

The GreB transcription cleavage factor of E. coli and the Gfh1 transcription elongation factor of T. thermophilus were cloned, expressed, purified and crystallized. Complete diffraction data sets were collected for the GreB and Gfh1 crystals to 2.6 and 2.8 Å resolution, respectively. Structure determination of these proteins is now in progress.
Keywords: transcription cleavage factors, GreB, anti-GreA factor, Gfh1
Abstract
The Escherichia coli gene encoding the transcription cleavage factor GreB and the Thermus thermophilus gene encoding the anti-GreA transcription factor Gfh1 were cloned and expressed and the purified proteins were crystallized by the sitting-drop vapor-diffusion technique. The GreB and Gfh1 crystals, which were improved by macroseeding, belong to space group P41212 (or P43212), with unit-cell parameters a = b = 148, c = 115.2 Å and a = b = 59.3, c = 218.9 Å, respectively. Complete diffraction data sets were collected for the GreB and Gfh1 crystals to 2.6 and 2.8 Å resolution, respectively. Crystals of the selenomethionine proteins were obtained by microseeding using the native protein crystals and diffract as well as the native ones. The structure determination of these proteins is now in progress.
1. Introduction
Recent developments in study of the transcriptional machinery and its regulation have highlighted the role of the RNA polymerase (RNAP) secondary channel in the control of such diverse processes as RNA-chain elongation (Marr & Roberts, 2000 ▶; Toulme et al., 2000 ▶), transcription through nucleosomes (Kireeva et al., 2005 ▶), transcriptional error correction (Erie et al., 1993 ▶; Shaevitz et al., 2003 ▶) and DNA repair (Trautinger et al., 2005 ▶). The number of both protein and small-molecule factors such as GreA, GreB, DksA, TFIIS, ppGpp etc. that have been shown to utilize the secondary channel (Artsimovitch et al., 2004 ▶; Kettenberger et al., 2003 ▶; Opalka et al., 2003 ▶; Perederina et al., 2004 ▶) continues to grow steadily, indicating the secondary channel’s versatility as a regulatory throughway. Structural analysis of GreA, TFIIS and DksA (Kettenberger et al., 2003 ▶; Perederina et al., 2004 ▶; Stebbins et al., 1995 ▶) revealed an architectural motif that is common to these protein regulators, namely an elongated domain that protrudes into the secondary channel towards the active site, with a few acidic residues on its tip that are capable of coordinating Mg2+ and thus remodeling the active site to change its catalytic propensities (Opalka et al., 2003 ▶; Sosunova et al., 2003 ▶). Beyond this observation, no generalization from such a limited data set appears to be possible, as the unrelated and structurally dissimilar GreA (coiled coil) and TFIIS (zinc-ribbon fold with a β-hairpin) exert comparable activation of endonucleolytic activities by their respective targets (Conaway et al., 2003 ▶), while the unrelated but structurally congruent GreA and DksA employ coiled coils tipped with acidic residues to confer dramatically different effects (Laptenko et al., 2003 ▶; Perederina et al., 2004 ▶; Sosunova et al., 2003 ▶). Moreover, the 35% identical proteobacterial paralogs GreA and GreB have distinctly different effects on endonucleolytic transcript cleavage (Fish & Kane, 2002 ▶), further stressing the need for more detailed high-resolution structural information in order to better understand the regulation of gene expression by the factors binding in the RNAP secondary channel. Another GreA paralog, a largely uncharacterized protein Gfh1 from Thermus thermophilus, appears to deviate even further from Gre-like function and instead of stimulating transcript cleavage, it inhibits several activities of RNAP (Hogan et al., 2002 ▶; Laptenko & Borukhov, 2003 ▶).
To elucidate the mechanism of action of GreB and Gfh1, we sought to determine their three-dimensional structures. To this end, we have cloned and expressed the Escherichia coli greB and T. thermophilus gfh1 genes. The purified GreB and Gfh1 proteins have been crystallized.
2. Experimental procedures and results
2.1. Cloning, expression and purification
The GreB expression vector pIA577 was constructed by cloning the E. coli greB gene into the NcoI and XhoI sites in the pET28b expression vector (Novagen). A genomic copy of the greB gene from the DH5α strain was amplified using Pfu DNA polymerase-driven high-fidelity PCR with primers IA414 (5′-GATCCCATGGGTATGAAAACGCCCCTGGTTAC) and IA415 (5′-CTAGCTCGAGCGGTTTCACGTACTCGATAGCA) that included recognition sites for the NcoI and XhoI restriction endonucleases, respectively.
