

Abstract
Transgenic mice carrying an Aγ gene construct containing a –382 5′ truncation of the Aγ gene promoter have a phenotype of hereditary persistence of fetal hemoglobin (HPFH) but, when the CACCC box of the –382Aγ promoter is deleted, there is no γ gene expression in the adult mice. We used this system to investigate the mechanism whereby human HPFH mutations result in γ gene expression in the adult. Introduction of the –198 T→C HPFH mutation into the CACCC-less Aγ gene construct re-established the HPFH phenotype, indicating that this mutation increases promoter strength, most probably by establishing a novel CACCC box sequence in the –198Aγ region. The HPFH phenotype was also re-established when the –117 C→T HPFH mutation was introduced into a –141Aγ promoter with a destroyed CACCC box, indicating that this mutation increases γ promoter strength in the absence of the CACCC motif. The T→A –175 HPFH mutation failed to re-establish the HPFH phenotype when the CACCC box was deleted, indicating that γ gene expression in this mutation is CACCC box dependent. These results provide the first in vivo experimental evidence in support of mechanistic heterogeneity of the non-deletion HPFH mutants.
Keywords: γ gene promoter/hemoglobin switching/heriditary persistence of fetal hemoglobin/transgenic mice
Introduction
Hereditary persistence of fetal hemoglobin (HPFH) is a condition characterized by continuation of fetal hemoglobin (HbF) synthesis in the adult stage of erythropoiesis. Three types of abnormality are responsible for this phenotype. In deletional HPFH, the mutations delete sequences from the 3′ end of the β-globin locus including the δ- and β-globin genes (reviewed in Stamatoyannopoulos and Grosveld, 2001). Levels of fetal hemoglobin in heterozygous carriers usually range from 20 to 30%, and HbF is distributed pancellularly among the red cells. The mechanism of activation of HbF in deletional HPFH has been explained by various hypotheses, including the activation of the γ-globin gene by enhancer elements that are normally located in the 3′ end of the locus but are juxtaposed to the γ genes as a result of the 3′ deletions (Feingold and Forget, 1989; Anagnou et al., 1995; Arcasoy et al., 1997). The non-deletional HPFH is due to mutations that are either linked or non-linked to the β locus. The molecular basis of non-linked to the β locus HPFH is unknown. Non-deletional HPFH linked to the β locus is due to mutations characterized by synthesis of either the Aγ globin chain (Aγ HPFH) or the Gγ globin chain (Gγ HPFH). Structural studies have shown that the Gγ and Aγ HPFHs are due to mutations of the Gγ or the Aγ gene promoters (reviewed in Stamatoyannopoulos and Grosveld, 2001). A large number of studies have been done to delineate how these mutations result in continuation of elevated γ gene transcription in adult life, but the molecular mechanisms responsible for these phenotypes remain unclear.
We have previously produced a series of γ gene promoter truncations and analyzed their effects in transgenic mice (Stamatoyannopoulos et al., 1993). We have shown that, when linked to a 2.5 kb µLCR (locus control region) cassette, an Aγ gene containing a promoter truncated to –382γ or to –201γ is expressed at high levels in the embryonic and fetal stages of development and at similarly high levels in the adult stage, indicating that these truncations result in loss of the developmental control of γ gene expression. When the Aγ promoter sequence is extended to –730γ, silencing of the γ gene occurs in several transgenic lines, suggesting that a silencer is present between –382Aγ and –730Aγ. The –382γ and the –201γ promoter truncations, therefore, escape the developmental control of globin genes and behave as mutants of HPFH. We have also found that the CACCC box of the γ gene is necessary for γ gene expression in definitive erythropoiesis and, when this regulatory motif is deleted in the context of the –382γ promoter, there is γ gene expression in embryonic erythropoiesis but only residual γ gene expression in adult stage erythropoiesis.
The two types of truncated promoter, i.e. the –382γ promoter and the –382γ promoter with the CACCC box deleted, produce very distinct phenotypes, i.e. either high levels of γ expression in the adult or only residual γ expression in the adult, and thus provide a model system for the investigation of the role of γ promoter motifs on γ gene activation or silencing. We decided to capitalize on these observations and use this system to investigate, in transgenic mice, the functional role of mutations causing non-deletional HPFHs. Mutations of regulatory motifs known to produce non-deletion HPFH were done in the context of a µLCR –382Aγ ΔCACCC box promoter, and it was asked, in transgenic mice, whether these mutations reverse the phenotype of the µLCR –382Aγ ΔCACCC mice from one of lack of γ expression in the adult to one of persistence of γ gene expression in the adult. The appearance of the latter phenotype was considered to indicate that the HPFH mutation up-regulates γ gene expression.
Results
Experimental approach
Transgenic mice carrying non-deletional HPFH mutants in the context of the µLCR –382Aγ or the µLCR –382Aγ ΔCACCC constructs were produced and levels of γ mRNA were quantitated by RNase protection in the embryonic [post-coitus (p.c.) days 10 and 12] and definitive stages of erythropoiesis. Although the definitive erythropoiesis of the fetal liver starts at p.c. day 10.5, the day 12 blood and yolk sac are composed predominantly of embryonic nucleated red cells and therefore provide information on globin gene expression in embryonic erythropoiesis. Results of γ-globin expression during the development of the µLCR –382Aγ mice were published before (Stamatoyannopoulos et al., 1993). As in all transgenic lines carrying human γ-globin genes, the highest γ mRNA levels were observed in the day 12 embryonic blood and yolk sac of the µLCR –382Aγ mice. There was no statistically significant difference in γ mRNA levels between the embryonic day 12 and adult blood (Tables I–III). Data from five transgenic lines carrying intact µLCR –382Aγ ΔCACCC constructs (copy numbers 4, 5, 7, 7 and 35, respectively) are summarized in the tables. The deletion of the CACCC box did not affect γ gene expression in the embryonic cells but significantly decreased the γ mRNA levels in the cells of definitive erythropoiesis. Thus, the levels of γ mRNA decreased by 50% in day 12 fetal liver and by 80% in day 16 fetal liver and blood, while γ mRNA was barely detectable in the adult blood. The levels of γ gene expression in the µLCR –382Aγ and the µLCR –382Aγ ΔCACCC mice were compared with the findings in mice carrying the non-deletional HPFH mutants.
