

Abstract
Studies of the interaction of the 16 residue fusion peptide domain of human immunodeficiency virus glycoprotein gp41 (gp41FD) with T lymphocytes are outlined. Fluorescence measurements of changes in the electrostatic surface and dipole potentials of the plasma membrane following the interaction with gp41FD are described. The results show that gp41FD interacts with heparan sulfate located on the cell surface. This interaction is blocked by interleukin-8 and abolished by pre-treating the cells with heparitinase. The specificity of the reaction was also assessed by observations that soluble heparan sulfate competes with the cell membrane interaction whereas soluble heparin (at the levels utilized) does not. Following binding to heparan sulfate, the interaction with the membrane seems to take place in a cooperative manner with the formation of gp41FD trimers. In simpler phospholipid membranes, however, a trimeric complex does not appear to be the dominant mode of interaction. Finally, by repeating some of these studies within an imaging regime, it appears that the gp41FD–T-cell interaction takes place within specific domains on the cell surface to similarly localized heparan sulfate moieties.
Keywords: cooperativity/fusion peptide/membrane raft/spatial imaging
Introduction
Entry of human immunodeficiency virus (HIV-1) into target T lymphocytes is known to be mediated by the viral envelope glycoproteins gp120/gp41 (Kowalski et al., 1987; Lasky et al., 1987; Bosch et al., 1989). gp120 binds to the CD4 receptor of prospective host-cell membranes. Interactions with members of the family of chemokine receptors (CCR5 and CXCR4), acting as co-receptors, are also known to be necessary for successful infection (Clapham et al., 1999). The viral interactions with the cell may also involve a number of other components of the host-cell membrane such as heparans and galactocerebrosides (Harouse et al., 1991; Roderiquez et al., 1995; Mondor et al., 1998; Ugolini et al., 1999) although their particular roles remain to be resolved.
Following T-cell receptor binding, the gp120–gp41 complex appears to undergo major conformational re-arrangements that are yet to be fully characterized but appear to result in the exposure of the ‘active’ form of gp41. This structure then promotes fusion of the viral and cellular membranes (Gallaher, 1987; Kowalski et al., 1987; Weissenhorn et al., 1999). From X-ray diffraction studies of crystals of the envelope glycoproteins, a trimeric gp41 structure has been hypothesized to be the active configuration. Such trimers have also been described for gp120 by Kwong et al. (1998) and Ugolini et al. (1999). Crystallographic studies, on the other hand, have confirmed that the core of gp41 forms an extended, triple-stranded α-helical coiled coil with the N-terminus at its tip (Chan et al., 1997; Tan et al., 1997; Weissenhorn et al., 1997). The last 16 residues at the N-terminus of the HIV-1 gp41, not present in the crystallographic structures, are known as the fusion domain peptide. This is a mostly hydrophobic sequence thought to be specifically responsible for triggering the membrane fusion process during insertion into a host-cell membrane (Durell et al., 1997; Weissenhorn et al., 1999).
During the last decade, a large body of work directed to the study of the molecular mechanisms of the membrane fusion process using synthetic peptides designed according to the N-terminal hydrophobic sequence of gp41 has been assembled (Martin et al., 1996; Durell et al., 1997; Pereira et al., 1997; Cladera et al., 1999). Many of these studies have been performed using model membranes but have led to conflicting interpretations, particularly with respect to the identity of the peptide secondary structures involved in the process, described as β-structures in some cases (Pereira et al., 1997; Peisajovich et al., 2000) and as obliquely oriented α-helices in others (Martin et al., 1996).
In the present paper, we report studies of the interaction of the 16 residue gp41 fusion peptide (gp41FD) with T-lymphocyte membranes. We show that the time evolution and overall extent of the interaction of gp41FD with T cells are accessible using a technique introduced by our laboratory (Wall et al., 1995a,b). Measurements rely on the fact that most proteins that interact with membranes possess a net charge and, once bound, result in changes of the electrostatic potential present on the membrane surface. Such changes influence the fluorescence yield of fluorescein phosphatidylethanolamine (FPE) previously added in very small amounts (1 mol FPE:800–1000 mol lipid) with the fluorescent indicator moiety precisely located at the membrane–solution interface. The underlying theory and application of this technique are outlined by Wall et al. (1995a,b), and it has been demonstrated to possess the additional virtue that it may be applied to model membrane systems as well as to living cells. These techniques facilitate real-time monitoring of the peptide– cell membrane interaction in a virtually non-invasive fashion.
These studies are complemented by the use of a fluorescent indicator, 1-(3-sulfonatopropyl)-4-[β[2-(di-n-octyl-amino)-6-naphthyl]vinyl]pyridinium betaine (di-8- ANEPPS), which responds to changes of the so-called membrane dipole potential (Gross et al., 1994; Cladera and O’Shea, 1998), the magnitude of which is particularly affected by peptide structures penetrating the lipidic bilayer (Cladera and O’Shea, 1998; Cladera et al., 1999). A number of parameters including the membrane affinity, peptide oligomerization, competitive ligand interactions and involvement of surface glycosaminoglycans (GAGs) are reported, and suggest a novel mechanism for the interactions of viral proteins with human cells.