The Gfh1 expression vector pVS26 was constructed by cloning the T. thermophilus gfhI gene into the Mva1269I and SalI sites of pTYB12 (New England Biolabs). A genomic copy of the gfh1 gene from T. thermophilus HB8 strain was PCR amplified using Pfu DNA polymerase with primers IA511 (5′-GGTGGTGGGAATGCTATGGCGCGCGAGGTGAAGCT) and IA512 (5′-GATTGTCGACTCAGCCGTGGATGGCCACCA) that included recognition sites for the Mva1269I and SalI restriction endonucleases, respectively. The sequences of the plasmids pVS26 and pIA577 were verified using automated sequencing at OSU Plant-Microbe Genomics Facility. For overexpression of the native proteins, pVS26 and pIA577 were transformed into E. coli strain BL21(λDE3). Production of GreB and Gfh1 was induced according to the Overnight Express protocol (Novagen).
For purification of the GreB protein (170 amino acids long; 20 kDa), cells were harvested and resuspended in lysis T buffer [50 mM Tris–HCl pH 6.9, 1.2 M NaCl, 5% glycerol, 1 mM 2-mercaptoethanol (ME), 0.1 mM PMSF] with Complete EDTA-free protease-inhibitor cocktail (Roche), 0.1% Tween 20 and 1 mg ml−1 lysozyme. The suspension was incubated on ice for 60 min with occasional swirling and followed by a brief sonication to disrupt the cells. The extract was cleared by centrifugation (27 000g, 15 min at 277 K). The cleared extract was combined with Ni–NTA agarose (Invitrogen) slurry in lysis T buffer and incubated with agitation for 30 min at 277 K. The slurry was poured into a disposable gravity-flow column and drained. The column was washed with ten volumes of lysis T buffer and ten volumes of the same buffer with 10 mM imidazole. Elution was carried out with five volumes of lysis T buffer with 500 mM imidazole. Fractions containing the protein of interest were combined, concentrated to approximately 3 ml using an Amicon Ultra-15 10 000 MW filter and loaded onto a HiLoad Superdex 75 16/60 column (GE Healthcare) using an AKTA Purifier system (GE Healthcare) at 0.5 ml min−1. The column was equilibrated and washed with GF buffer [10 mM Tris–HCl pH 7.8, 1 M NaCl, 2 mM dithiothreitol (DTT)]. The purified protein was concentrated to approximately 12 mg ml−1 and remained stable at 277 K without degradation for several weeks. The N-terminal His6 tag was not removed prior to crystallization. The yield was 50 mg of purified protein per litre of culture.
For purification of the Gfh1 protein (156 amino acids long; 17 kDa), cells were harvested and sonicated in ImpactCN500 buffer (50 mM Tris–HCl pH 8.8, 500 mM NaCl, 1 mM EDTA, 0.1 mM PMSF) with Complete EDTA-free protease-inhibitor cocktail (Roche) and 0.1% Tween 20. The extract was cleared by centrifugation (18 000g, 40 min at 277 K). The cell lysate was loaded onto a disposable gravity-flow column with chemically cross-linked chitin polymer (New England Biolabs) equilibrated with the same buffer. The column was washed with ten volumes of ImpactCN500 buffer. The column was finally equilibrated with three volumes of ImpactCN500 buffer with 50 mM ME and incubated overnight at room temperature to elicit the intein-mediated cleavage reaction. The cleaved-off protein was eluted with three column volumes of 50 mM Tris–HCl pH 6.9, 500 mM NaCl, 5% glycerol, 1 mM ME, 0.1 mM PMSF buffer. Fractions containing the protein of interest were combined and heated at 348 K for 25 min. A cleared suspension of the Gfh1 protein was concentrated to approximately 3 ml using an Amicon Ultra-15 10 000 MW filter and loaded onto a HiLoad Superdex 75 16/60 column (GE Healthcare) using an AKTA Purifier system (GE Healthcare) at 0.5 ml min−1. The column was equilibrated and washed with GF buffer. The purified protein was concentrated to approximately 10 mg ml−1 and remained stable at 277 K without degradation for several weeks. The yield was 5 mg of purified protein per litre of culture.
