

Abstract
Smad transcription factors mediate the actions of transforming growth factor-β (TGF-β) cytokines during development and tissue homeostasis. TGF-β receptor-activated Smad2 regulates gene expression by associating with transcriptional co-activators or co-repressors. The Smad co-repressor TGIF competes with the co-activator p300 for Smad2 association, such that TGIF abundance helps determine the outcome of a TGF-β response. Small alterations in the physiological levels of TGIF can have profound effects on human development, as shown by the devastating brain and craniofacial developmental defects in heterozygotes carrying a hypomorphic TGIF mutant allele. Here we show that TGIF levels modulate sensitivity to TGF-β-mediated growth inhibition, that TGIF is a short-lived protein and that epidermal growth factor (EGF) signaling via the Ras–Mek pathway causes the phosphorylation of TGIF at two Erk MAP kinase sites, leading to TGIF stabilization and favoring the formation of Smad2–TGIF co-repressor complexes in response to TGF-β. These results identify the first mechanism for regulating TGIF levels and suggest a potential link for Smad and Ras pathway convergence at the transcriptional level.
Keywords: EGF/Ras/Smad/TGF-β/TGIF
Introduction
The Smad proteins are central mediators of transforming growth factor-β (TGF-β) cytokine signals. Following receptor-mediated phosphorylation, Smad proteins translocate to the nucleus (Heldin et al., 1997; Massagué et al., 2000). Once in the nucleus, activated Smad complexes are recruited to DNA, in part via interactions with DNA-binding proteins. A DNA-bound Smad complex activates transcription, and it has been shown that interactions of Smad proteins with general co-activators, such as p300/CBP, play an important role in Smad transcriptional activation (Derynck et al., 1998; Massagué and Wotton, 2000). Within the nucleus, the transcriptional activity of nuclear Smad complexes can be modulated by TGIF (Wotton et al., 1999a,b), c-Ski and SnoN (Akiyoshi et al., 1999; Luo et al., 1999; Stroschein et al., 1999; Sun et al., 1999a,b), which act as Smad transcriptional co-repressors. These proteins bring both histone deacetylase (HDAC)-dependent and -independent transcriptional repression functions to the Smad complex (Luo et al., 1999; Wotton et al., 1999a,b).
The levels of TGIF, c-Ski and SnoN present within a cell set the maximal level to which TGF-β signaling can activate transcription. Increased co-repressor levels better repress Smad-activated transcription (Akiyoshi et al., 1999; Luo et al., 1999; Stroschein et al., 1999; Sun et al., 1999a,b; Wotton et al., 1999a), and decreased expression results in higher TGF-β-responsive transcriptional activation (Wotton et al., 1999a). SnoN can be upregulated by TGF-β itself, potentially establishing a negative feedback mechanism (Stroschein et al., 1999). TGIF is implicated as a Smad2 co-repressor in the TGF-β/Nodal/Activin pathways (Wotton et al., 1999a; Gripp et al., 2000). TGIF haploinsufficiency in humans causes holoprosencephaly (HPE), a genetic disorder affecting brain and craniofacial development (Gripp et al., 2000). TGIF mutations associated with HPE generally involve loss of a single copy of the TGIF gene or point mutations within one copy, resulting in only a partial loss of function (Gripp et al., 2000). Thus, a slight reduction in TGIF levels can have severe developmental consequences. However, to date, the normal cellular regulation of TGIF levels or activity remains unclear.
Also unclear is how TGIF levels affect the growth-inhibitory activity of TGF-β. TGF-β stimulation of cells that are sensitive to growth inhibition by TGF-β leads to a rapid increase in p15Ink4b expression (Hannon and Beach, 1994), which leads to the conversion of active p27Kip1–cyclin D–cdk4 complexes into inactive p15Ink4b–cdk4/6 (Reynisdóttir et al., 1995; Reynisdóttir and Massagué, 1997). Displaced from these kinases, p27Kip1 then associates with and inactivates cyclin E– cdk2 complexes (Koff et al., 1993; Polyak et al., 1994a,b; Reynisdóttir et al., 1995). As a Smad transcriptional co-repressor, TGIF could thus modulate gene response(s) critical for TGF-β-mediated growth inhibition.
In order to address this issue, TGIF was overexpressed and found to attenuate both TGF-β-mediated growth inhibition and p15Ink4b mRNA upregulation. These observations, together with the importance of TGIF levels during ventral forebrain induction in human, prompted us to search for stimuli that regulate the levels of TGIF in the cell. We provide evidence that the mitogen epidermal growth factor (EGF), via the Ras–Mek–Erk pathway, phosphorylates and stabilizes TGIF, enhancing the in vivo steady-state levels of TGIF and hence TGF-β-induced Smad–TGIF co-repressor complexes. These observations identify the Ras pathway as the first known regulator of TGIF and suggest a potential link between the Ras and Smad pathways at the transcriptional level.
Results
Overexpression of TGIF attenuates TGF-β-induced growth inhibition and p15Ink4b upregulation
One function of TGF-β critical in developmental regulation and tumor suppression is the inhibition of cellular proliferation (Massagué et al., 2000). The demonstrated role of TGIF as a Smad transcriptional co-repressor suggests that its physiological levels might differentially modulate the sensitivity of susceptible cells to TGF-β-induced growth inhibition by altering anti-proliferative gene responses. To test this hypothesis, stably transfected HaCaT human keratinocyte derivatives were generated expressing a human TGIF cDNA under negative control of the tetracycline trans activator (Gossen and Bujard, 1992). The immortalized but non-transformed human keratinocyte cell line HaCaT responds to TGF-β with a rapid increase in the expression of the cdk4 inhibitor p15Ink4b and cell cycle arrest (Hannon and Beach, 1994).
Two independent HaCaT derivatives (TT5 and TT6) that expressed exogenous TGIF upon induction in tetracycline-free medium (Figure 1A) were isolated to examine the effect of varying the TGIF protein levels on TGF-β induction of growth arrest and p15Ink4b gene expression. The results (Figure 1B) from both TT5 and TT6 consistently show that enhancing TGIF over the endogenous level attenuated the extent of TGF-β- mediated growth inhibition, as determined by iodinated deoxyuridine incorporation assays. TGIF overexpression not only conferred an increased resistance to TGF-β-mediated growth inhibition but also resulted in a reduction of TGF-β-induced p15Ink4b mRNA expression (Figure 1C). These findings are consistent with a role for the transcriptional repressor TGIF in antagonizing the TGF-β- dependent anti-proliferative response and underscore the importance of the in vivo regulation of TGIF levels.

