The solution was ice-cooled and MTBE (90 ml) was slowly added dropwise under vigorous stirring. A white powder was recovered after filtration through a por.3 Gooch, rapidly washed with ether and dried under vacuum, and stored in a dessicator over KOH pellets.
The presence of residual reagents or soluble by-products was checked by TLC (eluent acetone:ethyl acetate:water = 10:5:1, v/v/v). Under these conditions MPEG and MPEG-derivatives have Rf = 0. The crude product was suspended into EtOH (100 ml) and gently warmed up to 38°C; once solubilised, was slowly cooled from room temperature to 4-7°C, and left at 4-7°C for 1 h. The white cloudy powder formed was recovered by filtration through a por.3 Gooch, rapidly washed with ether and dried under vacuum, and stored in a dessicator over KOH pellets.
The degree of functionalisation was estimated spectrophotometrically: a weighed aliquot of product was dissolved in 60% perchloric acid:ethanol = 3:2 (v/v, 10 ml), the resulting orange solution was diluted 10- fold and the absorbance at 498 nm was measured. The nucleoside loading was calculated, in μmol/g, from the equation: [A498 (10 mm cell) x 10 x 14.3]/mg of weighed support.
Acetylation of 5'-DMT-dN1-3'-βCE-phosphite-MPEG (Capping)
1.0g of 5'-DMT-dN1-3'-βCE-phosphite-MPEG was dissolved in dry AcCN (5 ml) in a 250 ml round-bottom flask. 2,6-lutidine (0.5 ml), NMI (0.5 ml) and acetic anhydride (0.5 ml) were added following this order, under stirring. The solution was left to react, at room temperature for 3 min. DMT-dN1 -phosphate- MPEG was precipitated from the ice-cooled solution with MTBE (80 ml), which was slowly added dropwise. A white powder was recovered after filtration through a por.3 Gooch, extensively washed with ether and dried under vacuum and stored in a dessicator over KOH pellets. The product was stored at 4-7°C.
Synthesis of 5'-DMT-dN1-3'-βCE-phosphate-MPEG (Oxidation)
1.0g of 5'-DMT-dN1 -3'-βCE-phosphite-MPEG, placed in a 250 ml round-bottom flask, was dissolved in dry AcCN (10 ml) and the ice-cooled TBHP solution (0.6 ml) was added while stirring. The solution was stirred for 15 min. MTBE (90 ml) was then slowly added dropwise to the ice-cooled solution, under vigorous stirring. A white powder was recovered after filtration through a por.3 Gooch, washed with ether and dried under vacuum and stored in a dessicator over KOH pellets. The product was stored at 4- 7°C.
2. Synthesis Of MPEG-Oligonucleotide
1.0g of 5'-DMT-dN1-3'-bCE-phosphate-MPEG was dissolved in dry DCE (10 ml) in a 250 ml round-bottom flask. 6% TCA in DCE (w/v, 10 ml) was added dropwise to the ice-cooled solution, under vigorous stirring. After 15 min. MTBE (100 ml) was slowly added dropwise to the cooled orange solution. The mixture was filtered through a por.3 Gooch and the precipitate washed thoroughly with ether. The white powder was then dried under vacuum and stored in a dessicator over KOH pellets.
The extent of deblocking was controlled qualitatively by TLC (no orange colour developed by acidic spraying on spot with Rf = 0) and quantitatively by UV analysis. If the detritylation was not complete, the TCA treatment was repeated following the above procedure. In order to eliminate traces of TCA, the fully detritylated product was recrystallized from DCE/MTBE (10 ml/70 ml) and dried under vacuum over KOH pellets.
Coupling
1.0g of 5'-HO-dN1-3'-βCE-phosphate-MPEG, placed in a 250 ml three-necked round-bottom flask sealed with rubber caps and provided with a three-way stopcock, was coevaporated with dry AcCN (3 x 10 ml) and carefully dried under vacuum. Argon was flushed through the flask for 4-5 minutes. To the PEG-bound product, wetted with 1.0 ml of dry AcCN, 0.1 M 5'-DMT-dN2-3'-(βCE)(N,N-iPr2 )-phosphoramidite in dry AcCN solution (5.0 ml, 0.5 mmoles, 2.5 equivalents) and 0.5 M 1H-tetrazole in dry AcCN solution (4.0 ml, 2 mmoles, 10 equivalents) were then simultaneously added by syringes through the rubber septum. The mixture was stirred under argon atmosphere at room temperature for 5 min. The solution was ice-cooled and MTBE (90 ml) was slowly added dropwise under vigorous stirring. A white powder was recovered after filtration through a por.3 Gooch, rapidly washed with ether and dried under vacuum, and stored in a dessicator over KOH pellets. The product was stored at 4-7°C.