Table I. γ gene expressiona in the –198 HPFH transgenic mice.
| Construct | Line | Copy no. | Embryonic (primitive) erythropoiesis |
Definitive erythropoiesis |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Day 10b |
Day 12b |
Day 12b |
Day 16b |
Adult |
||||||
| Blood | Yolk sac | Blood | Yolk sac | Liver | Blood | Liver | Blood | |||
| µLCR –382Aγ | A | 5 | 6.9 ± 0.9 | 10.2 ± 1.1 | 21.6 ± 4.9 | 30.1 ± 4.7 | 23.7 ± 3.0 | 11.7 ± 0.8 | 12.4 ± 1.0 | 16.9 ± 1.1 |
| with –198 HPFH | B | 4 | 9.3 ± 0.3 | 10.7 ± 3.9 | 16.2 ± 4.0 | 30.5 ± 5.8 | 25.9 ± 2.8 | 19.8 ± 3.6 | 20.7 ± 2.3 | 15.1 ± 1.7 |
| mean | 8.1 | 10.5 | 18.9 | 30.3 | 24.8 | 15.8 | 16.6 | 16.0 | ||
| µLCR –382Aγ ΔCACCC | C | 12 | 7.6 ± 1.1 | 13.6 ± 1.3 | 28.6 ± 7.7 | 67.4 ± 14.5 | 30.1 ± 1.8 | 8.6 ± 0.9 | 10.2 ± 0.2 | 7.9 ± 1.5 |
| with –198 HPFH | D | 13 | 7.2 ± 0.5 | 10.4 ± 1.3 | 20.0 ± 3.7 | 51.6 ± 5.4 | 18.9 ± 2.8 | 11.9 ± 3.3 | 17.3 ± 5.2 | 13.8 ± 2.4 |
| E | 10 | 10.6 ± 0.6 | 19.1 ± 4.5 | 16.0 ± 1.1 | 69.7 ± 13.8 | 19.7 ± 1.3 | 11.9 ± 1.4 | 13.8 ± 1.7 | 10.4 ± 1.3 | |
| mean | 8.5 ± 1.9 | 14.4 ± 4.4 | 21.5 ± 6.4 | 62.9 ± 9.9 | 22.9 ± 6.2 | 10.8 ± 1.9 | 13.8 ± 3.6 | 10.7 ± 3.0 | ||
| µLCR –382Aγ | mean | 5.7 ± 6.3 | 5.2 ± 3.3 | 10.4 ± 9.5 | 21.0 ± 7.4 | 11.8 ± 2.8 | 13.4 ± 4.7 | 15.7 ± 4.8 | 16.5 ± 4.5 | |
| µLCR –382Aγ ΔCACCC | mean | 4.2 ± 0.6 | 6.1 ± 1.7 | 8.9 ± 4.3 | 13.9 ± 3.9 | 5.2 ± 3.0 | 2.9 ± 1.3 | 2.6 ± 0.9 | 0.9 ± 0.6 | |
aHuman γ mRNA is expressed as a percentage of murine α mRNA per copy. Mean values are shown in bold. Data in µLCR –382Aγ control mice are from Stamatoyannopoulos et al. (1993).
bDays post-coitus.
Table III. γ gene expressiona in the –117 HPFH transgenic mice.
| Construct | Line | Copy no. | Embryonic (primitive) erythropoiesis |
Definitive erythropoiesis |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Day 10b |
Day 12b |
Day 12b |
Day 16b |
Adult |
||||||
| Blood | Yolk sac | Blood | Yolk sac | Liver | Blood | Liver | Blood | |||
| µLCR –141Aγ | P | 8 | 6.0 ± 0.5 | 11.3 ± 0.7 | 10.4 | 14.9 | 10.0 | 11.5 ± 2.7 | 11.3 ± 3.6 | 6.4 ± 1.4 |
| with –117 HPFH | Q | 36 | 7.5 ± 0.8 | 11.3 ± 1.8 | 14.9 ± 1.0 | 24.0 ± 6.5 | 9.8 ± 1.2 | 14.4 ± 3.2 | 7.4 ± 0.3 | 5.3 ± 1.0 |
| R | 33 | 5.6 ± 0.9 | 12.4 ± 3.3 | 10.5 ± 1.8 | 17.7 ± 2.5 | 11.7 ± 0.6 | 7.9 ± 0.5 | 13.0 ± 2.4 | 6.7 ± 1.3 | |
| S | 22 | 8.5 ± 2.1 | 14.2 ± 1.5 | 15.0 ± 2.2 | 19.8 ± 4.4 | 11.7 ± 1.6 | 8.3 ± 1.5 | 8.2 ± 2.4 | 6.6 ± 0.5 | |
| mean | 6.9 ± 1.3 | 12.3 ± 1.4 | 12.9 ± 2.9 | 19.1 ± 3.8 | 10.8 ± 1.0 | 10.5 ± 3.0 | 10.0 ± 2.6 | 6.3 ± 0.6 | ||
| µLCR –141Aγ | mean | 2.4 ± 1.0 | 4.9 ± 1.0 | 10.6 ± 4.3 | 11.6 ± 2.1 | 4.3 ± 2.3 | 0.5 ± 0.3 | |||
aHuman γ mRNA is expressed as a percentage of murine α mRNA per copy. Mean values are shown in bold. Data in µLCR –141Aγ control mice are from Stamatoyannopoulos et al. (1993).
bDays post-coitus.