Fig. 1. Stable overexpression of TGIF inhibits TGF-β-induced growth inhibition and p15Ink4b upregulation. (A) Two stable Tet-TGIF HaCaT clones (TT5 and TT6) were induced to express human TGIF in the absence of tetracycline for 40 h. (B) Stable TGIF overexpression in TT5 (squares) and TT6 (circles) attenuates TGF-β-induced growth inhibition. Triplicate cultures were induced (tetracycline absent) or not induced (tetracycline present) to expressed TGIF for 24 h prior to treatment with the indicated concentrations of TGF-β for 18 h. [125I]deoxyuridine was added to the medium during the last 2 h of incubation. Percent growth inhibition reflects the percent reduction in [125I]deoxyuridine incorporation by cells treated with TGF-β as compared with those left untreated. (C) Stable TGIF overexpression in TT5 and TT6 inhibits TGF-β-induced upregulation of p15Ink4b mRNA levels. TT5 and TT6 were cultured in the absence of tetracycline for 40 h prior to TGF-β (200 pM) treatment (2 h) followed by RNA harvest for northern analysis. The p15Ink4b mRNA signals were quantified and normalized against the β-actin values.
EGF-activated Ras–Mek pathway enhances TGIF protein level
When TGIF was visualized by western immunoblotting, we noticed that both the endogenous and exogenously expressed protein generally migrate as a doublet of ∼30–35 kDa (Wotton et al., 1999a; Figure 2A). We were intrigued by the fact that the relative proportions of the two TGIF bands vary depending on the cell type and culture conditions (Figure 2A). Since cells cultured in the presence of serum-containing media have a higher proportion of the upper band (Figure 2A), we wanted to determine whether components of the signaling pathways activated by serum growth factors could mimic this effect. As shown in Figure 2B, HA epitope-tagged TGIF from serum-starved cells migrated predominantly as the lower form. However, treatment with EGF, an activator of the Ras pathway (Marshall, 1995) and an antagonist of the TGF-β growth-inhibitory response (Schwarz et al., 1988; Howe et al., 1993), resulted in an accumulation of the upper form and, to a lesser extent, an intermediate form of TGIF. A similar effect was observed on co-expression of vectors encoding constitutively active forms of Ras (RasV12) or its downstream mediator of Erk MAP kinase activation, Mek1 (Figure 2B). Co-expression of a vector encoding a dominant-negative form of Ras (RasN17) and addition of an inhibitor of Mek1 activation prior to EGF treatment prevented the accumulation of the upper form of TGIF, showing that this EGF effect is dependent on the activities of Ras and Mek1, respectively (Figure 2B).

Fig. 2. Effects of the Ras–Mek pathway on TGIF. (A) TGIF exists as distinct electrophoretic mobility forms. Cell extracts were subjected to anti-TGIF western immunoblotting. HepG2, human hepatoma cells; A549, human lung carcinoma cells; SW480, human colon carcinoma cells; K562, human erythroleukemic cells; HeLa, human cervical carcinoma cells; 293T, human embryonic kidney cells. A549 cells were compared after culture in the presence and absence of serum. (B) EGF stimulation results in an accumulation of the upper form of TGIF and this effect is mediated by activation of the Ras–Mek1 pathway. Vector encoding HA-tagged human TGIF was cotransfected into COS-1 cells with constructs encoding H-RasV12, constitutively activated Mek1 or H-RasN17. Twenty-four hours after transfection, the cells were either serum starved or treated with EGF for 18 h. An inhibitor of Mek activation (PD98058) was added 1 h prior to EGF addition. Cell lysates were then subjected to western immunoblotting with anti-HA antibodies. (C) The mouse mammary epithelial cell line EpH4 and its v-Ha-Ras-transformed derivative were serum starved for 18 h prior to cell lysis for western immunoblotting analysis using antibodies against TGIF. (D) In the human keratinocyte HaCaT cells, the levels of Mek-phosphorylated (active) Erk1 and Erk2 correlate with accumulation of the upper form of TGIF. HaCaT cells were serum starved for 24 h and treated either with EGF for 1 h or PD98058 for 6 h prior to cell lysis. The resulting lysates were analyzed by western immunoblotting with either antibodies against phosphorylated Erk1/2 or TGIF.
The abundance of the slower migrating form of endogenous TGIF was also enriched with oncogenic Ras stimulation, as shown by the relative ratio of the two TGIF forms in the mouse mammary epithelial cell line EpH4 and its v-Ha-Ras-transformed derivative EpRas (Oft et al., 1996; Figure 2C). Additionally, since our Tet-TGIF lines were derived from HaCaT keratinocytes, we examined the TGIF levels in these cells. EGF stimulation for 1 h following prolonged serum starvation leads to an increase in the endogenous level of TGIF (Figure 2D). Treatment with an inhibitor of Mek1 activation following serum starvation further reduced the residual levels of phosphorylated Erk1/2 and the levels of TGIF (Figure 2D). Thus, factors such as EGF induce an accumulation of the slower migrating form of TGIF via the Ras–Mek pathway, resulting in a net increase in TGIF protein level.
EGF treatment rapidly enhances Smad–TGIF–HDAC1 complex assembly
We have previously shown that TGIF is able to recruit HDACs to TGF-β-activated Smad proteins, that HDAC1, TGIF and Smad associate in the same complex, and that the recruitment of TGIF and HDAC activity plays a role in repressing Smad-activated transcription (Wotton et al., 1999a). To determine whether the EGF-induced enhancement of TGIF protein level could affect the formation of Smad complexes, COS-1 cells were transfected with Flag-tagged HDAC1, HA-tagged TGIF and Smad2. Following serum withdrawal, cells were treated with saturating TGF-β (200 pM) for 1 h in the presence or absence of EGF. At low concentrations of TGF-β, EGF stimulation has been shown to inhibit TGF-β-induced Smad2/3 nuclear accumulation (Kretzschmar et al., 1999). Thus, a saturating TGF-β concentration was chosen, since EGF does not attenuate Smad2 nuclear accumulation at high TGF-β concentrations (Kretzschmar et al., 1999; Figure 3A). Proteins were then precipitated via the Flag epitope present on HDAC1 and complexes analyzed for the presence of TGIF and Smad2. As shown in Figure 3B, in cells treated with EGF, HDAC1-associated TGIF is clearly detectable. Without EGF treatment, the level of TGIF present in the cell is lower and consequently less HDAC1–TGIF complexes are present. The addition of TGF-β resulted in an increase in the amount of Smad2 associated with HDAC1, with TGIF bridging this interaction (Wotton et al., 1999a; Figure 3B). Importantly, this TGF-β-induced association of HDAC1 with Smad2 was further enhanced when cells were treated with EGF in addition to TGF-β (Figure 3B). Therefore, it appears that EGF signaling results in a rapid increase in TGIF protein level, which leads to an enhancement of the recruitment of HDAC to TGF-β-activated Smad complexes.