The presence of residual reagents or soluble by-products was ascertained by TLC [eluent acetone:ethyl acetate:water = 10:5:1 (v/v/v)]. Under these conditions MPEG and MPEG-derivatives have Rf = 0. The crude product was suspended into EtOH (100 ml) and gently heated to 38°C; once solubilised, it was slowly cooled from room temperature to 4-7°C, and left at 4-7°C for 1 h. The white cloudy powder formed was recovered by filtration through a por.3 Gooch, rapidly washed with ether and dried under vacuum, and stored in a dessicator over KOH pellets.
The coupling yield was monitored spectrophotometrically: a weighed aliquot of product was dissolved in 60% perchloric acid:ethanol = 3:2 (v/v, 10 ml), the resulting orange solution was diluted 10-fold and the absorbance at 498 nm was measured. The nucleoside loading was calculated (in μmol/g) from the equation: [A498 (10 mm cell) x 10 x 14.3]/mg of weighed support.
Capping
1.0g of 5'-DMT-dN2-3'-βCE-phosphite-dN1-3'-βCE-phosphate-MPEG was dissolved in dry AcCN (5 ml) in a 250 ml round-bottom flask. 2,6-lutidine (0.5 ml), NMI (0.5 ml) and acetic anhydride (0.5 ml) were added following this order, under stirring. The solution was left to react at room temperature for 3 min. 5'-DMT-dN2-phosphate-dN1-MPEG was precipitated from the ice-cooled solution with MTBE (80 ml) which was slowly added dropwise. A white powder was recovered after filtration through a por.3 Gooch, extensively washed with ether and dried under vacuum, and stored in a dessicator over KOH pellets. The product was stored at 4-7°C.
Oxidation
1.0g of 5'-DMT-dN2-3'-βCE-phosphite-dN1-3'-βCE-phosphate-MPEG, placed in a 250 ml round-bottom flask, was dissolved in dry AcCN (10 ml) and the ice-cooled TBHP solution (0.6 ml) was added under stirring. The solution was stirred for 15 min. MTBE (90 ml) was then slowly added dropwise to the ice-cooled solution, with vigorous stirring. A white powder was recovered after filtration through a por.3 Gooch, washed with ether and dried under vacuum, and stored in a dessicator over KOH pellets. The product was stored at 4-7°C.
These steps were repeated until the desired length of the oligonucleotide chain had been obtained.
3. Deprotection Of Oligonucleotide-MPEG
Deprotection of phosphate backbone and N-deacylation of nucleobases
The following treatment removes the cyanoethyl-protecting group from phosphates and the N-isobutyryl- and N-benzoyl-protecting groups from guanosines, cytidines and adenosines. DMT-oligonucleotide- MPEG was dissolved in 30% NH3 (5 ml x 20 mg) in a tightly closed glass container to avoid ammonia leakage, and left in an oven, without stirring, at 60°C overnight. The solution was transferred into a large round-bottom flask in order to avoid loss of product caused by foam, and evaporated to dryness, rinsed with water and evaporated, and the procedure repeated until an ammonia odourless solution was obtained. The residue was then dissolved in water (5 ml) and extracted with ether (4 x 5 ml). The aqueous layer was freeze-dried and the residue stored at -20°C, ready for HPLC analysis and purifications.
Deprotection of terminal 5'-hydroxyl group
This treatment removes the dimethoxytrityl-protecting group from 5'-terminal hydroxyl-group. DMT-oligonucleotide- MPEG previously treated with ammonia was dissolved in glacial acetic acid:water = 4:1 (v/v, 5 ml x 20 mg), in a 100 ml round-bottom flask, and stirred at room temperature for 30 min. The resulting yellow solution was extracted with ether (5 x 5 ml) and the aqueous layer evaporated to dryness. The residue was rinsed with water and evaporated and the procedure repeated until an acetic acid odorless solution was obtained. The residue was dissolved in water and freeze-dried and stored at -20°C, ready for HPLC analysis and purification.