The –198 T→C HPFH mutation up-regulates γ gene transcription
The British variant of non-deletional HPFH is due to a T→C transition at position –198 of the γ-globin promoter (Tate et al., 1986). To test whether this mutation activates γ-globin gene expression, the –198 T→C transition was introduced into the µLCR –382Aγ construct and the µLCR –382Aγ (–198 HPFH) mutant was used for production of transgenic mice. Two transgenic mouse lines carrying correct copies of the construct were established. The presence of the –198 T→C transition resulted in elevated γ gene expression in the day 10 and 12 embryonic cells and the day 12 fetal liver cells of the µLCR –382Aγ (–198 HPFH) mice (Table I and Figure 1). In the adult µLCR –382Aγ (–198 HPFH) mice, the level of γ mRNA (16% of murine α) was similar to that of the –382Aγ controls (16.5% of murine α).

Fig. 1. Human γ-globin gene expression in transgenic mice carrying the –198Aγ HPFH mutant. Lines A and B carry the construct µLCR –382Aγ (–198 HPFH) while lines C, D and E carry the construct µLCR –382Aγ ΔCACCC (–198 HPFH). RNase protection assays were done using the total RNAs from erythroid cells from various stages of development. d10 y/s, day 10 yolk sac; d12 y/s, day 12 yolk sac; d16 f/l, day 16 fetal liver; ad bl, adult blood. Hu γ indicates the protected fragment (170 bp) from exon 2 of the human γ mRNA. Mu ζ is the protected fragment (151 bp) from exon 1 of the murine ζ mRNA. Mu α is the protected fragment (128 bp) from exon 1 of the murine α mRNA. Notice that the –198 HPFH mutation increases the γ mRNA level.
To test whether the –198 T→C mutation up-regulates γ gene expression in the absence of the γCACCC box, we deleted the CACCC box from the µLCR –382Aγ (–198 HPFH) construct. Three transgenic lines carrying correct copies of the µLCR –382Aγ ΔCACCC (–198 HPFH) construct were established. As shown in Table I (lines C, D and E), the introduction of the –198 C→T mutation in the –382Aγ ΔCACCC construct increased the level of γ mRNA in all stages of development. Thus, compared with the µLCR –382Aγ mice with a deleted CACCC box, in the mice that had a deleted CACCC box and the –198 C→T HPFH mutation, γ mRNA levels were 2- to 3-fold higher in embryonic cells, 4- to 7-fold higher in fetal liver erythroid cells, and at least 10-fold higher in the adult blood. These results provide direct evidence that the –198 C→T mutation up-regulates γ gene expression in vivo.
Previous studies have suggested that the introduction of the –198 C→T mutation creates a motif that can play a role similar to that of the γCACCC box (Ronchi et al., 1989; Fischer and Nowock, 1990; Gumucio et al., 1991). To test whether the motif created by the –198 HPFH mutant and the γCACCC box bind a similar set of proteins, gel retardation assays were performed. A 24 bp oligonucleotide encompassing the γCACCC box was 32P labeled and incubated with nuclear extracts from MEL cells. Two major and one minor shift bands could be seen on the gel (Figure 2, lane 1). The binding of all complexes was specific because it was competed away completely by the cold probe itself (lanes 2 and 3). None of the retarded bands could be competed out by the cold oligonucleotide encompassing the wild-type sequence between –210 and –185 of the γ promoter (lanes 6 and 7). The retarded bands in lane 7 became fainter when a higher concentration of the cold competitor (500-fold) was applied. In contrast, when the –198 C→T mutation was introduced into the oligonucleotide, all the bands were competed away completely (compare lanes 4 and 5 with lanes 6 and 7). When the oligonucleotide with the –198 C→T mutation was used as probe, the same shift pattern was observed and all the bands could be competed away by the γCACCC box (data not shown). These results suggest that the –198 HPFH mutation creates a motif that behaves like a novel γCACCC box.
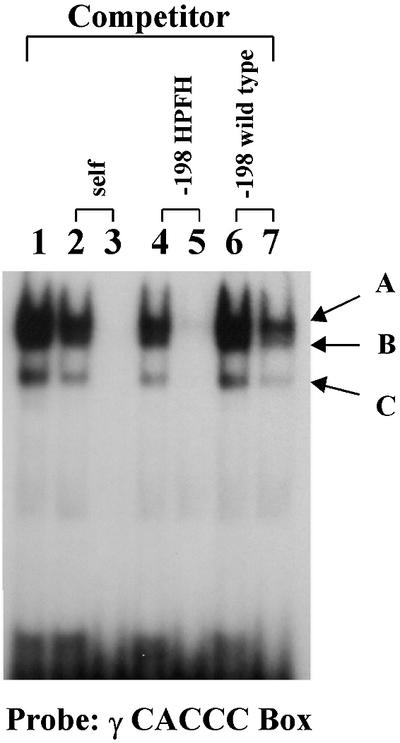
Fig. 2. Proteins bound to the –198 C→T HPFH region are related to those bound to the γ gene CACCC box. A 32P-labeled oligonucleotide encompassing the γCACCC box was used in the electrophoretic mobility shift assay. Nuclear extracts were prepared from MEL cells. Lane 1, the probe without competitor; lanes 2 and 3, competed with the cold probe; lanes 4 and 5, competed with an oligonucleotide corresponding to the sequence –185 to –210 of the γ gene promoter having a T→C mutation at position –198γ; lanes 6 and 7, competed with an oligonucleotide having the wild-type –185 to –210 sequence of the γ gene promoter. The concentration of the cold competitor was 30-fold higher than that of the hot probe in lanes 2, 4 and 6, and 50-fold higher in lanes 3, 5 and 7.