Results
The interaction of the fusion domain of gp41 with phospholipid membranes
The experimental traces shown in Figure 1 indicate that there is a clear interaction of gp41FD with phospholipid membranes made up of phosphatidylcholine supplemented with a virtually non-invasive amount of FPE (0.2 mol%). The increment of the fluorescence signal following exposure of the membranes to gp41FD is consistent with binding of the positively charged N-terminus of the peptide to the membrane surface. The cumulative change of fluorescence following complete titrations of the membranes with gp41FD leads to a binding profile that is clearly not a hyperbolic binding isotherm. The simplest ‘best-fit’ analysis of this profile was found to be described by equation 1 (see Materials and methods) and yields a cooperativity coefficient (i.e. the so-called Hill coefficient) (n) close to 1.5.

Fig. 1. (A) Time course of the interaction of gp41FD with 100 nm diameter unilamellar phosphatidylcholine vesicles as revealed by fluorescence of FPE-labelled membranes, with (1) and without (2) 700 nM IL-8 present in the medium. (B) Amplitude of the fluorescence variation as a function of peptide concentration following normalization by background subtraction. Data were fitted to equation 1 (see Materials and methods). Lipid concentration, 200 µM; temperature, 37°C; buffer, 280 mM sucrose, 10 mM Tris pH 7.4. Excitation wavelength: 490 – emission (λem) = 520 nm.
Figure 1 also indicates the effect of supplementing the membranes with 700 nM interleukin (IL)-8 prior to exposure to gp41FD. Under these conditions, no effects of the chemokine on the membrane interactions of gp41FD were noted; these observations are of some importance, as outlined during the course of the discussion below.
The membrane penetration of gp41FD as revealed by di-8-ANEPPS
The use of di-8-ANEPPS as an indicator of membrane penetration by gp41FD is shown in Figure 2. This analytical system is more complicated than that offered by FPE, as a spectral shift is the principal response of the dye system to the penetration of membranes by certain types of macromolecule. Difference spectra shown in Figure 2 reflect a very similar spectral shift of di-8-ANEPPS following exposure of either model or cell membranes to gp41FD. As in the case of the simian immunodeficiency virus (SIV) fusion peptide (Cladera et al., 1999), the shift may be interpreted as a decrease in the magnitude of the membrane dipole potential caused by the peptide. The intensity of the difference spectrum for both of these hydrophobic peptides was ∼10 times larger than that of the difference spectrum of hydrophilic peptides that have no tendency to penetrate the lipidic bilayer (Figure 2).

Fig. 2. Fluorescence difference spectra obtained by subtracting the excitation spectrum at the indicated wavelengths (λem = 580 nm) of di-8-ANEPPS-labelled membranes from the spectrum of labelled membranes plus gp41FD: (a) Jurkat cells (2 × 104 cells/ml); (b) model membranes (200 µM phosphatidylcholine). gp41FD concentration was 10 µM and temperature was 37°C. The buffer was 280 mM sucrose, 10 mM Tris pH 7.4. The traces are compared with that obtained from the interaction of the hydrophilic peptide bacitracin with phosphatidyl choline membranes (c) in conditions for which the peptide has been found to interact with the surface of the membranes (positive FPE response in phosphate buffer pH 6.5).
The interaction of gp41FD with T lymphocytes; effect of IL-8 and heparitinase treatment on the binding process
We have shown previously that it is possible to label cell membranes with FPE in order to study the interactions of many types of molecule with living cells (e.g. Wall et al., 1995b). In a similar manner to the experiments shown in Figure 1, Figure 3 illustrates studies of the interactions of gp41FD with the Jurkat T-lymphocyte cell line. The experimental traces shown in Figure 3 indicate that there is a clear interaction of gp41FD with the T-lymphocyte cell surface. In contrast to the simple phospholipid membranes described in Figure 1, however, it is also shown that treatment of the T cells with IL-8 completely abolishes their interaction with gp41FD (Figure 3).

Fig. 3. (A) Time course of the interaction of gp41FD with Jurkat cells as revealed by fluorescence of FPE-labelled membranes. (B) Amplitude of the fluorescence variation as a function of peptide concentration following normalization by background subtraction in the absence (open circles) and presence (closed triangles) of 700 nM IL-8. Data were fitted to equation 1 (see Materials and methods). Cell concentration was 2 × 104 cells/ml. Other experimental conditions as in Figure 1.
The experimental data shown in Figure 3 also do not fit well to a simple binding isotherm, but are best described as a sigmoidal process (equation 1, Materials and methods) with n close to 3. The cooperativity inherent in the membrane interaction described by the Hill coefficient may be interpreted to indicate the formation of a trimeric molecular complex of gp41FD (see below).
Heparitinase treatment of the Jurkat T-cell line, which removes heparan sulfate from the cell surface, may be undertaken without any obvious consequences on cell viability or integrity. Following this treatment, similar studies to those described in Figure 3 result in the virtual abolition of interaction of gp41FD with the T cells (Figure 4).

Fig. 4. Amplitude of the fluorescence variation as a function of gp41FD concentration following normalization by background subtraction: Jurkat cells (open triangles) and Jurkat cells treated with heparitinase (open circles). Experimental conditions as in Figure 3.
Together with the effect of IL-8, previously suggested to have an affinity for heparan sulfate (Webb et al., 1993; Spillmann et al., 1998), these observations imply that gp41FD may interact specifically with heparan sulfate. The specificity of this mutual interaction is addressed in the following section.