Fig. 3. EGF treatment rapidly enhances Smad–TGIF–HDAC1 complex formation. (A) EGF does not attenuate Smad2/3 nuclear accumulation induced by a high concentration of TGF-β (200 pM). HaCaT and COS-1 cells were treated with EGF for 1 h before fixation and/or TGF-β for 30 min before fixation, and the endogenous Smad2/3 were visualized by immunofluorescence. (B) COS-1 cells were transfected with Smad2, HA-tagged TGIF and Flag-tagged HDAC1. Following 6 h of serum withdrawal, cells were treated for 1 h with either EGF alone or TGF-β (200 pM) in the presence or absence of EGF co-treatment. Complexes were precipitated on Flag–agarose and analyzed by western blotting for the presence of TGIF and Smad2. A portion of the lysate was analyzed for total expression levels of Smad2, HA-tagged TGIF and Flag-tagged HDAC1.
EGF stimulation rapidly leads to TGIF phosphorylation
The switch to the upper form of TGIF is a rapid event and was clearly detectable after 10 min of treatment with EGF, whether looking at endogenous (Figure 4A) or exogenous (Figure 4B) TGIF. In addition, treatment with EGF rapidly (within 10 min) led to an overall increase in TGIF protein levels (Figure 4A and B), strongly suggesting that this EGF-upregulated TGIF protein level occurs post-transcriptionally. Early in the time course, this increase affected predominantly the upper form of TGIF, suggesting that this modified form of TGIF may be more stable. Late in the time course, an increase in the lower form of TGIF also became evident.

Fig. 4. EGF treatment results in rapid phosphorylation of TGIF. EGF stimulation rapidly leads to an accumulation of the upper form of both endogenous (A) and exogenous (B) TGIF. The lower form also accumulates with longer EGF treatment, resulting in a clear increase in the total steady-state amount of TGIF. Untransfected A549 cells and COS-1 and L-17 cells transfected with vectors encoding HA-tagged human TGIF were serum starved for 18 h followed by EGF treatment for the indicated durations. The lysates were analyzed by western immunoblotting with antibodies against either TGIF (A) or the HA epitope (B). The expression level of HA-TGIF in L-17 cells is considerably lower than that in COS-1 cells. (C) The lower form of TGIF gives rise to the upper form in response to EGF stimulation. In COS-1 cells, transfected HA-tagged human TGIF was radiolabeled with [35S]methionine/cysteine in cells that had been serum starved for 18 h. The electrophoretic mobility of the radiolabeled lower form of TGIF was then followed as a function of time with EGF stimulation. (D) The upper form of TGIF is an EGF-inducible hyperphosphorylated species. Following transfection of COS-1 cells with vector encoding HA-tagged human TGIF and serum starvation for 18 h, EGF was added for 1 h, as indicated, prior to cell lysis. Phosphatase treatment of the lysates converted the upper form of TGIF into the lower form, and co-treatment with the phosphatase inhibitor sodium vanadate prevented this effect.
To demonstrate that the upper form of TGIF is derived from the lower form, cells were transfected with HA-TGIF, serum starved and pulse labeled with radiolabeled methionine and cysteine. Labeled amino acids were removed, and the cells were incubated in the presence of EGF. As shown in Figure 4C, within 10–20 min of EGF addition, a significant proportion of the labeled lower form of TGIF had shifted to the upper form, demonstrating that the EGF-induced upper form of TGIF is derived from the lower form. Furthermore, the EGF-induced transition from the lower to the upper form of TGIF was lost when cell extracts were incubated with alkaline phosphatase, whereas in the presence of the phosphatase inhibitor sodium vanadate, phosphatase treatment had no effect on the mobility of TGIF (Figure 4D). Thus, the EGF-induced change in the electrophoretic mobility of TGIF is due to phosphorylation.
Identification of the MAP kinase phosphorylation sites in TGIF
Of the two pairs of potential Erk MAP kinase phosphorylation sites (PXS/TP motifs) present within TGIF, the more C-terminal sites appeared to be responsible for the mobility shift based on deletion analysis (Figure 5A). To investigate this possibility, we created a series of hydroxyl amino acid mutant forms of TGIF (Figure 5B). Cells were transfected with wild-type or mutant TGIF and, following serum starvation, treated briefly with EGF in the presence of radiolabeled phosphate. As shown in Figure 5C, in the absence of EGF, a small amount of phosphate is present in both forms of TGIF. However, on addition of EGF, a clear increase in the amount of phosphorylated upper form of TGIF was observed (Figure 5C, upper panel). This effect coincided with an accumulation of the upper form of TGIF (Figure 5C, lower panel). Comparison of the level of 32P labeling and protein abundance (Figure 5C, upper versus lower panels) showed that the slower migrating form of TGIF was more highly phosphorylated than the faster form. Mutation of the central pair of sites slightly but reproducibly reduced the EGF-stimulated increase in phosphorylation but did not alter the EGF-induced mobility shift (Figure 5C, lanes 5 and 6). In contrast, mutating the C-terminal MAP kinase sites (T235 and T239) essentially abolished the EGF-stimulated increase in phosphorylation and prevented the EGF-induced mobility shift (Figure 5C, lanes 7 and 8). When one of the two C-terminal sites was mutated, the other was phosphorylated only to a limited degree in response to EGF (Figure 5C, lanes 9–12). The full mobility shift appears to require phosphorylation of these two sites, although a shift to an intermediate position was observed in the T235V mutant (Figure 5C, lanes 9 and 10).

Fig. 5. Identification of the MAP kinase phosphorylation sites in TGIF. (A) Deletion of the C-terminal pair of potential Erk MAP kinase sites correlates with loss of the upper form of TGIF. Lysates from COS-1 cells transfected with HA-tagged deletion constructs of TGIF were subjected to western immunoblotting with anti-HA antibodies. (B) Creation of single and combination point mutations within both the central and C-terminal pairs of potential MAP kinase sites. (C) EGF-induced phosphorylation and accumulation of the upper form of TGIF are dependent on the presence of the C-terminal pair of MAP kinase sites (top). Twenty-four hours after transfection with the indicated HA-tagged TGIF constructs, L-17 cells were serum withdrawn for 18 h, incubated in phosphate-free media for 1 h, and pulsed with [32P]orthophosphate for 2 h. EGF was added as indicated for 30 min prior to cell lysis, and the phosphorylation level of transfected TGIF was determined by anti-HA immunoprecipitation. TGIF expression level was monitored by anti-HA immunoblotting of cell lysates from parallel transfections. Elimination of the C-terminal MAP kinase phosphorylation sites abolishes the EGF-induced shift to the upper form of TGIF and results in higher expression levels independently of EGF stimulation (bottom).