The T→A –175 HPFH mutation fails to compensate for the γCACCC box deletions
A T→A mutation at position –175 of the γ-globin gene is associated with a phenotype of HPFH characterized by levels of HbF ranging from 20 to 40% in heterozygous carriers (Ottolenghi et al., 1988; Surrey et al., 1988; Stoming et al., 1989). To test whether this mutation up-regulates the γ gene in transgenic mice, the –175 T→A transversion was introduced into the µLCR –382Aγ construct. Five transgenic lines carrying correct copies of the construct were established and developmental studies were carried out. Quantitative results are summarized in Table II; RNase protections are shown in Figure 3. There was a large variation in γ mRNA levels among the –175 HPFH lines and, in one line (line J), γ mRNA levels were very low in all embryonic tissues. Except for this line, γ mRNA was increased in the embryonic tissues and in all the definitive erythroid tissues studied (Table II). Thus, compared with the µLCR –382Aγ transgenics, the mice carrying the µLCR –382Aγ construct with the –175 T→A mutation had from 1.5- to 2.0-fold higher levels of γ mRNA in embryonic cells, 2- to 3-fold higher in fetal erythroid cells, and 1.3-fold higher in adult cells. Collectively, these data suggest that the –175 T→A mutation increases γ gene promoter strength in vivo. We next examined whether the –175 HPFH mutation, like the –198 HPFH mutation, could reinstate γ gene expression in adult mice with a deleted γCACCC box. For this purpose, we deleted the CACCC box in the context of the –175 HPFH promoter and produced the construct µLCR –382Aγ ΔCACCC (–175 HPFH). Quantitative data in five lines are given in Table II and RNase protection profiles in Figure 3. As shown in the table, there is essentially no difference in γ mRNA levels between the µLCR –382Aγ ΔCACCC mice and the µLCR –382Aγ ΔCACCC mice carrying the –175 T→A mutation, indicating that this HPFH mutation cannot compensate for the deleted γCACCC box.
Table II. γ gene expressiona in the –175 HPFH transgenic mice.
| Construct | Line | Copy no. | Embryonic (primitive) erythropoiesis |
Definitive erythropoiesis |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Day 10b |
Day 12b |
Day 12b |
Day 16b |
Adult |
||||||
| Blood | Yolk sac | Blood | Yolk sac | Liver | Blood | Liver | Blood | |||
| µLCR –382Aγ | F | 3 | 6.0 ± 1.4 | 10.9 ± 2.7 | 13.7 ± 1.5 | 17.6 ± 3.0 | 15.8 ± 2.7 | 17.5 ± 2.8 | 31.7 ± 1.3 | 21.8 ± 4.5 |
| with –175 HPFH | G | 4 | 10.2 ± 2.3 | 21.2 ± 5.0 | 22.6 ± 2.4 | 49.6 ± 14.5 | 46.1 ± 10.8 | 49.8 ± 9.9 | 54.6 ± 10.8 | 25.9 ± 2.2 |
| H | 10 | 1.8 ± 0.7 | 3.2 ± 0.5 | 5.7 ± 2.0 | 8.5 ± 2.7 | 20.0 ± 4.8 | 15.1 ± 6.0 | 24.8 ± 4.2 | 16.9 ± 3.9 | |
| I | 6 | 10.2 ± 3.9 | 15.5 ± 2.6 | 27.9 ± 5.5 | 25.9 ± 3.8 | 58.2 ± 4.8 | 48.7 ± 1.0 | 45.3 ± 5.9 | 25.3 ± 1.1 | |
| J | 8 | 1.2 ± 0.4 | 1.8 ± 0.3 | 2.0 ± 1.4 | 3.9 ± 1.7 | 9.6 ± 2.7 | 5.0 ± 1.1 | 5.5 ± 0.1 | 4.9 ± 2.7 | |
| mean | 5.8 ± 4.4 | 10.5 ± 8.2 | 14.8 ± 10.9 | 21.1 ± 18.0 | 29.9 ± 21.0 | 27.2 ± 20.7 | 32.4 ± 19.0 | 19.1 ± 8.6 | ||
| µLCR –382Aγ ΔCACCC | K | 11 | 7.0 ± 1.2 | 9.3 ± 1.8 | 16.2 ± 3.5 | 23.9 ± 3.1 | 14.0 ± 2.1 | 7.5 ± 2.0 | 7.3 ± 0.6 | 4.7 ± 0.7 |
| with –175 HPFH | L | 2 | 0.44 ± 0.1 | 0.75 ± 0.1 | 0.44 ± 0.1 | 1.1 ± 0.2 | 0.70 ± 0.3 | 0.75 ± 0.2 | 0.61 ± 0.4 | 0.70 ± 0.3 |
| M | 58 | 2.0 ± 0.3 | 3.0 ± 0.3 | 4.9 ± 1.9 | 29.7 ± 8.0 | 9.5 ± 1.6 | 7.4 ± 2.0 | 6.7 ± 0.7 | 2.7 ± 0.8 | |
| N | 58 | 1.5 ± 0.2 | 3.8 ± 0.1 | 3.4 ± 0.2 | 20.2 ± 4.2 | 7.6 ± 0.6 | 2.8 ± 0.1 | 3.9 ± 1.0 | 1.1 ± 0.2 | |
| O | 3 | 1.1 ± 0.2 | 2.3 ± 0.2 | 3.4 ± 1.2 | 5.5 ± 1.8 | 1.1 ± 0.1 | 0.47 ± 0.1 | 0.47 ± 0.1 | 0.22 ± 0.1 | |
| mean | 2.4 ± 2.6 | 3.8 ± 3.3 | 5.7 ± 6.1 | 16.1 ± 12.2 | 6.6 ± 5.9 | 3.8 ± 3.5 | 3.8 ± 3.2 | 1.9 ± 1.8 | ||
| µLCR –382Aγ | mean | 5.7 ± 6.3 | 5.2 ± 3.3 | 10.4 ± 9.5 | 21.0 ± 7.4 | 11.8 ± 2.8 | 13.4 ± 4.7 | 15.7 ± 4.8 | 16.5 ± 4.5 | |
| µLCR –382Aγ ΔCACCC | mean | 4.2 ± 0.6 | 6.1 ± 1.7 | 8.9 ± 4.3 | 13.9 ± 3.9 | 5.2 ± 3.0 | 2.9 ± 1.3 | 2.6 ± 0.9 | 0.9 ± 0.6 | |
aHuman γ mRNA is expressed as a percentage of murine α mRNA per copy. Mean values are shown in bold. Data in µLCR –382Aγ control mice are from Stamatoyannopoulos et al. (1993).
bDays post-coitus.