The specificity of the gp41FD–GAG interaction; effects of soluble GAGs on the T-cell interaction
It was considered worthwhile to explore the selectivity of gp41FD for particular GAGs. In particular, to determine whether such reagents in soluble form could compete with the binding to the cell surface or alternatively whether soluble heparan sulfate could augment the binding reaction of gp41FD to the T cell. Some of the results of such an analysis are shown in Figure 5 in which the interaction of gp41FD with T cells in the presence of soluble heparan sulfate or heparin is illustrated. It is clear from this study that the presence of 5 mg/ml heparin has little effect on the overall interaction of gp41FD with the T cell, as the same signal end point is reached. The presence of 1 mg/ml heparan sulfate, however, has a dramatic effect, causing a reduction in the interaction of gp41FD with the T cells. Whilst further studies of this phenomenon are in progress, the implications of these observations are that there is a significant specificity for the interaction of gp41FD with heparan sulfate.

Fig. 5. Effect of heparin and heparan sulfate (HS) on the interaction of gp41FD with Jurkat cells as revealed by fluorescence of FPE-labelled membranes. Cell concentration, 2 × 104 cells/ml; heparin concentration, 5 mg/ml; HS, heparan sulfate concentration, 1 mg/ml; other experimental conditions as in Figure 1.
On this basis it appears that there is no enhancement of the gp41FD–T-cell interaction due to the presence of heparan sulfate or heparin when they are free in solution. These molecules in soluble form appear able to interact with gp41FD and, in the case of heparan sulfate, prevent the binding reaction to the T cell. With heparin, however, it is not clear whether it is able to interact with gp41FD or not, as there is little effect on the total reaction with the T cell. Up to this stage, however, we had noticed that heparin affected the light scattering properties of the peptides in solution (i.e. during routine correction of fluorescence signals) and thus indicated that some form of interaction was likely to be taking place. This problem is addressed further as Fourier transform infrared (FTIR) spectra were taken of gp41FD in the absence and presence of heparin (Figure 6). By making use of the attenuated total reflectance (ATR) facility, FTIR offers the possibility of determining directly the interaction of gp41FD with heparin. The IR spectrum of gp41FD in D2O in the amide-I region shown in Figure 6 exhibits a characteristic band centred at 1625 cm–1. This feature has previously been assigned to intermolecular β-sheet structure and is indicative of peptide aggregates (Nieva et al., 1994). Such studies of gp41FD have been carried out previously (Cladera et al., 1999) and illustrate that preparations do indeed exhibit some level of peptide aggregation in aqueous solution. Spectra of gp41FD taken in the presence of heparin, but with removal of the spectroscopic contribution of heparin may reveal any interaction of heparin with gp41FD. Following this, a lack of such a band or at least a dramatic reduction in the ATR-FTIR spectral band corresponding to the peptide in the presence of heparin implies disassembly of such aggregates upon interaction of the peptide with heparin (Figure 6). Thus, heparin appears able to interact with and, at least at the concentrations utilized (i.e. 5× greater than that of heparan sulfate), unable to prevent the interaction of gp41FD with the T cell. Consequently, these observations imply that the interaction of gp41FD takes place with a specific heparan sulfate binding domain. This domain must be different to the portion of gp41FD that interacts with heparin and, therefore, the cellular binding is unaffected by the presence of soluble heparin.

Fig. 6. ATR-FTIR spectra of gp41FD (100 µM) in D2O in the absence (dotted line) and presence (solid line) of heparin (30 mg/ml).
The spatial localization of the interaction of gp41FD on the T-lymphocyte cell surface
In addition to studying the interactions of molecules with ensembles of cells as illustrated in Figures 3 and 4, it is also possible to acquire FPE fluorescence images of single cells (Cladera and O’Shea, 2000) following their exposure to (macro)molecules of interest. Provided that the fluorescence detection system is sensitive enough, this latter approach has the additional virtue of yielding spatial information on the interaction of molecules with the cell surface. Such studies were carried out with the gp41FD–T-cell interaction (Figure 7).
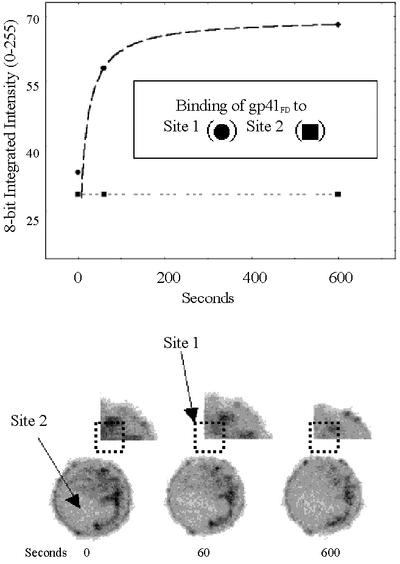
Fig. 7. Confocal laser scanning microscopy images of Jurkat cells following exposure to 5 µM gp41FD. The lower series of images include at 0 s a confocal fluorescence image of a single T cell labelled with FPE. Images of the same cell at 60 and 600 s indicate the effects of challenge with the gp41FD peptide. No laser irradiation between the indicated times of image acquisition took place and so photobleaching was negligible. The upper series of images indicates an enlarged section of the T cell from the top right-hand corner, and site 1 and 2 represent matched regions from each image. The upper plot indicates any change of localized fluorescence intensity at sites 1 and 2 following treatment of the T cell with gp41FD at the indicated times. The values on the ordinate scale are at 8 bit resolution (0–255).