Analysis and Purification
Analytical RP-HPLC
Reversed-phase HPLC was performed using a Vydac C18 column. Samples (4 mg each) were dissolved in start buffer (1 ml), filtered through a 0.22 μm sterile filter and degassed by centrifugation. 10 l of the resulting solution were injected. Mobile phase A: 0.1 M TEAAc (pH 7.0). Mobile phase B: 0.1 M TEAAc (pH 7.0):AcCN = 20:80 (v/v) Linear gradient: 40-90% B in 50 min. Flow: 1.0 ml/min. Temperature: ambient, UV detector: 254 nm.
Analytical IE-HPLC
Ion-exchange HPLC was performed using a Pharmacia Mono Q HR 5/5 column. Samples (4 mg each) were dissolved in water (1.0 ml), filtered through a 0.22 μm sterile filter and degassed by centrifugation. 20 l of the resulting solution were injected. Mobile phase A: 0.05 M NaCl (pH 12.0) Mobile phase B: 0.5 M NaCl (pH 12.0). Elution gradient: 0% B 2 min. hold, 0-85% B in 23 min., 85-100% B in 5 min., 100% B 1 min. hold. Flow: 0.6 ml/min. Temperature: ambient, UV detector: 254 nm.
Purification of crude MPEG-conjugated oligonucleotide by anion-exchange HPLC-IE
Purification of the final crude MPEG-oligonucleotide was carried out using a Pharmacia Mono Q HR 5/5 column. Sample (10 mg) was dissolved in water (1.0 ml), filtered through a 0.22 μm sterile filter and degassed by centrifugation. Sample was eluted by injection of 300 l each time. Mobile phase A: 0.05 M NaCl (pH 12.0) Mobile phase B: 0.5 M NaCl (pH 12.0). Elution gradient: 30-100% B in 13 min., 100% B 5 min. hold. Flow: 1.0 ml/min. Temperature: ambient UV detector: 254 nm.
Desalting of purified MPEG-conjugated oligonucleotide by gel-filtration
Desalting of purified MPEG-oligonucleotide was carried out by gel-filtration using a 20 x 100 mm column of Sephadex G25-F resin. Sample (50 mg) was dissolved in water (1 ml). Mobile phase: water Millipore grade. Elution: isocratic. UV detector: 254 nm. Alternatively, the excess of salt can be removed by a selective precipitation: MPEG-oligonucleotide afforded from IE-HPLC (0.2 g) was suspended in acetone or methanol (5 ml) and left, under stirring for 30 min. The residue remained undissolved, was filtered and the solution with the product was evaporated to dryness. The amount of MPEG-oligonucleotide was ascertained by UV and TLC. A further possibility is given by a selective extraction: MPEG-oligonucleotide afforded from IE-HPLC (1.0 g) was dissolved in the minimum amount of water (4-5 ml) and extracted with a large volume of dichloromethane (4 x 100 ml). The organic layers with the product were pooled, dried over Na2SO4 and evaporated to dryness. The residue was checked by UV and TLC.
Discussion
The general procedure for the liquid-phase synthesis of oligonucleotides conjugated with high-molecular mass PEGs can be schematized by the flow-chart.
As clearly indicated in the original article (19), the HELP-derived procedure described offers many advantages for the production of these conjugates, even in a large scale. Moreover, the recent development of an automated instrument for the liquid-phase synthesis opens new possibilities for the industrial production of oligomers and biomolecules when their pharmacological features are improved by conjugation with PEG.
Fig. 1.
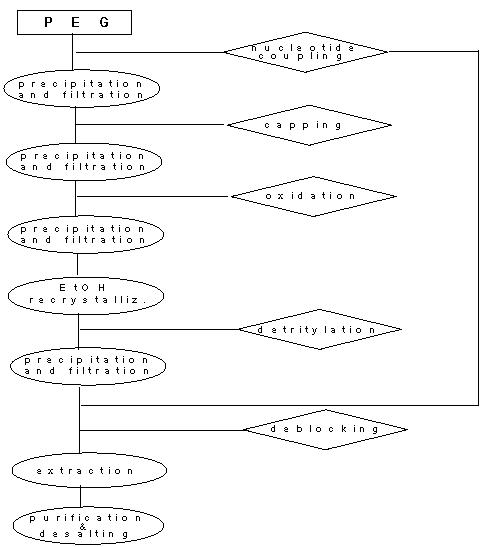
Flow-chart of the HELP process
The detailed procedures described above are easily reproducible on the bench on condition that some special precautions are taken. In particular :
A careful dehydration of the starting PEG and of their oligonucleotide-bound intermediates must always be achieved, especially before the coupling steps, owing to the hygroscopicity of the polymer.