Phosphorylation-induced stabilization of the TGIF protein
Incubation with EGF rapidly results in a clear increase in the level of TGIF protein, with much of this change being due to an accumulation of the hyperphosphorylated form of TGIF (Figure 4A and B). To determine whether this was due to a phosphorylation-induced increase in the stability of TGIF, we performed an analysis of the half-life of transfected TGIF. As shown in Figure 6A, the half-life of the upper form of TGIF was 1 h, whereas the lower form appeared to be much less stable, with a half-life of <30 min. It should be noted that the metabolic chase in this experiment was carried out in the absence of serum or EGF stimulation. Therefore, the difference in the apparent half-lives of these two forms of TGIF may be underestimated due to conversion of the upper form into the lower form by dephosphorylation. Substitution of threonine with glutamic acid residues within the two C-terminal MAP kinase sites also yielded a more stable form of TGIF, mimicking the phosphorylation-induced stabilization (Figure 6B). Surprisingly, several amino acid substitutions within these sites, including the threonine to valine substitutions that prevented phosphorylation, resulted in a constitutively stable protein (data not shown). These results suggest that recognition of this region by a proteolytic system may be extremely sensitive to structural changes. The degradative system may target only wild-type TGIF devoid of phosphorylation at T235 and T239.
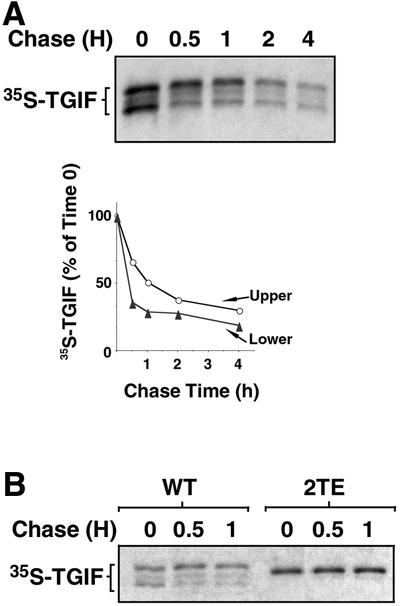
Fig. 6. Phosphorylation-induced stabilization of TGIF. (A) The upper form of TGIF is metabolically more stable than the lower form. COS-1 cells were transfected with HA-tagged TGIF, grown in high (10%) serum, subjected to a pulse metabolic labeling with [35S]methionine/cysteine, and chased for the indicated amounts of time in low (0.2%) serum. HA-TGIF was immunoprecipitated, subjected to gel electrophoresis and the autoradiographic signals were quantified. (B) Substitution of the C-terminal threonines with glutamic acids yields a metabolically more stable TGIF. Analysis of pulse-labeled proteins was carried out as described for (A).
Discussion
The TGF-β/Smad pathway is prototypic of growth-inhibitory signals. The sensitivity of susceptible cells to TGF-β-mediated G1/S growth arrest is, however, subject to modulation by the cellular context. We have shown that the cellular levels of the Smad transcriptional co-repressor TGIF can be one such modulator of TGF-β-induced growth inhibition. This and the extreme sensitivity of human brain development to TGIF levels prompted a search for a regulator of TGIF levels in vivo. We have found that activation of the Ras–Mek–Erk pathway by EGF results in rapid phosphorylation of TGIF. Phosphorylation at the C-terminal T235 and T239 Erk MAP kinase sites increases the half-life of TGIF, thereby enhancing its steady-state levels within the cell. Increased TGIF levels may lead to diminished activation of TGF-β-inducible genes, as shown in the case of the p15Ink4b gene response, or, potentially, to enhanced TGF-β-dependent transcriptional repression (Figure 7).
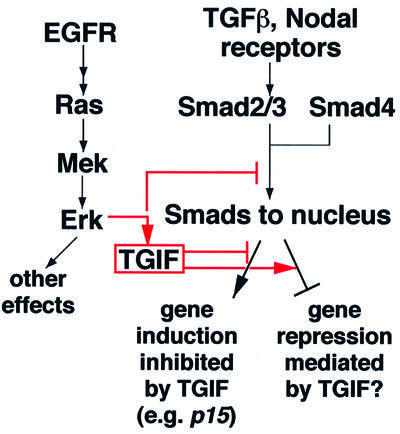
Fig. 7. A partial depiction of the TGF-β and EGF pathways and their cross-talk mechanisms impacting on Smad activities. Activation of the Ras/MAP kinase pathway leads to phosphorylation-induced stabilization of the TGIF protein. Altering the TGIF levels can modulate the TGF-β-induced antiproliferative p15Ink4b gene response and sensitivity of the cell to growth inhibition by TGF-β. These findings thus suggest a novel link for cross-talk between the Ras/MAP kinase and TGF-β pathways at the transcriptional level. TGIF recruitment into Smad complexes decreases transcriptional activation responses and may also act as a mediator of the negative effects of the Smad pathway on gene expression. Arrows denote upregulation; tee bars, downregulation.
TGIF at the intersection of Ras and Smad pathways
It has been shown previously that expression of oncogenic Ha-Ras inhibits G1 cell cycle arrest by saturating concentrations of TGF-β (Schwarz et al., 1988; Howe et al., 1993). In this context, we have demonstrated here that activation of the Mek pathway, whether by EGF stimulation, expression of a constitutively active Ras, or expression of an activated Mek, leads to a rapid increase in the level of the TGIF protein, whereas pharmacological inhibition of activated Mek blocks the EGF-induced increase in TGIF level. This enhancement in TGIF level occurs by accumulation of a phosphatase-sensitive, hyperphosphorylated TGIF form, which has a retarded electrophoretic mobility. The increase in phosphorylation of this upper form of TGIF in response to EGF requires a pair of Erk MAP kinase consensus sites near the C-terminus of TGIF. In addition, this upper TGIF form has a longer metabolic half-life than the lower TGIF form, leading to an overall build-up in the steady-state level of TGIF itself and hence its increased assembly with activated Smad and HDAC, forming co-repressor complexes. Thus, the effect of the Ras–Mek pathway on TGIF protein stability described here suggests a novel mechanism for modulating TGF-β signaling at the transcriptional level.