On a qualitative basis, two points are evident from Figure 7. The first is that following correction for any spatial heterogeneity of FPE, the cell surface electrostatic potential is not evenly delocalized about the cell surface. This property appears to be common in most eukaryotic cell types (Cladera and O’Shea, 2000). Secondly, the interactions of gp41FD are also spatially heterogeneous. The implication of the latter observation is that there are ‘preferred sites’ on the cell surface of the T cell at which gp41FD interacts. It is also possible to make quantitative assessments of the affinity of the respective sites on the cell surface, i.e. the affinity of the localized site (site 1) is clearly much greater than that represented by site 2 (see Discussion).
Discussion
A number of comments on the nature of the interaction of gp41FD with membranes appear possible based on the experimental studies described. The first is that the binding profile of gp41FD with simple phospholipid membranes is characterized, as shown in Figure 1, by a cooperativity (i.e. Hill) coefficient of 1.5, and could be taken as evidence to indicate that a dimeric peptide complex has been formed on the membrane surface. Analogous studies with T cells shown in Figure 3, however, indicate that the membrane interaction is characterized by a cooperativity coefficient consistent with a trimeric molecular complex of gp41FD appearing to offer the most stable membrane-associated structure. The latter is consistent with reports in the literature that gp41 exists as a coiled-coil structure on the basis of X-ray diffraction studies of crystals of the ecto-domain of gp41 (Chan et al., 1997; Tan et al., 1997; Weissenhorn et al., 1999). The present study, however, represents the first clear indication that this state of affairs may also exist in the presence of membranes.
We have tried to emphasize in earlier publications that the membrane interactions of many types of peptide with analogous properties to gp41FD are complicated by the presence of peptide aggregates that occur in aqueous preparations (Cladera et al., 1999). Whilst this does not compromise the overall interpretation that the membrane interactions take place (as outlined by Cladera et al., 1999), we are less confident that some specific molecular details can be assigned in an unequivocal manner unless care is taken to account for any contribution from such aggregates (particularly any structural features). Thus, this behaviour may underlie the conflicting views as to the secondary structural characteristics of such peptides when associated with membranes, given that such structural studies require much larger concentrations of peptide and cannot be undertaken with living cell preparations. This latter comment appears important as according to the present study T cells exhibit significant differences to artificial membranes.
Treatment of the Jurkat T-cell line with IL-8 or heparitinase virtually abolished the gp41FD–membrane interaction (Figures 3 and 4). This behaviour was not found in similar studies with phospholipid membranes (Figure 1). Thus, by using the phospholipid membrane– gp41FD interaction as an indicator of the availability of gp41FD, from Figure 1 it is possible to state that IL-8 does not bind directly to gp41FD and prevent the binding reaction simply by making it unavailable to bind.
It is known that the Jurkat T-cell line employed in this study does not possess appropriate chemokine (IL-8) receptors (Jones et al., 1997), but it has been reported that IL-8 may bind to heparan sulfate resident on the T-cell surface (Webb et al., 1993; Spillmann et al., 1998). Similarly, it has been claimed that the envelope glycoprotein of HIV may bind to heparan sulfate (Harouse et al., 1991; Roderiquez et al., 1995; Mondor et al., 1998; Ugolini et al., 1999). The latter suggestion prompted our studies on the possibility that heparan sulfate may represent a specific gp41FD‘receptor’. The fact that this interaction appears to be abolished by removal of heparan sulfate from the cell surface and is blocked by IL-8 is also consistent with this latter possibility.
The specificity and attendant mechanism of the gp41FD–heparan sulfate interaction are clearly important considerations given that a number of GAGs feature in cellular behaviour. A comprehensive survey is beyond the scope of the present article, but it was considered worthwhile to compare the effects of soluble heparan sulfate and heparin on the membrane interactions of gp41FD. Thus, the question of whether soluble heparan sulfate could augment or prevent the membrane interaction of gp41FD was considered of sufficient merit to address in the present study. This is illustrated in Figure 5 and shows that soluble heparan sulfate competes with those molecules present on the cell surface. In other words, soluble heparan sulfate does not promote the membrane interaction of gp41FD. This conclusion may aid in the elucidation of the specific mechanism of their mutual intermolecular interaction and hence possibly in the development of therapeutic agents to prevent intracellular infection by viruses that depend on such fusion mechanisms.
Heparin was also found to interact with gp41FD, as indicated by the ATR-FTIR studies shown in Figure 6; thus, in a complementary study also shown in Figure 5, heparin was included when T cells were challenged with gp41FD. In this experimental system, however, virtually no effect was observed at levels of heparin 5-fold in excess of those of soluble heparan sulfate at which a clear effect was observed. This appears to indicate that there is a high level of selectivity by gp41FD for the heparan sulfate moiety on the cell surface that is not overcome by excessive levels of heparin but may be by soluble heparan sulfate. Perhaps it is also worth emphasizing that as a corollary to these observations, heparan sulfate appears to act as a specific receptor for IL-8, in accordance with the views of Webb et al. (1993).
The data obtained using di-8-ANEPPS, on the other hand, illustrate how gp41FD may penetrate the cell membrane, as revealed by changes in the magnitude of the membrane dipole potential. We have reported previously that in the case of artificial phospholipid membranes, the di-8-ANEPPS technique may be employed to indicate membrane penetration of electroneutral peptides (Cladera and O’Shea, 1998). This approach was particularly revealing with studies of the gp41FD–membrane interaction where the di-8-ANEPPS measurements were consistent with a substantial electroneutral portion of peptide becoming inserted into the membrane during the course of the membrane interaction (Cladera et al., 1999). Such peptide penetration had been measured previously using ATR-FTIR spectroscopy (Martin et al., 1996) and by fluorescence quenching with brominated phospholipids (Agirrea et al., 2000). The interaction of peptides with very hydrophilic (and non-amphipathic) sequences yields di-8-ANEPPS difference spectra that are >10× less intense than those of hydrophobic or amphipathic peptides (see Figure 2).