During the coupling step it is imperative to avoid any unnecessary dilution of the reaction mixture to avoid a rapid decrease in yield. Thus, it is strongly suggested always to operate at the highest concentration allowed by the solubility of the reaction components.
Regarding the capping procedure, as already underlined in the usual solid-phase approaches, it is preferable to carry it out before the oxidation, to destroy any side-product that, on the contrary, would be irreversibly stabilized by the oxidation step.
A further strong recommendation regards the need for the cooling down of the organic solution before its treatment with the acidic mixture demanded for the detritylation step, to avoid, or to reduce as much as possible any depurination of the product. A high percentage of acid and repeated treatments at the beginning of the synthesis when the ratio between the PEG and the oligonucleotide is disadvantageous, are demanded to completely remove the DMT protecting groups due to the ether units of this support, easily protonated, that reduce the effective acid concentration.
A critical point of the proposed procedure is the amount of polymer-bound product recovered at the end of the overall process because of several precipitation/filtration steps required during the intermediate purification. For a maximum yield it is customary to add the ether solution slowly, under vigorous stirring of the cooled reaction mixture. This procedure must take at least 30 minutes to be efficiently completed. It is not convenient to save time by speeding up this step, since any residual impurity will be entrapped into the precipitated polymer-conjugate and will be transferred to the following steps.
A TLC analysis must be routinely performed before any new synthetic step. If there were evidence that the ether precipitation did not purify the product completely, an additional crystallization from an EtOH solution would be strongly recommended: the PEG-bound crude product dissolves quite easily into a 10% solution ( w/v ) at 37C, and precipitates quantitatively at temperature below 20°C. This purification step must be introduced as a rule at the end of each growing cycle.
The final deblocking procedure is almost identical to that commonly adopted for similar solid-phase processes. The purification of the crude, deblocked product is better achieved by ionic exchange chromatography. In fact, the lipophylic properties of the PEG moiety hamper any useful separation of the conjugates by the standard reverse-phase procedure, even if the terminal DMT group is left on the chain: a single peak was always observed in our chromatographic conditions, in which are present all the different PEG-bound oligonucleotides obtained at the various steps of synthesis. On the other hand, the polymer moiety does not mask the oligonucleotide part from its binding to the strong anion-exchange support, and the different chains are efficiently separated on the basis of their charge.
The desalting of the purified PEG-conjugate can be safely performed by the gel-filtration procedure. As an alternative, a selective solvent-cosolvent precipitation of the PEG-bound product can also be used owing to the solubility of these conjugates in organic solvents where the inorganic salts are insoluble. Furthermore, a selective extraction of the conjugates from a very concentrated aqueous solution with large volumes of halogenated hydrocarbons can be also taken into account. In any case it is convenient to compare the results of the different procedures, verified on a small scale, before deciding which apply to the entire batch.
The procedure here reported may be in principle extended to other high-molecular weight polymeric supports whose properties are similar to the one of PEG.