The interplay between the TGF-β and EGF/Ras signal transduction pathways occurs at other levels as well. These include Ras inhibition of TGF-β receptor expression and of Smad accumulation in the nucleus (Filmus et al., 1992; Zhao and Buick, 1995; Kretzschmar et al., 1999; Massagué and Chen, 2000). EGF stimulation via Ras activation has been shown to diminish nuclear accumulation of TGF-β-activated Smad proteins. However, at high levels of TGF-β signaling, EGF addition or transformation by an oncogenic H-ras allele is unable to prevent Smad entry into the nucleus (Kretzschmar et al., 1999; Figure 3A), even though it can profoundly alter the cellular response to TGF-β (Oft et al., 1996). The subcellular distribution of Smad in the cell is a function of its interactions with protein partners in the cytoplasm and nucleus. Smad proteins have intrinsic nuclear import activity that in the basal state is negated by contacts with SARA (Smad anchor for receptor activation) (Xu et al., 2000). Likewise, overexpression of a nuclear partner of Smad, namely the Smad DNA binding co-factor FAST1, leads to Smad2 nuclear accumulation in the absence of receptor activation (Hoodless et al., 1999). Receptor-mediated Smad phosphorylation diminishes the affinity of Smad for SARA, which results in Smad movement to the nucleus and association with various protein partners (Tsukazaki et al., 1998; Xu et al., 2000). In light of these insights, attenuation of Smad nuclear accumulation by Ras–Mek signaling could result not only from direct effects on Smad nuclear import and/or export machinery, but also from effects of Ras–Mek signaling on Smad interactions with protein partners.
Ras signaling has long been known to act as a modifier of cellular responsiveness to TGF-β. During embryo development, many processes are cooperatively stimulated by TGF-β and Ras signaling (Whitman, 1998). In principle, this cooperativity could be achieved by Ras modulating gene activation or repression by Activin, Nodal and other TGF-β-like signals. Smad complexes activated by these factors can associate with either general co-activators, such as p300/CBP, or co-repressors like TGIF that specifically target nuclear Smad proteins (Massagué and Wotton, 2000). Regulation of co-activator activity by mitogenic signals, such as EGF, may result in general transcriptional upregulation. Increased TGIF activity in response to the same signals provides a mechanism to repress a specific subset of gene responses. Hence, regulation of TGIF levels by Ras signaling allows an effective and selective way to adjust the level of Smad-activated transcription in vivo. TGIF thus provides a potential link within the nucleus between signals that activate the Ras pathway and TGF-β morphogens that exert different effects on gene expression at different levels of signal.
Likewise, during tumorigenesis, transformation by disregulation of Ras or EGFR and related tyrosine kinases in various types of epithelial cells modifies their responsiveness to TGF-β by conferring resistance to growth inhibition by TGF-β, while allowing other responses to TGF-β, including extracellular matrix production, cellular motility and stimulation of angiogenesis (Schwarz et al., 1988; Houck et al., 1989; Valverius et al., 1989; Welch et al., 1990; Filmus et al., 1992; Longstreet et al., 1992; Cui et al., 1996; Yin et al., 1999). In fact, TGF-β collaborates with oncogenic Ras to bring about metastatic and invasive phenotypic alterations in Ras-transformed mammary epithelial cells (Oft et al., 1996, 1998). Thus, oncogenic Ras signaling can attenuate certain TGF-β responses while allowing or even enabling others. Our results suggest that stabilization of TGIF provides a mechanism for the modification of Smad responses by Ras–Mek signaling. In this context, it is noteworthy that a recently identified form of human TGIF, TGIF2, has been found to be amplified and overexpressed in a third of ovarian cancer cell lines (Imoto et al., 2000). TGIF and TGIF2 are highly conserved in the C-terminus containing the EGF-inducible phosphorylation sites.
The critical nature of TGIF levels
Our interest in identifying regulators of TGIF also stemmed from the recent evidence that single point mutations within one copy of the TGIF gene, which result in a partial loss of function, can cause HPE, a human developmental anomaly leading to severe forebrain and craniofacial malformation (Gripp et al., 2000). In the most severe forms, a single brain ventricle is present without evidence of an interhemispheric fissure, and the facial anomalies may include cyclopia (Muenke and Beachy, 2000). In the zebrafish, mutations in the Nodal-related gene cyclops and the Nodal receptor cofactor one-eyed pinhead (oep) can also lead to cyclopia (Feldman et al., 1998; Rebaglianti et al., 1998; Sampath et al., 1998; Gritsman et al., 1999). The phenotypic effects of the cyclops and oep mutations can be rescued by the expression of Smad2 (Gritsman et al., 1999; Dick et al., 2000). Furthermore, in mice doubly heterozygous for null alleles in both Nodal and Smad2, cyclopia was observed in 56% of the embryos (Nomura and Li, 1998).
That haploinsufficiency in human TGIF leads to severe craniofacial defects including cyclopia suggests a potential role for TGIF in modulating Nodal/TGF-β signaling during midline structure development. The expression of TGIF is essentially ubiquitous during early embryonic development (Bertolino et al., 1996), consistent with a role for TGIF in modulating Nodal signaling. It is important to note that HPE is caused by a <50% decrease in the effectiveness of TGIF (Gripp et al., 2000). The fact that deficiencies in both Nodal and TGIF lead to similar phenotypes suggests that TGIF may engage Nodal-activated Smad proteins and mediate Nodal-induced downregulation of gene expression (Figure 7). Alternatively, TGIF might tone down Nodal-induced gene activation such that it confers a narrow window of Nodal/Smad signaling, neither deficient nor excessive, required for correct anterior neural and craniofacial midline structure formation (Figure 7). The low penetrance of the HPE phenotype caused by TGIF mutations in humans suggests the existence of modifiers that may bring the levels of TGIF over the threshold needed for proper midline development. Activators of the Ras–Mek pathway would be candidates for this modifier role. Interestingly, among the known activators of the Ras pathway, members of the fibroblast growth factor (FGF) family participate in the early regional patterning of the developing forebrain (Vaccarino et al., 1999). FGF8, for instance, is expressed in a morphogenetic center during neurogenesis in the ventral region between the telencephalic vesicles, the commissural plate or anterior neural ridge (Crossley and Martin, 1995). The action of FGF8 may be complemented by other FGFs expressed diffusely throughout the dorsal telencephalic region; for instance, FGF2 is required for the proper growth of the telencephalic epithelium (Vaccarino et al., 1999). Control of TGIF by FGF or other activators of the Ras–Mek pathway could thus play a critical role in the outcome of TGF-β/Nodal signal inputs, thereby dramatically altering the developmental program specified by these factors.