We report for the first time that di-8-ANEPPS may also be used with T cells to indicate membrane interactions as shown in Figure 2. The spectral shifts of the gp41FD–T-cell interaction shown in Figure 2 indicate that the membrane interactions are similar to those observed with artificial membranes (see also Cladera et al., 1999). The simplest explanation of this observation is that the peptide orientation and/or depth of penetration of gp41FD is similar in the T-cell membrane and the simpler phospholipid membrane. An appropriate question to be addressed, therefore, concerns the additional role of the heparan sulfate. In other words, there is a simple phospholipid bilayer of the type present in the phospholipid vesicles (PLVs) present as part of the T-cell plasma membrane, and as gp41FD is able to interact with this membrane what does heparan sulfate do to augment the membrane interaction? It is clear, however, that following removal of the heparan sulfate binding sites (either by their physical removal or blocking with IL-8 or soluble heparan sulfate) there seems to be little apparent interaction of gp41FD with the T-cell phospholipid membrane. One solution to this apparent anomaly is that the amount of phospholipid membrane presented by the T-cell suspension, and therefore available to bind gp41, is insignificant compared with that offered by the PLV suspension. Thus, by assuming that a typical single phospholipid occupies an area of 74 Å2, the PLV diameter is 100 nm and the T-cell diameter is 10 µm (and assuming the total surface represented by phospholipid), the respective phospholipid surface area is at least 104 times smaller in the T-cell experimental system. This ratio is diminished much further if the T-cell surface area is also corrected for the presence of proteins, etc. Thus, in order for the gp41–phospholipid membrane interaction to be emphasized or dominate, it would be necessary for the T-cell population to be expanded to become closer to that represented by the artificial membrane system. A potential role of the heparan sulfate, by binding the gp41 fusion domain and bringing it into close proximity with the T-cell plasma membrane, is that it significantly increases the possibility of the membrane interaction of the fusion peptide >104–5-fold over and above that presented by a simple phospholipid membrane. Secondly, and just as importantly, it seems that the presence of heparan sulfate promotes the formation of a gp41 trimer rather than a dimer. The former structure seems to be the preferred structure to promote fruitful fusion between the viral and host-cell membranes. However, this structure, with a high degree of membrane affinity, does not appear to be formed in the presence of soluble heparan sulfate (Figure 5).
Previous work describes the interaction between heparan sulfate and the V3 loop of gp120 protein, which would be dependent on the oligomerization state of the complex gp120–gp41 (Roderiquez et al., 1995). These results may be interpreted in terms of heparan sulfate participating in the process of attachment of the virus to the cell surface since gp120 does not participate in the posterior fusion events in a direct way (although it may well do so via the induction of conformational changes that affect gp41). Our results, however, imply a more direct involvement of heparan sulfate with the structures directly implicated in the membrane fusion process. A similar situation has been described for the fusion protein of vaccinia virus (Hsiao et al., 1998; Vazquez and Esteban, 1999). In a similar manner to gp41, the 14 kDa vaccinia virus protein consists of a triple-stranded coiled-coil region. The protein contains an internal, hydrophobic fusion peptide very similar to gp41FD close to the protein N-terminus. A short hydrophilic sequence adjacent to, and partially overlapping with the fusion peptide, which contains positively charged Lys residues, has been identified as the part of the protein involved in post-virus binding interactions with heparan sulfate (Hsiao et al., 1998; Vazquez and Esteban, 1999). Such an electrostatic interaction between the peptide and heparan sulfate could also take place in the case of gp41FD used in the present studies, via the positively charged N-terminus. In the case of the native gp41 protein, on the other hand, Lys residues present in the 16-residue-long stretch adjacent to the fusion peptide (Peisajovich et al., 2000) would appear to be good candidates to interact with the negatively charged molecules on the cell surface.
The final comment to make from the present study concerns the possibility that the interaction of gp41FD with the T-cell surface takes place within specific domains on the cell surface. Thus, the role of heparan sulfate during the interaction of gp41FD appears to be further complicated by spatial localization about the T-cell surface according to the extension of the use of the electrostatic sensing within an imaging regimen as illustrated in Figure 5. In accordance with this possibility, it is known that heparan sulfate proteoglycans regulate internalization and clearance of ligands via both clathrin-coated pits and membrane rafts associated with caveolae (Bernfield et al., 1999). Syndecans act as internalizing receptors for lipoprotein lipase and its bound ligands via a non-coated pit pathway leading to lysosomal degradation (Martinho et al., 1996; Fuki et al., 1997; Williams and Fuki, 1997). Glypicans, on the other hand, are thought to be linked to glycosyl-phosphatidylinositol (GPI). In this form, the GPI-anchored proteins in mammalian cells are believed to associate with ordered regions of lipid (i.e. rafts) (Simons and Ikonen, 1997). Thus, the images shown in Figure 5 indicate that the interactions of a virus such as HIV/SIV on a target T-cell membrane would take place at ‘preferred’ localized sites. This has the effect of providing a platform for the easy formation of trimeric fusion structures that facilitate infection. If there were no spatial localization and concentration, virus binding to the other families of receptors (i.e. CD4/chemokine) might not be compromised, but cellular penetration and infection would be highly unlikely and, therefore, would not be favoured from an evolutionary point of view.