Fig. 3. γ-globin gene expression in transgenic mice carrying the –175 HPFH mutant. Lines F, G, H, I and J carry the construct µLCR –382Aγ (–175 HPFH). Lines K, L, M, N and O carry the same construct from which the γCACCC box was deleted. Abbreviations are as in Figure 1.
The –117 G→A HPFH mutation can compensate for the absence of the γCACCC box
We next examined whether the –117 G→A HPFH mutation could restore γ gene expression that is abolished by the deletion of the γCACCC box. This mutation results in the phenotype of Greek HPFH, which is characterized by production of 10–20% HbF in adult carriers (Stamatoyannopoulos and Grosveld, 2001). Since a truncation of the Aγ promoter to position –141Aγ destroys the γCACCC box (Stamatoyannopoulos et al., 1993), the –117 G→A HPFH mutant was introduced into the µLCR –141Aγ construct. Results in the four transgenic mouse lines we established are shown in Table III and Figure 4. The –117 G→A HPFH mutation produced a significant increase in γ mRNA levels in the cells of definitive erythropoiesis. Thus, compared with the µLCR –141Aγ mice, the levels of γ mRNA were 2-fold higher in the 12 day fetal liver of the µLCR –141Aγ mice carrying the –117 HPFH mutation, and 10-fold higher in the adult erythroid cells.

Fig. 4. The –117Aγ HPFH up-regulates γ expression in the absence of the γ gene CACCC box. Lines P, Q, R and S carry the construct –141Aγ (–117 HPFH). Abbreviations are as in Figure 1.
Discussion
The initial observations suggesting heterogeneity of the HPFH mutants were made ∼30 years ago when the deletional and non-deletional variants of HPFH were distinguished purely on the basis of biochemical criteria. When the non-deletional HPFHs were analyzed molecularly, a large number of mutants were found to be located at various motifs of the Aγ or Gγ gene promoters. Although several mechanisms have been proposed to explain the effect of these mutations, in vivo evidence for mechanistic heterogeneity is not available. In the present study, we used transgenic mice for testing whether three non-deletional HPFH mutants up-regulate γ-globin gene expression in vivo. Our experimental approach is based on the observation that a truncation of the γ gene promoter to position –382γ produces an HPFH phenotype while a γCACCC box deletion in the context of the –382 truncation results in severe reduction or absence of γ gene expression in the adult. By introducing an HPFH mutation in a γ gene promoter lacking the γ gene CACCC box, we could ascertain whether the HPFH mutation can restore γ gene expression and produce an HPFH phenotype in the adult mice.
The –117 G→A HPFH mutation affects the distal CCAAT box of the Aγ gene and has been the subject of a large number of in vitro and in vivo studies. Several studies have focused on the identification of proteins binding to the CCAAT boxes of the γ-globin genes. Thus, CP-1 (NF-Y), CDP, C/EBP, GATA-1, NF-E3, NF-E4, NF-E6 and an isoform of GATA-2 have been identified as γCCAAT-binding proteins (Chodosh et al., 1988; Superti-Furga et al., 1988; Mantovani et al., 1989; Berry et al., 1992; Ronchi et al., 1996; Partington and Patient, 1998; Zafarana et al., 2000; Zhou et al., 2000). The –117 HPFH mutation results in increased CP-1 and CDP binding (Superti-Furga et al., 1988; Berry et al., 1992), while it decreases GATA-1 and NF-E3 binding (Mantovani et al., 1989; Berry et al., 1992; Ronchi et al., 1996). Two mechanisms have been proposed to explain the HPFH phenotype: the –117Aγ HPFH mutation either increases transcription by increasing promoter strength or it allows γ gene expression in the adult by interfering with γ gene silencing. Studies using transfections of cell lines suggested that the –117Aγ HPFH mutation fails to increase or only slightly increases γ promoter strength (Dixon and Gelinas, 1988; Superti-Furga et al., 1988; Ulrich and Ley, 1990). Studies in transgenic mice carrying either –117Aγ HPFH cosmids or –117Aγ HPFH yeast artificial chromosomes (YACs) have reproduced the phenotype of elevated γ gene expression in adult animals (Berry et al., 1992; Peterson et al., 1995). Results in transgenic mice were interpreted to suggest that the –117Aγ HPFH mutation affects a GATA-1 site implicated in γ gene silencing (Berry et al., 1992). This hypothesis, however, was not supported by later studies, which also excluded an in vivo suppressive role of binding of NF-E3 near the distal CCAAT box (Ronchi et al., 1996). In the system we used, the –117Aγ HPFH mutation up-regulated γ expression in the cells of definitive erythropoiesis and this effect was independent of the γ gene CACCC box. Since the –117 G→A mutation changed the phenotype of a silenced γ promoter (the –141Aγ promoter) to the phenotype of an HPFH promoter, it is unlikely that the mechanism whereby the –117Aγ HPFH increases γ gene expression is inhibition of γ gene silencing. At least in the system we used, the most likely mechanism of action of this HPFH is the increase in γ gene promoter strength.