Materials and methods
Expression vectors
Human TGIF expression constructs were created within a modified pCMV5 containing an initiation codon together with two copies of an HA epitope. Untagged Smad2 was expressed from within pCMV5 and Myc-Fast2 was from pCS2. Flag-tagged HDAC1 has been described (Yang et al., 1997). TGIF mutations were made by a PCR-based strategy as described previously (Lo et al., 1998). All PCR-generated fragments were subcloned into wild-type TGIF and sequence verified.
Cell culture, transfection and immunoassays
Mink lung epithelial L-17, HaCaT and COS-1 cells were maintained as previously described (Hata et al., 1997; Lo and Massagué, 1999). All serum starvation was performed by incubating the cells in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 0.2% fetal bovine serum (FBS) for 16–24 h prior to cell lysis. For direct immunoblotting, COS-1 and L-17 cells were transfected by the DEAE–dextran method as previously described (Lo et al., 1998) and, as indicated, treated with EGF (10 nM) for 16 h or pretreated with the MEK1 inhibitor PD98059 (New England Biolabs) for 1 h prior to EGF stimulation. To examine the levels of phosphorylated Erk1/2 and TGIF in HaCaT cells, cells were serum starved and, prior to cell lysis, either left untreated, treated with the MEK1 inhibitor PD98059 (New England Biolabs) for 6 h, or treated with EGF (10 nM) for 1 h. The cells were lysed in TNE buffer (10 mM Tris–HCl pH 7.8, 150 mM NaCl, 1 mM EDTA, 1% NP-40) containing protease and phosphatase inhibitors. A fraction of the soluble lysate was then separated by SDS–PAGE and transferred to PVDF membranes (Immobilon-P; Millipore). For in vitro phosphatase treatment, the transfected COS-1 cells were serum starved for 18 h, treated with EGF (10 nM) for 1 h and lysed in a buffer containing 50 mM Tris–HCl, 5 mM dithiothreitol, 2 mM MgCl2, 0.1% Triton X-100. A fraction of the soluble lysate was then mixed with alkaline phosphatase (40 U) (Boehringer Mannheim) in dephosphorylation buffer and the mix incubated at 30°C for 45 min with or without the phosphatase inhibitor sodium vanadate (2 mM). The reactions were stopped by addition of 4× Laemmli buffer. HA-tagged TGIF constructs were detected using anti-HA monoclonal antibody 12CA5 (Boehringer Mannheim), followed by donkey anti-mouse antibody conjugated with horseradish peroxidase (Sigma) and enhanced chemiluminescence (ECL; Amersham). Endogenous TGIF were detected by a rabbit polyclonal anti-TGIF antibody and phosphorylated Erk1/2 by a mouse p-Erk1/2-specific antibody (Santa Cruz), followed by the appropriate species-specific secondary antibody conjugated with horseradish peroxidase (Sigma) and enhanced chemiluminescence (ECL Plus; Amersham).
For co-immunoprecipitation experiments, COS-1 cells were transfected using Lipofectamine (GIBCO-BRL) according to the manufacturer’s instructions. Twenty-four hours post-transfection, cells were serum starved for 6 h, treated with TGF-β (200 pM) for 1 h alone or together with EGF (10 nM) under reduced serum. The cells were then washed, resuspended in LSLD buffer (50 mM HEPES pH 7.4, 50 mM NaCl, 0.1% Tween-20, 10% glycerol) supplemented with protease and phosphatase inhibitors and lysed by sonication. The lysates were then subjected to immunoprecipitation using M2 Flag monoclonal antibody (Kodak). After SDS–PAGE and transfer to PVDF membranes, HA-tagged TGIF was detected with an anti-HA monoclonal antibody and Smad2 detected with a rabbit polyclonal antiserum raised against the corresponding glutathione S-transferase–Smad fusion, together with the appropriate secondary antibody conjugated with horseradish peroxidase and chemiluminescence.
The human TGIF cDNA was cloned into the XbaI site of the pUHD 10-3 vector (Reynisdóttir et al., 1995). The HaCaT-tTA cell line was maintained in DMEM supplemented with 10% FBS plus 0.5 mg/ml G418. HaCaT-tTA cells were transfected with pUHD 10-3 hygromycin using Lipofectamine (GIBCO-BRL) and TGIF-inducible clones were selected as described (Reynisdóttir et al., 1995).
In vivo phosphorylation
For in vivo [32P]orthophosphate labeling, L-17 cells were transiently transfected with the indicated HA-tagged TGIF constructs by the DEAE–dextran method. Twenty-four hours after transfection, cells were serum starved for 18 h. Following pre-incubation with phosphate-free media for 1 h, the cells were then exposed to 1 mCi/ml [32P]phosphate for 2 h at 37°C. As indicated, some cells were treated with 10 nM EGF during the last 30 min. Subsequently, the cells were lysed in TNE buffer containing protease and phosphatase inhibitors, and the lysates subjected to immunoprecipitation with an anti-HA monoclonal antibody. Protein expression of the HA-TGIF constructs was determined by direct western blotting from parallel transfections. Immunoprecipitates were visualized by SDS–PAGE followed by autoradiography.
Pulse–chase experiments
For experiments examining the metabolic stability of HA-TGIF constructs, COS-1 cells were transfected by the DEAE–dextran method, serum starved for 18 h where indicated, pre-incubated for 1 h in methionine/cysteine-deficient medium, and pulsed for 30 min with 200 mCi/ml [35S]methionine/cysteine. The cells were then washed extensively with DMEM and chased for the indicated lengths of time in DMEM/0.2% fetal calf serum with or without EGF (10 nM) as indicated. The cells were then lysed in TNE buffer containing protease inhibitors, and the lysates pre-cleared with protein A–Sepharose beads and subjected to anti-HA immunoprecipitation. The immunoprecipitates were subjected to SDS–PAGE, fixed and enhanced with 1 M sodium salicylate and visualized by autoradiography.
Growth inhibition assay
Triplicate cultures were plated at low confluence (20%) and induced (tetracycline absent in the medium) or not induced (tetracycline present in the medium) to express TGIF for 24 h prior to treatment with the indicated concentrations of TGF-β in 0.2% FBS for 18 h. [125I]deoxyuridine was added to the medium during the last 2 h of incubation. The cells were then fixed, and the acid-insoluble [125I]deoxyuridine was counted to measure the amount of incorporation.
Northern analysis
Total RNA was prepared using an RNAqueous™ kit (Ambion); fractionation, transfer and cross-linking of total RNA were performed using NorthernMax-Gly™ (Ambion). Pre-hybridization was carried out at 42°C for 4–6 h in ULTRAhyb™ (Ambion). Hybridization was then performed overnight at 42°C after addition of denatured human p15Ink4b DNA. The probes were labeled with psoralen–biotin using the BrightStar™ non-isotopic labeling kit (Ambion) and post-hybridization signals detected using BrightStar™ BioDetect™ (Ambion). To control for loading, the northern blots were probed against the β-actin housekeeping gene.