2.2. Crystallization and data collection
The Hampton Crystal Screen method (Jancarik & Kim, 1991 ▶) was used to determine the initial crystallization conditions for the GreB and Gfh1 proteins. Crystallization was carried out by the sitting-drop vapor-diffusion technique at 293 and at 277 K. The Crystal Screen precipitant solutions were diluted eightfold with GF buffer in the initial screening for GreB and fivefold with GF buffer for Gfh1. Drops containing 1 µl of the diluted precipitant solutions and 1 µl of the protein solution in GF buffer were equilibrated against 0.5 ml of the same diluted precipitant solutions with additional 1 M NaCl. After 5 d of equilibration at 293 K, small hexagonal crystals of GreB protein had occasionally grown in Crystal Screen I solution No. 15. The crystals were subjected to macroseeding using drops prepared under the same conditions. After a few days of equilibration, the crystals reached typical dimensions of 0.45 × 0.45 × 0.45 mm (Fig. 1 ▶ a) and diffracted to beyond 2.8 Å resolution at the in-house X-ray generator. They appeared to belong to space group P41212 (or P43212), with unit-cell parameters a = b = 146.4, c = 114.78 Å. Well diffracting GreB crystals were obtained by mixing 2 µl protein solution at 12 mg ml−1 in GF buffer with 2 µl of solution containing 3.75% PEG 8000, 25 mM ammonium sulfate, 12.5 mM sodium cacodylate pH 6.5, 8 mM Tris–HCl pH 7.8, 800 mM NaCl, 1.6 mM DTT.
Figure 1.

Crystals of the GreB protein (a) and Gfh1 protein (b).
Small bipyramidal crystals of Gfh1 protein grew in Crystal Screen I solution No. 45 at 277 K over the same time interval. The crystals were subjected to microseeding and then to macroseeding using drops prepared under the same conditions except that Crystal Screen I solution No. 45 was diluted fivefold with water. After a few days of equilibration, the crystals reached typical dimensions of 0.1 × 0.1 × 0.35 mm (Fig. 1 ▶ b) and diffracted to beyond 2.8 Å resolution at the in-house X-ray generator. They also appeared to belong to space group P41212 (or P43212), with unit-cell parameters a = b = 59.3, c = 218.9 Å. Well diffracting Gfh1 crystals were obtained by mixing 1 µl protein solution at 10 mg ml−1 in GF buffer with 1 µl of solution containing 3.6% PEG 8000, 40 mM zinc acetate, 20 mM sodium cacodylate pH 6.5.
The data for GreB and Gfh1 were collected on the high-intensity beamline NW12, Photon Factory, Tsukuba, Japan and on an R-AXIS IV++ imaging-plate detector mounted on a MicroMAX-007HR rotating-anode X-ray generator (Rigaku, Japan), respectively (Table 1 ▶). The data were collected at 100 K using the mother-liquor solution with a higher concentration of NaCl as a cryoprotectant (about 4 M) and 15% glycerol for GreB crystals and 12% ethylene glycol for Gfh1 crystals. All data were processed using the programs DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶).
Table 1. Data-collection statistics for the GreB and Gfh1 proteins.
Data for the highest resolution shell are shown in parentheses.
| GreB | Gfh1 | |
|---|---|---|
| Space group | P41212/P43212 | P41212/P43212 |
| Unit-cell parameters | ||
| a (Å) | 148.0 | 59.28 |
| b (Å) | 148.0 | 59.28 |
| c (Å) | 115.12 | 218.9 |
| Source | Synchrotron† | X-ray generator‡ |
| Wavelength (Å) | 1.0 | 1.5418 |
| Temperature (K) | 100 | 100 |
| Molecules in asymmetric unit | 3–6 | 1–2 |
| Solvent content (%) | 80–50 | 77–54 |
| Resolution (Å) | 40–2.6 (2.69–2.6) | 35–2.8 (2.9–2.8) |
| Observations | 245848 | 43900 |
| Unique reflections | 38590 | 10262 |
| Multiplicity | 6.4 (3.0) | 4.3 (3.4) |
| Rmerge§ | 0.057 (0.498) | 0.050 (0.375) |
| Completeness (%) | 97.1 (93.4) | 99.2 (98.2) |
| I/σ(I) | 30.7 (2.8) | 33.9 (3.97) |
Beamline NW12, Photon Factory, Tsukuba, Japan.