The –198 T→C mutation produces the phenotype of British variant of HPFH and it is associated with synthesis of 5–10% HbF in heterozygotes. The –198 HPFH mutant increases Sp-1 binding and creates novel binding for another ubiquitous protein (Ronchi et al., 1989; Fischer and Nowock, 1990; Gumucio et al., 1991). The –198 HPFH mutation increases γ gene expression by 4- to 5-fold in stable or transient transfections of erythroid cell lines (Ronchi et al., 1989; Fischer and Nowock, 1990). This enhancement is not observed in non-erythroid cell lines (Ronchi et al., 1989). The results of our study indicate that the –198 T→C HPFH mutation up-regulates γ gene transcription even when the γCACCC box is deleted. The protein-binding experiments (Ronchi et al., 1989; Fischer and Nowock, 1990; Gumucio et al., 1991; this study) suggest that the –198 HPFH mutation produces a CACCC-like motif. The presence of this motif is perhaps responsible for the increase in the strength of the –382 (–198 HPFH) promoter. It is reasonable to assume that the presence of a CACCC box in its natural position stabilizes the interaction between the γ gene promoter and the LCR. When the γCACCC box is deleted, the interaction with the LCR is mediated through the CACCC box produced by the –198 mutation. The same mechanism may account for the HPFH phenotype in humans. Normally, in the adult, the CACCC box may become inactivated, contributing to the down-regulation of γ gene expression, but the –198 T→C mutation produces an alternative CACCC box that allows the interaction of the γ promoter with the LCR, resulting in the phenotype of HPFH.
The investigation of the molecular basis of the –175 T→A HPFH mutation led Orkin and colleagues to discover GATA-1 (Martin et al., 1989). The motif that is disrupted by the –175 HPFH mutant contains two GATA-1 sites overlapping an octamer-binding site. In spite of extensive biochemical investigation, it is still unknown how these motifs are related to the control of γ gene expression and how the –175 T→A mutation produces the phenotype of HPFH. In transient and stable transfection assays, the –175 HPFH mutation augments the strength of the γ promoter ∼4-fold (Lloyd et al., 1989; Martin et al., 1989; Nicolis et al., 1989; Ronchi et al., 1989; Gumucio et al., 1990). The mutation destroys Oct-1 binding, but does not influence GATA-1 binding as judged by gel retardation assays (Martin et al., 1989; Gumucio et al., 1990; Langdon and Kaufman, 1998). A mutation that destroyed the Oct-1 motif did not reproduce the HPFH phenotype, excluding the possibility that Oct-1 acts as a repressor in the γ gene promoter (Martin et al., 1989; Ronchi et al., 1989). Although the –175 HPFH mutation fails to alter GATA-1 binding, a mutation that eliminates the GATA-1 sites diminishes γ gene expression, indicating that GATA-1 is involved in creating the HPFH phenotype (Martin et al., 1989; McDonagh et al., 1991). The –175 HPFH mutation also decreases binding of HMG-1, a ubiquitous chromatin structural protein (Magis and Martin, 1995), but the physiological meaning of this decline is unclear. Our results show that this mutation up-regulates the γ gene promoter in all stages of development. It is likely that the mechanism whereby the mutation produces the HPFH phenotype requires the presence of a γ gene CACCC box. We base this conclusion on the fact that the –175 HPFH mutant fails to increase γ gene expression in the adult cells when the γCACCC box is deleted.
The experimental system we used may prove useful in the analysis of the developmental role of other motifs of the γ gene promoter. Several transcriptional factors have been reported, on the basis of in vitro assays, to interact with motifs of the γ gene promoter (reviewed in Stamatoyannopoulos and Grosveld, 2001). Mutations that disrupt these interactions can be introduced in the –382Aγ and –382Aγ ΔCACCC constructs and their effects analyzed in transgenic mice. It is likely that such studies will provide new insights into the in vivo functional relevance of proteins that bind to regulatory motifs of the γ gene promoter.
Materials and methods
Plasmid constructs
A StuI–EcoRI fragment was released from the construct pµLCRAγ or pµLCRAγ (ΔCACCC), both of which have a –382 truncated promoter. The fragments were recloned in pAlter-1 (Promega, Madison, WI). Oligonucleotide-directed site mutagenesis was performed on the pAlter-1 vector according to the manufacturer’s manual to introduce a T→C point mutation at position –198γ and T→A at position –175γ. The mutations were confirmed by DNA sequencing. The mutated fragment was released from the pAlter-1 vector and placed back in the parental pµLCRAγ construct, generating pµLCR –382Aγ (–175 HPFH), pµLCR –382Aγ (–198 HPFH), pµLCR –382Aγ ΔCACCC (–175 HPFH) and pµLCR –382Aγ ΔCACCC (–198 HPFH). The plasmid containing the Aγ-globin gene with the –117 mutation (a gift of Dr K.Peterson) was subjected to partial digestion with NcoI and recloned in pBluescript (Stratagene, CA). A clone containing the –117 mutation in a –141 truncated Aγ gene was produced and confirmed by DNA sequencing. The µLCR was placed 5′ to the –141Aγ gene to produce µLCR –141Aγ (–117 HPFH).
Transgenic mice and developmental studies
Purified DNA fragments were injected into fertilized mouse eggs (B6/C3F1) and then transferred to pseudopregnant foster mothers (B6/D2F1). Founder animals were identified by slot blotting with an HS3 probe. F1 progeny were obtained by breeding founder animals with non-transgenic mice (B6/D2F1) and were screened for correct integration and for exclusion of mosaicism in the founders. To study the developmental pattern of human γ gene expression, staged pregnancies were interrupted on p.c. days 10, 12 and 16. Samples from blood and yolk sac were collected from day 10 and 12 embryos, which contain mostly the cells from the primitive erythropoiesis; day 12 fetal liver, day 16 fetal liver and adult blood contain mostly the cells from definitive erythropoiesis.