Materials and methods
Egg phosphatidylcholine, phosphatidylethanolamine and heparitinase were purchased from Sigma Chemical Company (St Louis, MO). FPE was synthesized as previously described according to Wall et al. (1995a). Di-8-ANEPPS was purchased from Molecular Probes. Purified recombinant (expressed in Escherichia coli) human IL-8 was a generous gift from Dr Dave Layfield, Astra-Zeneca, Loughborough, UK.
HPLC-purified synthetic peptides prepared with the C-terminus in amide form were purchased from Quality Controlled Biochemical, Inc. (Hopkinton, MA). Stock solutions of these peptides were made up in dimethyl sulfoxide (DMSO), typically at a concentration of 4 mg/ml. Purified heparin, heparitinase and heparan sulfate were obtained from Sigma Chemical Co., Poole, Dorset, UK.
Membrane preparations and labelling with FPE and di-8-ANEPPS
Phospholipids dissolved in chloroform, di-8-ANEPPS (when required) and the appropriate additive (6-ketocholestanol or phloretin in methanol) were mixed in a round-bottom flask and the solution was dried under a stream of nitrogen to deposit a thin lipid film on the inside of a glass tube. PLVs were prepared by hydrating the dried lipid film with the sucrose buffer (280 mM sucrose, 10 mM Tris pH 7.5), then repeatedly freezing and thawing the suspension five times, and finally extruding it 10× through two polycarbonate filters of pore size 0.1 µm (Nuclepore Corp., Pleasanton, CA) using an extruder (Lipex Biomembranes Inc., Vancouver, Canada) according to the extrusion procedure of Mayer et al. (1986). PLVs were labelled exclusively in the outer bilayer leaflet with FPE as described by Wall et al. (1995a). Briefly, PLVs were incubated with FPE dissolved in ethanol (never >0.1% of the total aqueous volume) at 37°C for 1 h in the dark. Any remaining unincorporated FPE was removed by gel filtration on a PD10 Sephadex column equilibrated with the appropriate buffer. Such a procedure leads to the incorporation of 30–50% of the externally added FPE into the preformed PLV. Furthermore, there was no observed transmembrane ‘flipping’ of the FPE, at least over time scales of 1 week. The FPE-liposomes were stored at 4°C until use.
Jurkat T-lymphocyte culture, labelling with FPE and di-8-ANEPPS and treatment with heparitinase
Jurkat cells were cultured and labelled with FPE as described in Wall et al. (1995b) and Cladera and O’Shea (2000). The cells were cultured in RPMI 1640 medium with NaHCO3 (Sigma) and supplemented with 10% fetal calf serum, 2 mM glutamine and 2% (v/v) Bash at 37°C and 5% CO2. The cells were harvested and washed by two centrifugation steps at 2500 g for 5 min in sucrose buffer (280 mM sucrose, 10 mM Tris pH 7.4). The cells were counted using a haemocytometer and the trypan blue exclusion technique. A volume of FPE in chloroform–methanol at a ratio of 10 µg of FPE:3 × 106 cells was placed in a tube, and the organic solvent was removed under a stream of argon followed by resolvation with 15 µl of ethanol. The cell suspension was then added to the FPE suspension and the mixture was incubated for 45 min at 37°C. After this time, unincorporated FPE was removed by centrifugation (2500 g, 5 min) and resuspension in sucrose-based buffer.
Cells were labelled with di-8-ANEPPS as follows: 0.5 µM dye was added to a suspension containing 20 000 cells/ml. The mixture was incubated for 2.5 h at 36°C. After this period, very small increments in the intensity of the excitation spectra could still be detected as a consequence of the dye still being incorporated into the membrane. This variation, however, did not lead to any spectral shift that could compromise the difference spectra of the kind originating from the peptide after normalization (see Figure 2 and legend).
Cells (3 × 105 cells/ml) were treated with heparitinase (1 international mU) after labelling with FPE for 1 h at 37°C and then exposed to HIV gp41FD. After enzyme treatment, the FPE response emission spectrum and the dye response to calcium ions were checked in order to ensure that the labelling had not been affected by the enzymatic treatment and was equivalent to that of untreated cells.
Fluorescence measurements
Fluorescence time courses were obtained by adding the desired amount of peptide to 2 ml lipid suspensions (200 µM lipid or 20 000 cells/ml) on a SLM-AMINCO model spectrofluorometer. For FPE experiments, excitation and emission wavelengths were set at 490 and 518 nm, respectively.
Di-8-ANEPPS spectra were obtained by exciting the samples as indicated in Figure 2 and measuring the intensity at 580 nm (Gross et al., 1994; Cladera et al., 1999).
In FPE experiments, the contribution of light scattering to the signal was corrected by recording it using vesicles or cell suspensions without the fluorescent dye at the same vesicle concentration, and subtracting the trace from that obtained with the dye present.
The total amplitudes of the FPE fluorescence signals plotted against peptide concentration were found to be sigmoidal. These data were analysed according to equation 1 and yielded values for the Hill coefficient (n):
observed signal = 100% signal × [peptide]n/Kd + [peptide]n(1)
where Kd is the affinity of the peptide for the membrane in concentration units.