Immunofluorescence
HaCaT and COS-1 cells were first treated as indicated and fixed. Endogenous Smad 2/3 proteins were visualized with affinity-purified anti-Smad2/3 antibodies, biotin-conjugated anti-rabbit antibody and fluorescein isothiocyanate-conjugated streptavidin (Jackson Immuno Research). All slides were counterstained with DAPI to visualize nuclei (data not shown).
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We would like to thank members of the Massagué laboratory for helpful discussion and J.Seoane for the HaCaT-tTA cell line. R.S.L. is grateful for the help of S.H.Roan and would like to dedicate this work to his parents. This work was supported by a National Institutes of Health grant to Memorial Sloan-Kettering Cancer Center and by a National Institutes of Health Medical Scientist Training Program (MSTP) grant to R.S.L. J.M. is an investigator of the Howard Hughes Medical Institute.
References
- Akiyoshi S., Inoue,H., Hanai,J., Kusanagi,K., Nemoto,N., Miyazono,K. and Kawabata,M. (1999) c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with Smads. J. Biol. Chem., 274, 35269–35277. [DOI] [PubMed] [Google Scholar]
- Bertolino E., Wildt,S., Richards,G. and Clerc,R.G. (1996) Expression of a novel murine homeobox gene in the developing cerebellar external granular layer during its proliferation. Dev. Dyn., 205, 410–420. [DOI] [PubMed] [Google Scholar]
- Crossley P.H. and Martin,G.R. (1995) The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development, 121, 439–451. [DOI] [PubMed] [Google Scholar]
- Cui W., Fowlis,D.J., Bryson,S., Duffie,E., Ireland,H., Balmain,A. and Akhurst,R.J. (1996) TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell, 86, 531–542. [DOI] [PubMed] [Google Scholar]
- Derynck R., Zhang,Y. and Feng,X.-H. (1998) Smads: transcriptional activators of TGF-β responses. Cell, 95, 737–740. [DOI] [PubMed] [Google Scholar]
- Dick A., Mayr,T., Bauer,H., Meier,A. and Hammerschmidt,M. (2000) Cloning and characterization of zebrafish smad2, smad3 and smad4. Gene, 246, 69–80. [DOI] [PubMed] [Google Scholar]
- Feldman B., Gates,M.A., Egan,E.S., Dougan,S.T., Rennebeck,G., Sirotkin, H.I., Schier,A.F. and Talbot,W.S. (1998) Zebrafish organizer development and germ-layer formation require nodal-related signals. Nature, 395, 181–185. [DOI] [PubMed] [Google Scholar]
- Filmus J., Zhao,J. and Buick,R.N. (1992) Overexpression of H-ras oncogene induces resistance to the growth-inhibitory action of transforming growth factor β1 (TGF-β1) and alters the number and type of TGF-β1 receptors in rat intestinal epithelial cell clones. Oncogene, 7, 521–526. [PubMed] [Google Scholar]
- Gossen M. and Bujard,H. (1992) Tight control of gene expression in mammalian cells by tetracyclin-responsive promoters. Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp K.W. et al. (2000) Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nature Genet., 25, 205–208. [DOI] [PubMed] [Google Scholar]
- Gritsman K., Zhang,J., Cheng,S., Heckscher,E., Talbot,W.S. and Schier,A.F. (1999) The EGF-CFC protein One-Eyed Pinhead is essential for nodal signaling. Cell, 97, 121–132. [DOI] [PubMed] [Google Scholar]
- Hannon G.J. and Beach,D. (1994) p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature, 371, 257–261. [DOI] [PubMed] [Google Scholar]
- Hata A., Lo,R.S., Wotton,D., Lagna,M. and Massagué,J. (1997) Mutations increasing autoinhibition inactivate the tumour suppressors Smad2 and Smad4. Nature, 388, 82–86. [DOI] [PubMed] [Google Scholar]
- Heldin C.-H., Miyazono,K. and ten Dijke,P. (1997) TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature, 390, 465–471. [DOI] [PubMed] [Google Scholar]
- Hoodless P.A., Tsukazaki,T., Nishimatsu,S., Attisano,L., Wrana,J.L. and Thomsen,G.H. (1999) Dominant-negative Smad2 mutants inhibit activin/Vg1 signaling and disrupt axis formation in Xenopus. Dev. Biol., 207, 364–379. [DOI] [PubMed] [Google Scholar]
- Houck K.A., Michalopoulos,G.K. and Strom,S.C. (1989) Introduction of a Ha-ras oncogene into rat liver epithelial cells and parenchymal hepatocytes confers resisitance to the growth inhibitory effects of TGF-β. Oncogene, 4, 19–25. [PubMed] [Google Scholar]
- Howe P.H., Dobrowolski,S.F., Reddy,K.B. and Stacey,D.W. (1993) Release from G1 growth arrest by transforming growth factor β1 requires cellular ras activity. J. Biol. Chem., 268, 21448–21452. [PubMed] [Google Scholar]
- Imoto I., Pimkhaokham,A., Watanabe,T., Saito-Ohara,F., Soeda,E. and Inazawa,J. (2000) Amplification and overexpression of TGIF2, a novel homeobox gene of the TALE superclass, in ovarian cancer cell lines. Biochem. Biophys. Res. Commun., 276, 264–270. [DOI] [PubMed] [Google Scholar]
- Koff A., Ohtsuki,M., Polyak,K., Roberts,J.M. and Massagué,J. (1993) Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-β. Science, 260, 536–539. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M., Doody,J., Timokhina,I. and Massagué,J. (1999) A mechanism of repression of TGFβ/Smad signaling by ongenic ras. Genes Dev., 13, 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo R.S. and Massagué,J. (1999) Ubiquitin-dependent degradation of TGF-β-activated Smad2. Nature Cell Biol., 1, 472–478. [DOI] [PubMed] [Google Scholar]
- Lo R.S., Chen,Y.G., Shi,Y.G., Pavletich,N. and Massagué,J. (1998) The L3 loop: a structural motif determining specific interactions between SMAD proteins and TGF-β receptors. EMBO J., 17, 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longstreet M., Miller,B. and Howe,P.H. (1992) Loss of transforming growth factor β1 (TGF-β1)-induced growth arrest and p34cdc2 regulation in ras-transfected epithelial cells. Oncogene, 7, 1549–1556. [PubMed] [Google Scholar]