MicroMAX-007HR (Rigaku, Japan) with R-AXIS IV++ imaging-plate detector.
R
merge = 
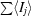 , where Ij is the intensity of reflection j and 〈Ij〉 is the average intensity of reflection j.
, where Ij is the intensity of reflection j and 〈Ij〉 is the average intensity of reflection j.
To phase the diffraction data, we prepared GreB and Gfh1 proteins containing selenomethionine (SeMet). SeMet GreB and Gfh1 crystals were grown under the same conditions as the native proteins using microseeding from the native crystals. For microseeding, the large native crystals were crushed by a needle, which was then transferred into the crystallization drop containing SeMet protein. Since the SeMet-substituted crystals are of the same diffraction quality as those of the native protein, we will undertake the structure solution of GreB and Gfh1 by multi-wavelength anomalous diffraction (MAD) methods using a number of the Se atoms incorporated into the Met residues of each molecule.
Acknowledgments
We thank Drs N. Igarashi, N. Matsugaki and M. Suzuki from the laboratory of Professor S. Wakatsuki (Photon Factory, Tsukuba, Japan) for assistance during data collection at the NW12 synchrotron beamline. This work was supported in part by grants from the National Institute of Health GM74252 and GM74840 (to DGV) and GM67153 (to IA).
References
- Artsimovitch, I., Patlan, V., Sekine, S., Vassylyeva, M. N., Hosaka, T., Ochi, K., Yokoyama, S. & Vassylyev, D. G. (2004). Cell, 117, 299–310. [DOI] [PubMed] [Google Scholar]
- Conaway, R. C., Kong, S. E. & Conaway, J. W. (2003). Cell, 114, 272–274. [DOI] [PubMed] [Google Scholar]
- Erie, D. A., Hajiseyedjavadi, O., Young, M. C. & von Hippel, P. H. (1993). Science, 262, 867–873. [DOI] [PubMed] [Google Scholar]
- Fish, R. N. & Kane, C. M. (2002). Biochim. Biophys. Acta, 1577, 287–307. [DOI] [PubMed] [Google Scholar]
- Hogan, B. P., Hartsch, T. & Erie, D. A. (2002). J. Biol. Chem.277, 967–975. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kettenberger, H., Armache, K. J. & Cramer, P. (2003). Cell, 114, 347–357. [DOI] [PubMed] [Google Scholar]
- Kireeva, M. L., Hancock, B., Cremona, G. H., Walter, W., Studitsky, V. M. & Kashlev, M. (2005). Mol. Cell, 18, 97–108. [DOI] [PubMed] [Google Scholar]
- Laptenko, O. & Borukhov, S. (2003). Methods Enzymol.371, 219–232. [DOI] [PubMed] [Google Scholar]
- Laptenko, O., Lee, J., Lomakin, I. & Borukhov, S. (2003). EMBO J.22, 6322–6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr, M. T. & Roberts, J. W. (2000). Mol. Cell, 6, 1275–1285. [DOI] [PubMed] [Google Scholar]
- Opalka, N., Chlenov, M., Chacon, P., Rice, W. J., Wriggers, W. & Darst, S. A. (2003). Cell, 114, 335–345. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Perederina, A., Svetlov, V., Vassylyeva, M. N., Tahirov, T. H., Yokoyama, S., Artsimovitch, I. & Vassylyev, D. G. (2004). Cell, 118, 297–309. [DOI] [PubMed] [Google Scholar]
- Shaevitz, J. W., Abbondanzieri, E. A., Landick, R. & Block, S. M. (2003). Nature (London), 426, 684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunova, E., Sosunov, V., Kozlov, M., Nikiforov, V., Goldfarb, A. & Mustaev, A. (2003). Proc. Natl Acad. Sci. USA, 100, 15469–15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins, C. E., Borukhov, S., Orlova, M., Polyakov, A., Goldfarb, A. & Darst, S. A. (1995). Nature (London), 373, 636–640. [DOI] [PubMed] [Google Scholar]
- Toulme, F., Mosrin-Huaman, C., Sparkowski, J., Das, A., Leng, M. & Rahmouni, A. R. (2000). EMBO J.19, 6853–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautinger, B. W., Jaktaji, R. P., Rusakova, E. & Lloyd, R. G. (2005). Mol. Cell, 19, 247–258. [DOI] [PubMed] [Google Scholar]