IR spectroscopy
ATR-IR spectra of gp41FD in D2O in the absence and presence of heparin were acquired on a Brucker IFS88 spectrometer equipped with an MCT detector and a benchmark ATR flowthrough crystal, operational at an instrumental resolution of 2 cm–1. Typically, 2000 scans were averaged at room temperature, apodized with a triangle function, and Fourier transformed. Peptides were solubilized in 2 ml of D2O supplemented with 30 µl of DMSO containing the peptide (i.e. identical conditions to those used with the fluorescence studies of the membranes). To obtain the pure spectra of the peptide, spectra of the solvent (and the solvent with heparin when required) were collected under identical conditions and subtracted. There was no contribution to the amide-I region from the DMSO and any residual water vapour bands were also subtracted using a previously stored water vapour spectrum (Cladera and O’Shea, 1998).
Confocal microscopy
Confocal images were obtained using a Bio-Rad MRC600 Confocal laser scanning microscope (Bio-Rad Laboratories Ltd, Hemel Hempstead, UK). The fluorescein system of filters was utilized, which are very close to the excitation–emission properties of FPE. FPE-labelled Jurkat T cells were placed on a coverslip and a series of images were taken before and after addition of gp41FD using oil immersion. The settings of the photomultipliers (gain and black level) were constant for the series of images. Care was taken that any photobleaching did not compromise the interpretation, and laser irradiation and other illumination was prevented between acquisitions. The disposition of FPE about the cell membrane was corrected as described by Cladera and O’Shea (2000). The confocal images were obtained at 8 bit resolution (i.e. with an intensity range from 0 to 255).
Acknowledgments
Acknowledgements
We are grateful to other members of our research group, Bushra Sim and Tanong Asawakarn for their careful assistance with the Jurkat cell culture, Chris Gregory was also most generous in providing additional stocks of the Jurkat cell line, Dave Barrett and Adam Watkins for help with the ATR-FTIR studies, and Nancy Awad and Kate Dodson for the studies with the T cells. Simon Jones (Cardiff) was also helpful with advice about the Jurkat cell line and chemokines in general.
References
- Agirrea A., Flachb,C., Goni,F.M., Mandelsohnb,R., Valpuestac,J.M., Wub,F. and Nieva,J.L. (2000) Interactions of the HIV-1 fusion peptide with large unilamellar vesicles and monolayers. A cryo-TEM and spectroscopic study. Biochim. Biophys. Acta, 1467, 153–164. [DOI] [PubMed] [Google Scholar]
- Bernfield M., Gotte,M., Park,P.W., Reizes,O., Fitzgerald,M.L., Lincecum,J. and Zako,M. (1999) Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem., 68, 729–777. [DOI] [PubMed] [Google Scholar]
- Bosch M.L., Earl,P.L., Fargnoli,K., Picciafuoco,S., Giombini,F., Wong-Staal,F. and Franchini,G. (1989) Identification of the fusion peptide of primate immunodeficiency viruses. Science, 244, 694–697. [DOI] [PubMed] [Google Scholar]
- Chan D.C., Fass,D., Berger,J.M. and Kim,P.S. (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell, 89, 263–273. [DOI] [PubMed] [Google Scholar]
- Cladera J. and O’Shea,P. (1998) Intramembrane molecular dipoles affect the membrane insertion and folding of a model amphiphilic peptide. Biophys. J., 74, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cladera J. and O’Shea,P. (2000) Generic techniques for fluorescence measurements of protein–ligand interactions; real-time kinetics and spatial imaging. In Harding,S.E. and Chowdery,B. (eds), Protein–Ligand Interactions. Oxford University Press, Oxford, UK, pp. 169–200.
- Cladera J., Martin,I., Ruysschaert,J.M. and O’Shea,P. (1999) Characterization of the sequence of interactions of the fusion domain of the simian immunodeficiency virus with membranes. J. Biol. Chem., 274, 29951–29959. [DOI] [PubMed] [Google Scholar]
- Clapham P.R., Reeves,J.D., Simmons,G., Dejucq,N., Hibbitts,S. and McNight,A. (1999) HIV co-receptors, cell tropism and inhibition by chemokine receptor ligands. Mol. Membr. Biol., 16, 49–55. [DOI] [PubMed] [Google Scholar]
- Durell S.R., Martin,I., Ruysschaert,J.M., Shai,Y. and Blumenthal,R. (1997) What studies with fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion. Mol. Membr. Biol., 14, 97–112. [DOI] [PubMed] [Google Scholar]
- Fuki I.V., Kuhn,K.M., Lomazov,I.R., Rothman,V.L., Tuszynski,G.P., Lozzo,R.V., Swenson,T.L., Fisher,E.A. and Williams,K.J. (1997) The syndecan family of proteoglycans. Novel receptors mediating internalization of atherogenic lipoproteins in vitro. J. Clin. Invest., 100, 1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallaher W.R. (1987) Detection of a fusion peptide sequence in the transmembrane protein of HIV. Cell, 50, 327–328. [DOI] [PubMed] [Google Scholar]
- Gross E., Bedlack,R.S. and Loew,L.M. (1994) Dual-wavelength ratiometric fluorescence measurement of the membrane dipole potential. Biophys. J., 67, 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harouse J.M., Bhat,S., Spitalnik,S.L., Laughlin,M., Stefano,K., Silberberg,D.H. and Gonzalez-Scarano,F. (1991) Inhibition of entry of HIV-1 in neural cell lines by antibodies against galactosyl ceramide. Science, 253, 320–323. [DOI] [PubMed] [Google Scholar]