- Luo K., Stroschein,S.L., Wang,W., Chen,D., Martens,E., Zhou,S. and Zhou,Q. (1999) The ski oncoprotein interacts with the smad proteins to repress TGFβ signaling. Genes Dev., 13, 2196–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall C.J. (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell, 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Massagué J. and Chen,Y.-G. (2000) Controlling TGF-β signaling. Genes Dev., 14, 627–644. [PubMed] [Google Scholar]
- Massagué J. and Wotton,D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J., 19, 1745–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J., Blain,S.W. and Lo,R.S. (2000) TGF-β signaling in growth control, cancer and heritable disorders. Cell, 103, 295–309. [DOI] [PubMed] [Google Scholar]
- Muenke M. and Beachy,P.A. (2000) Genetics of ventral forebrain development and holoprosencephaly. Curr. Opin. Genet. Dev., 10, 262–269. [DOI] [PubMed] [Google Scholar]
- Nomura M. and Li,E. (1998) Smad2 role in mesoderm formation, left–right patterning and craniofacial development. Nature, 393, 786–790. [DOI] [PubMed] [Google Scholar]
- Oft M., Peli,J., Rudaz,C., Schwarz,H., Beug,H. and Reichmann,E. (1996) TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev., 10, 2462–2477. [DOI] [PubMed] [Google Scholar]
- Oft M., Heinz,K.-H. and Beug,H. (1998) TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol., 8, 1243–1252. [DOI] [PubMed] [Google Scholar]
- Polyak K., Kato,J.-Y., Solomon,M.J., Sherr,C.J., Massagué,J., Roberts, J.M. and Koff,A. (1994a) p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev., 8, 9–22. [DOI] [PubMed] [Google Scholar]
- Polyak K., Lee,M.-H., Erdjument-Bromage,H., Koff,A., Roberts,J.M., Tempst,P. and Massagué,J. (1994b) Cloning of p27kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell, 78, 59–66. [DOI] [PubMed] [Google Scholar]
- Rebaglianti M.R., Toyama,R., Haffter,P. and Dawid,I. (1998) Cyclops encodes a Nodal-related factor involved in midline signaling. Proc. Natl Acad. Sci. USA, 95, 9932–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisdóttir I. and Massagué,J. (1997) The subcellular location of p15Ink4b and p27Kip1 coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev., 11, 492–503. [DOI] [PubMed] [Google Scholar]
- Reynisdóttir I., Polyak,K., Iavarone,A. and Massagué,J. (1995) Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev., 9, 1831–1845. [DOI] [PubMed] [Google Scholar]
- Sampath K., Rubinstein,A.L., Cheng,A.M.S., Liang,J.O., Fekany,K., Solnica-Krezel,L., Korzh,V., Halpern,M.E. and Wright,C.V.E. (1998) Induction of the zebrafish ventral brain and floorplate requires cyclops/nodal signalling. Nature, 395, 185–189. [DOI] [PubMed] [Google Scholar]
- Schwarz L.C., Gingras,M.C., Goldberg,G., Greenberg,A.H. and Wright, J.A. (1988) Loss of growth factor dependence and conversion of transforming growth factor-β1 inhibition to stimulation in metastatic H-ras-transformed murine fibroblasts. Cancer Res., 48, 6999–7003. [PubMed] [Google Scholar]
- Stroschein S.L., Wang,W., Zhou,S., Zhou,Q. and Luo,K. (1999) Negative feedback regulation by TGF-β signaling by the SnoN oncoprotein. Science, 286, 771–774. [DOI] [PubMed] [Google Scholar]
- Sun Y., Liu,X., Eaton,E., Lane,W.S., Lodish,H.F. and Weinberg,R. (1999a) Interaction of the Ski oncoprotein with Smad3 regulates TGF-β signaling. Mol. Cell, 4, 499–509. [DOI] [PubMed] [Google Scholar]
- Sun Y., Liu,X., Ng-Eaton,E., Lodish,H.F. and Weinberg,R.A. (1999b) SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor-β signaling. Proc. Natl Acad. Sci. USA, 56, 12442–12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukazaki T., Chiang,T.A., Davison,A.F., Attisano,L. and Wrana,J.L. (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell, 95, 779–791. [DOI] [PubMed] [Google Scholar]
- Vaccarino F.M., Schwartz,M.L., Raballo,R., Rhee,J. and Lyn-Cook,R. (1999) Fibroblast growth factor signaling regulates growth and morphogenesis at multiple steps during brain development. Curr. Top. Dev. Biol., 46, 179–200. [DOI] [PubMed] [Google Scholar]
- Valverius E.M., Walker-Jones,D., Bates,S.E., Stampfer,M.R., Clark,R., McCormick,F., Dickson,R.B. and Lippman,M.E. (1989) Production and responsiveness to transforming growth factor-β in normal and oncogene-transformed human mammary epithelial cells. Cancer Res., 49, 6269–6274. [PubMed] [Google Scholar]
- Welch D.R., Fabra,A. and Nakajima,M. (1990) Transforming growth factor β stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc. Natl Acad. Sci. USA, 87, 7678–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman M. (1998) Smads and early developmental signaling by the TGFβ superfamily. Genes Dev., 12, 2445–2462. [DOI] [PubMed] [Google Scholar]
- Wotton D., Lo,R.S., Lee,S. and Massagué,J. (1999a) A Smad transcriptional corepressor. Cell, 97, 29–39. [DOI] [PubMed] [Google Scholar]
- Wotton D., Lo,R.S., Swaby,L.C. and Massagué,J. (1999b) Multiple modes of repression by the Smad transcriptional corepressor TGIF. J. Biol. Chem., 274, 37105–37110. [DOI] [PubMed] [Google Scholar]
- Xu L., Chen,Y.-G. and Massagué,J. (2000) The nuclear import function of Smad2 is masked by SARA and unmasked by TGFβ-dependent phosphorylation. Nature Cell Biol., 2, 559–562. [DOI] [PubMed] [Google Scholar]
- Yang W.-M., Yao,Y.-L., Sun,J.-M., Davie,J.R. and Seto,E. (1997) Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem., 272, 28001–28007. [DOI] [PubMed] [Google Scholar]
- Yin J.J., Selander,K., Chirwing,J.M., Dallas,M., Grubbs,B.G., Wieser,R., Massagué,J., Mundy,G.R. and Guise,T.A. (1999) Blockade of transforming growth factor β signaling inhibits parathyroid hormone-related protein secretion by breast cancer cells and the development of bone metastases. J. Clin. Invest., 103, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. and Buick,R.N. (1995) Regulation of transforming growth factor β receptors in H-ras oncogene-transformed rat intestinal epithelial cells. Cancer Res., 55, 6181–6188. [PubMed] [Google Scholar]