DNA analysis
DNA from fetal brain or carcasses of F2 progeny in each line were isolated by standard procedures. At least three DNA samples were obtained from each line. Individual samples were then digested with a restriction enzyme. A 10 µg aliquot of DNA from each enzyme reaction performed on samples from a given line was loaded onto a 0.8% agarose gel, and DNA fragments were resolved by electrophoresis. Southern blot hybridization was performed with a BamHI–EcoRI probe derived from the large intron of the γ-globin gene. Signals were quantitated on a PhosphorImager. The blot was then striped and rehybridized with a mouse α-globin gene probe and the intensity of the signals in each lane was used for DNA loading correction. Copy numbers were determined by comparing the signal from a given transgenic line with that of human genomic DNA. In cases in which the computed value was not an integer, the copy number was rounded to the nearest integer in standard fashion.
Globin expression analysis
Total RNA was prepared from the tissues containing the primitive erythrocytes (p.c. day 10 and 12 blood and yolk sac) and the tissues containing the definitive erythrocytes (p.c. day 12 and 16 fetal liver and adult blood). RNA samples were isolated separately from three or more transgenic individuals from each time point. The human γ-globin and murine α- and ζ-globin mRNA was detected by RNase protection assay and quantified by a PhosphorImager. To minimize experimental error, samples from individual animals were quantified independently and multiple measurements were performed in RNase protection. Copy number-corrected globin mRNA levels were expressed as human γ mRNA/γ copy number/(murine ζ mRNA/2 + murine α mRNA/4).
Electrophoretic mobility shift assay (EMSA)
MEL nuclear extracts were prepared as described by Andrews and Faller (1991). In detail, ∼3 × 107 logarithmic-phase MEL cells were harvested and washed once with cold phosphate-buffered saline (PBS). The cells were then incubated in 2 ml of cold buffer A [10 mM HEPES–KOH pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT), 0.2 mM phenylmethylsulfonyl fluoride (PMSF)] for 10 min on ice. The suspension was centrifuged at 10 000 g for 10 s. The pellet was resuspended in 500 µl of cold buffer C (20 mM HEPES–KOH pH 7.9, 25% glycerol, 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 0.5 mM DTT, 0.2 mM PMSF) and incubated on ice for 30 min. The crude nuclear extract was obtained after the cellular debris was removed by centrifugation at 12 000 g for 30 min at 4°C. Protein concentration was determined by using the Bio-Rad protein assay kit. The labeled γCACCC box probes, 5′-TGGCTAAACTCCACCCATGGGTTG-3′ (1 × 104 c.p.m.), were incubated with 5 µg of MEL nuclear extracts for 20 min at room temperature, in the binding buffer containing 10 mM Tris–HCl pH 7.5, 50 mM NaCl, 0.5 mM DTT, 10% glycerol, 1 µg of poly(dI–dC), 0.05% NP-40 and competitors of various concentrations. Samples were electrophoresed in a 4.5% polyacrylamide gel in 0.5× Tris-borate/EDTA (TBE) buffer containing 4 mM Mg2+ at 4°C. The competitors used in the assay are oligonucleotides with the wild-type –198 sequence (5′-TTGGGGGCCCCTTCCCCACACTATCT-3′) or with the –198 HPFH mutation (5′-TTGGGGGCCCCTCCCCCACACTATCT-3′).
Acknowledgments
Acknowledgements
We thank Alex Rohde and Hemei Han for excellent technical assistance. This research was supported by NIH grants HL20899 and DK45365.
References
- Anagnou N.P., Perz-Stable,C., Gelinas,R., Costantini,F., Liapaki,K., Costantopoulou,M., Kosteas,M., Moschonas,N.K., and Stamatoyannopoulos,G. (1995) Sequences located 3′ to the breakpoint of the hereditary persistence of hemoglobin-3 deletion exhibit enhancer activity and can modify the developmental expression of the human fetal A γ-globin gene in transgenic mice. J. Biol. Chem., 270, 10256–10263. [DOI] [PubMed] [Google Scholar]
- Andrews N.C. and Faller,DV. (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res., 19, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcasoy M.O., Romana,M., Fabry,M.E., Skarpidi,E., Nagel,R.L. and Forget,B.G. (1997) High levels of human γ-globin gene expression in adult mice carrying a transgene of deletion-type hereditary persistence of fetal hemoglobin. Mol. Cell. Biol., 17, 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M., Grosveld,F. and Dillon,N. (1992) A single point mutation is the cause of the Greek form of hereditary persistence of fetal hemoglobin. Nature, 358, 499–502. [DOI] [PubMed] [Google Scholar]
- Chodosh L.A., Baldwin,A.S., Carthew,R.W. and Sharp,P.A. (1988) Human CCAAT-binding proteins have heterologous subunits. Cell, 53, 11–24. [DOI] [PubMed] [Google Scholar]
- Dixon M.W. and Gelinas,R.E. (1988) A fetal globin gene mutation in Aγ nondeletion hereditary persistence of fetal hemoglobin increases promoter strength in nonerythroid cell. Mol. Cell. Biol., 8, 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold E.A. and Forget,B.G. (1989) The breakpoint of a large deletion causing hereditary persistence of fetal hemoglobin occurs within an erythroid DNA domain remote from the β-globin gene cluster. Blood, 74, 2178–2186. [PubMed] [Google Scholar]
- Fischer K.-D. and Nowock,J. (1990) The T→C substitution at –198 of the Aγ-globin gene associated with the British form of HPFH generates overlapping recognition sites for two DNA-binding proteins. Nucleic Acids Res., 18, 5685–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumucio D.L., Lockwood,W.K., Weber,J.L., Saulino,A.M., Delgrosso,K., Surrey,S., Schwartz,E., Goodman,M. and Collins,F.S. (1990) The –175 T→C mutation increases promoter strength in erythroid cells: correlation with evolutionary conservation of binding sites for two trans-acting factors. Blood, 75, 756–761. [PubMed] [Google Scholar]