- Hsiao J.C., Chung,C.S. and Chang,W. (1998) Cell surface proteoglycans are necessary for A27L protein-mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J. Virol., 72, 8374–8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.A., Dewald,B., Clark-Lewis,I. and Baggiolini,M. (1997) Chemokine antagonists that discriminate between interleukin-8 receptors. J. Biol. Chem., 272, 16166–16169. [DOI] [PubMed] [Google Scholar]
- Kowalski M. et al. (1987) Functional regions of the envelope glycoprotein of human immunodeficiency virus type-1. Science, 237, 1351–1355. [DOI] [PubMed] [Google Scholar]
- Kwong P.D., Wyatt,R., Robinson,J., Sweet,R.W., Sodroski,J. and Hendrickson,W.A. (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature, 393, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky L.A., Nakamura,G., Smith,D., Fennie,C., Shimlasaki,C. Patzer,E., Berman,T., Gregory,T. and Capon,D. (1987) Delineation of a region of the HIV-1 gp120 glycoprotein critical for interaction with CD4 receptor. Cell, 50, 975–985. [DOI] [PubMed] [Google Scholar]
- Martin I., Schaal,H., Scheid,A. and Ruysschaert,J.-M. (1996) Lipid membrane fusion induced by the human immunodeficiency virus type 1 gp41 N-terminal extremity is determined by its orientation in the lipid bilayer. J. Virol., 70, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinho R.G., Castel,S., Urena,J., Fernandez-Borja,M., Makiya,R., Olivecrona,G., Reina,M., Alonso,A. and Vilaro,S. (1996) Ligand binding to heparan-sulfate proteoglycans induces their aggregation and distribution along actin cytoskeleton. Mol. Biol. Cell, 7, 1771–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer L.D., Hope,M.J. and Cullis,P.R. (1986) Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta, 858, 161–168. [DOI] [PubMed] [Google Scholar]
- Mondor I. et al. (1998) Interactions among HIV gp120, CD4 and CXCR4: dependence on CD4 expression level, gp120 viral origin, conservation of the gp120 COOH- and NH2-termini and V1/V2 and V3 loops and sensitivity to neutralizing antibodies. Virology, 248, 394–432. [DOI] [PubMed] [Google Scholar]
- Nieva J.L., Nir,S., Muga,A., Goni,F.M. and Wilschut,J. (1994) Interaction of the HIV-1 fusion peptide with phospholipid vesicles: different structural requirements for fusion and leakage. Biochemistry, 33, 3201–3209. [DOI] [PubMed] [Google Scholar]
- Peisajovich S.G., Epand,R.F., Pritsker,M., Shai,Y. and Epand,R.M. (2000) The polar region consecutive to the HIV fusion peptide participates in membrane fusion. Biochemistry, 39, 1826–1833. [DOI] [PubMed] [Google Scholar]
- Pereira F.B., Goñi,F.M., Muga,A. and Nieva,J.L. (1997) Perme abilization and fusion of uncharged lipid vesicles induced by the HIV-1 fusion peptide adopting an extended conformation: dose and sequence effects. Biophys. J., 73, 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderiquez G., Oravecz,T., Yanagishita,M., Bou-Habib,D.C., Mostowski,H. and Norcross,M.A. (1995) Mediation of human immunodeficiency virus type I binding by interaction of cell surface heparan sulfate proteoglycans with the V3 region of envelope gp120–gp41. J. Virol., 69, 2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. and Ikonen,E. (1997) Functional rafts in cell membranes. Nature, 387, 569–572. [DOI] [PubMed] [Google Scholar]
- Spillmann D., Witt,D. and Lindahl,U. (1998) Defining the interleukin-8-binding of heparan sulfate. J. Biol. Chem., 273, 15487–15493. [DOI] [PubMed] [Google Scholar]
- Tan K., Liu,J., Wang,J., Shen,S. and Lu,M. (1997) Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl Acad. Sci. USA, 94, 12303–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolini S., Mondor,I. and Sattendau,Q.J. (1999) HIV-1 attachment: another look. Trends Microbiol., 7, 144–149. [DOI] [PubMed] [Google Scholar]
- Vazquez M.I. and Esteban,M. (1999) Identification of functional domains in the 14-kilodalton envelope protein (A27L) of vaccinia virus. J. Virol., 73, 9098–9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall J., Golding,C., van Veen,M. and O’Shea,P. (1995a) The use of fluoresceinphosphatidylethanolamine as a real time probe of peptide–membrane interactions. Mol. Membr. Biol., 12, 183–192. [DOI] [PubMed] [Google Scholar]
- Wall J., Ayoub,F. and O’Shea,P. (1995b) The interactions of macromolecules with the mammalian cell surface. J. Cell Sci., 108, 2673–2682. [DOI] [PubMed] [Google Scholar]
- Webb L.M., Ehrengruber,M.U., Clark-Lewis,I., Baggiolini,M. and Rot,A. (1993) Binding to heparan sulfate or heparin enhances neutrophil responses to interleukin-8. Proc. Natl Acad. Sci. USA, 90, 7158–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen,A., Harrison,S.C., Skehel,J.J. and Wiley,D.C. (1997) Atomic structure of the ectodomain from HIV-1 gp41. Nature, 387, 426–430. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen,A., Calder,L.J., Harrison,S.C., Skehel,J.J. and Wiley,D.C. (1999) Structural basis for membrane fusion by enveloped viruses. Mol. Membr. Biol., 16, 3–9. [DOI] [PubMed] [Google Scholar]
- Williams K.J. and Fuki,I.V. (1997) Cell-surface heparan-sulfate proteoglycans: dynamic molecules mediating ligand catabolism. Curr. Opin. Lipidol., 8, 253–262. [DOI] [PubMed] [Google Scholar]