References
- Heidenreich O, Kang SH, Xu X, Nerenberg M. Application of Antisense to Therapeutics. Molecular Medicine Today. 1995;1:128–133. doi: 10.1016/s1357-4310(95)80090-5. [DOI] [PubMed] [Google Scholar]
- De Mesmaeker A, Hr R, Martin P, Moser HE. Antisense Oligonucleotides. Acc Chem Res. 1995;28:366–374. [Google Scholar]
- Thuong NT, Hélène C</lead> . Sequence-Specific Recognition and Modification of Double-helical DNA by Oligonucleotides. Angew Chem , Int Ed Engl. 1993;32:666–690. [Google Scholar]
- Ravikumar VT, Andrade M, Wyrzykievicz T, Scozzari A, Cole DL. Large-scale Synthesis of Oligodeoxyribonucleotide Phosphorotioate Using Controlled-Pore Glass as Support. Nucleosides & Nucleotides. 1995;14:1219–1222 . [Google Scholar]
- Bayer E, Mutter M. Liquid Phase Synthesis of Peptides. Nature. 1972;237:512–513. doi: 10.1038/237512a0. [DOI] [PubMed] [Google Scholar]
- Bonora GM. Polyethylene Glycol. A High-Efficiency Liquid Phase (HELP) for the Large-scale Synthesis of Oligonucleotides. Appl Biochem Biotechnol. 1995;54:3–17. [Google Scholar]
- Bagno A, Bicciato S, Di Bello C, Bonora G M. Automation of the Liquid-Phase Synthesis of Biopolymers. J Chem Tech Biotechnol. 1998;71:77–83. [Google Scholar]
- Dan Cook P. Medicinal Chemistry Strategies for Antisense Research in Antisense Research and Applications Crooke, S.T. and Lebleu B. Eds., CRC Press Inc. Boca Raton (USA); 1993. pp.149-188.
- Sanghvi TYS, Cook PD. Eds. Carbohydrate Modifications in Antisense Research. ACS Symposium Series 580, Maple Press, York, PA; 1994.
- Shea RG, Marsters JC, Bischofberger N. Synthesis, Hybridization Properties and Antiviral Acitivity of Lipid-Oligonucleotide Conjugates. Nucleic Acids Res. 1990;18:3777–3783. doi: 10.1093/nar/18.13.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutorin A, Guskova L, Ivanova E, Kobetz N, Zarytova V, Ryte A, Yurchenko L, Vlassov V. Synthesis of Alkylating Oligonucleotide Derivatives Containing Chloresterol and Phenazinium Residues at their 3'-Terminus and their Interaction with DNA within Mammalian Cells. FEBS Lett. 254:129–132. doi: 10.1016/0014-5793(89)81023-3. [DOI] [PubMed] [Google Scholar]
- Manoharan M, Johnson L, Bennet CF, Vickers TA, Ecker DJ, Cowsert LM, Freier SM, Cook PD. Cholic Acid Oligonucleotide Conjugates for Antisense Application. Bioorg Med Chem Lett. 1994;4:1053–1060 . [Google Scholar]
- Robles J, Maseda M, Beltran M, Concerneau M., Pedroso E, Grandas A. Synthesis and Enzymatic Stability of Phosphodiester-Linked Peptide-Oligonucleotide Hybrids. Bioconjugate Chem. 1997;8:785–788. doi: 10.1021/bc970051u. [DOI] [PubMed] [Google Scholar]
- Harris JM. Ed. Poly(ethylene Glycol) Chemistry. Biotechnical and Biomedical Applications. Plenum Press, New York; 1992.
- Zalipsky S. Chemistry of Polyethylene Glycol Conjugates with Biologically Active Molecules. Adv Drug Delivery Rev. 1995;16:157–182. [Google Scholar]
- Efimov VA, Pashkova I.N, Kalinkina AL, Chakhmakhcheva OG. Synthesis of Conjugates of Oligonucleotides with Polyethyleneglycol. Bioorg Khimi. 1983;19:800–804. [Google Scholar]
- Jäschke A, Fürste JP, Nordhoff E, Hillenkamp F, Cech D, Erdmann VA. Synthesis and Properties of Oligodeoxyribonucleotides-Polyethylene Glycol Conjugates. Nucleic Acids Res. 1994;22:4810–4817. doi: 10.1093/nar/22.22.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T, Asakana H, Tashiro Y, Juni K, Sueishi T. Stability, Specific Binding Activity, and Plasma Concentration in Mice of an Oligodeoxynucleotide Modified at 5'-Terminal with Poly(ethylene Glycol). Biol Pharm Bull. 1995;18:474–476. doi: 10.1248/bpb.18.474. [DOI] [PubMed] [Google Scholar]
- Bonora GM, Ivanova E, Zarytova V, Burcovich B, Veronese FM. Synthesis and Characterization of High-Molecular Mass Polyethylene Glycol-Conjugated Oligonucleotides. Bioconjugate Chem. 1997;8:793–797. doi: 10.1021/bc970082p. [DOI] [PubMed] [Google Scholar]