- Gumucio D.L., Rood,K.L., Blanchard-McQuate,K.L., Gray,T.A., Saulino,A. and Collins,F.S. (1991) Interaction of Sp1 with the human γ globin promoter: binding and transactivation of normal and mutant promoters. Blood, 78, 1853–1863. [PubMed] [Google Scholar]
- Langdon S.D. and Kaufman,R.E. (1998) γ-globin gene promoter elements required for interaction with globin ehhancers. Blood, 91, 309–318. [PubMed] [Google Scholar]
- Lloyd J.A., Lee,R.F. and Lingrel,J.B. (1989) Mutation in two regions upstream of the Aγ globin gene canonical promoter affect gene expression. Nucleic Acids Res., 17, 4339–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magis W. and Martin,D.I.K. (1995) HMG-1 binds to GATA motifs: implications for an HPFH syndrome. Biochem. Biophys. Res. Commun., 214, 927–933. [DOI] [PubMed] [Google Scholar]
- Mantovani R., Superti-Furga,G., Gilman,J. and Ottolenghi,S. (1989) The deletion of the distal CCAAT box region of the Aγ-globin gene in black HPFH abolishes the binding of the erythroid specific protein NFE3 and of the CCAAT displacement protein. Nucleic Acids Res., 17, 6681–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.I.K., Tsai,S.-F. and Orkin,S.H. (1989) Increased γ-globin expression in a nondeletion HPFH mediated by an erythroid-specific DNA binding factor. Nature, 338, 435–438. [DOI] [PubMed] [Google Scholar]
- McDonagh K.T., Lin,H.J., Lowrey,C.H., Bodine,D.M. and Nienhuis,A.W. (1991) The upstream region of the human γ-globin gene promoter. J. Biol. Chem., 266, 11965–11974. [PubMed] [Google Scholar]
- Nicolis S., Ronchi,A., Malgaretti,N., Mantovani,R., Giglioni,B. and Ottolenghi,S. (1989) Increased erythroid-specific expression of a mutated HPFH γ-globin promoter requires the erythroid factor NFE-1. Nucleic Acids Res., 17, 5509–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolenghi S. et al. (1988) Sardinian Gγ-HPFH: a T→C substitution in a conserved ‘octamer’ sequence in the Gγ-globin promoter. Blood, 71, 815–817. [PubMed] [Google Scholar]
- Partington G.A. and Patient,R.K. (1998) Factor binding to the human γ-globin gene distal CCAAT site: candidates for repression of the normal gene or activation of HPFH mutations. Br. J. Haematol., 102, 940–951. [DOI] [PubMed] [Google Scholar]
- Peterson K.R., Li,Q., Clegg,C.H., Furukawa,T, Navas,P.A., Norton,E.J., Kimbrough,T.G. and Stamatoyannopoulos,G. (1995) Use of yeast artificial chromosomes (YACs) in studies of mammalian development: production of β-globin locus YAC mice carrying human globin developmental mutants. Proc. Natl Acad. Sci. USA, 92, 5655–5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi A., Nicolis,S., Santoro,C. and Ottolenghi,S. (1989) Increased Sp1 binding mediates erythroid-specific overexpression of a mutated (HPFH) γ-globin promoter. Nucleic Acids Res., 17, 10231–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi A., Berry,M., Raguz,S., Iman,A., Yannoutsos,N., Ottolenghi,S., Grosveld,F. and Dillon,N. (1996) Role of the duplicated CCAAT box region in γ-globin gene regulation and hereditary persistence of fetal hemoglobin. EMBO J., 15, 143–149. [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos G. and Grosveld,F. (2001) Hemoglobin switching. In Stamatoyannopoulos,G., Majerus,P., Perlmutter,R. and Varmus,H. (eds), The Molecular Basis of Blood Diseases. W.B.Saunders, Philadelphia, PA, pp. 135–182.
- Stamatoyannopoulos G., Josephson,B., Zhang,J.-W. and Li,Q. (1993) Developmental regulation of human γ-globin genes in transgenic mice. Mol. Cell. Biol., 13, 7636–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoming T.A., Stoming,G.S., Harris,H.F., Lanclos,K.D., Fei,Y.J., Allay,C., Kutlar,F. and Huisman,T.H. (1989) An Aγ type of nondeletional hereditary persistence of fetal hemoglobin with a T→C mutation at position –175 to the cap site of the Aγ globin gene. Blood, 73, 329–333. [PubMed] [Google Scholar]
- Superti-Furga G., Barberis,A., Schaffner,G. and Busslinger,M. (1988) The –117 mutation in Greek HPFH affects the binding of three nuclear factors to the CCAAT region of the γ-globin gene. EMBO J., 7, 3099–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surrey S., Delgrosso,K., Malladi,P. and Schwartz,E. (1988) A single base change at position –175 in the 5′-flanking region of the Gγ-globin gene from a black with Gγβ+-HPFH. Blood, 71, 807–810. [PubMed] [Google Scholar]
- Tate V.E., Wood,W.G. and Weatherall,D.J. (1986) The British form of hereditary persistence of fetal hemoglobin results from a single base pair mutation adjacent to an S1 hypersensitive site 5′ to the Aγ globin gene. Blood, 68, 1389–1393. [PubMed] [Google Scholar]
- Ulrich M.J. and Ley,T.J. (1990) Function of normal and mutated γ-globin gene promoters in electroporated K562 erythroleukemia cells. Blood, 75, 990–999. [PubMed] [Google Scholar]
- Zafarana G., Rottier,R., Grosveld,F. and Philipsen,S. (2000) Erythroid overexpression of C/EBPγ in transgenic mice affects γ-globin expression and fetal liver erythropoiesis. EMBO J., 19, 5856–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Clouston,D.R., Wang,X., Cerruti,L., Cunningham,J.M. and Jane,S.M. (2000) Induction of human fetal globin gene expression by a novel erythroid factor, NF-E4. Mol. Cell. Biol., 20, 7662–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]