

Abstract
The Escherichia coli periplasmic peptidyl-prolyl isomerase (PPIase) SurA is involved in the maturation of outer membrane porins. SurA consists of a substantial N-terminal region, two iterative parvulin-like domains and a C-terminal tail. Here we show that a variant of SurA lacking both parvulin-like domains exhibits a PPIase-independent chaperone-like activity in vitro and almost completely complements the in vivo function of intact SurA. SurA interacts preferentially (>50-fold) with in vitro synthesized porins over other similarly sized proteins, leading us to suggest that the chaperone-like function of SurA preferentially facilitates maturation of outer membrane proteins.
Keywords: chaperone/outer membrane proteins/periplasm/prolyl isomerase/SurA
Introduction
The periplasm, located between the inner and the outer membrane of the Gram-negative bacterial cell, communicates directly with the external environment through pores in the outer membrane. As small molecules and some substrates exchange freely through these pores (Nikaido, 1994), periplasmic conditions (e.g. pH, redox potential, metals, solvents and toxic molecules) fluctuate with those of the external environment (Wülfing and Plückthun, 1994). Periplasmic and outer membrane proteins (OMPs) are the first targets of potentially deleterious changes that could affect both membrane integrity and periplasmic protein function. Moreover, these proteins also fold in the periplasm, creating high demand in this compartment for protein folding catalysts and chaperones.
Both classes of protein folding catalysts, i.e. the disulfide bond reductases and isomerases (Dsb proteins), which catalyze the formation and rearrangement of disulfide bonds, and the peptidyl-prolyl isomerases (PPIases), which catalyze the cis↔trans conversion of peptidyl proline bonds, are copiously represented in the periplasmic space of Escherichia coli. At least six Dsb proteins have been identified (Bardwell et al., 1991, 1993; Missiakas et al., 1994; Rietsch et al., 1996). In addition, representatives of the three major families of PPIase have been found in the periplasm: one cyclophilin, PpiA or RotA (Liu and Walsh, 1990; Hayano et al., 1991), an FKBP-like protein FkpA (Horne and Young, 1995; Missiakas et al., 1996) and two parvulin homologs PpiD (Dartigalongue and Raina, 1998) and SurA (Lazar and Kolter, 1996; Missiakas et al., 1996; Rouvière and Gross, 1996).
Despite the obvious demand for accessory proteins performing chaperone functions in the periplasm, few periplasmic chaperones have been identified. As the periplasm lacks ATP (Wülfing and Plückthun, 1994), periplasmic chaperones must be mechanistically distinct from their cytoplasmic counterparts, most of which use ATP to drive their remodeling cycle (Bukau and Horwich, 1998). The best studied periplasmic chaperone is PapD, the prototype of pilus-specific chaperones (Hultgren et al., 1993; Soto et al., 1998). The Skp protein has been suggested to affect general periplasmic protein folding (Bothmann and Plückthun, 1998; Hayhurst and Harris, 1999) and to act as a chaperone in the biogenesis of OMPs (Chen and Henning, 1996; Missiakas et al., 1996; de Cock et al., 1999; Schäfer et al., 1999). However, additional periplasmic chaperones that facilitate folding and prevent off-pathway reactions are likely to exist. In particular, chaperones are likely to be important for the quite complex maturation of the trimeric outer membrane porins, which must fold, and in an order still to be determined, assemble and insert into the outer membrane.
One possibility is that chaperone functions in the periplasm are carried out by the chaperone-like activities of the copious protein folding catalysts found in this compartment of the cell. Indeed, the DsbA and DsbC proteins have been reported to possess chaperone-like activities (Jacob-Dubuisson et al., 1994; Zheng et al., 1997; Chen et al., 1999), and the PPIase FkpA has recently been shown to prevent protein aggregation (Ramm and Plückthun, 2000). In this report, we investigate whether the PPIase activity of SurA is the primary biochemical activity required for its biological role in the periplasm.
SurA is involved in the maturation of outer membrane porins (Lazar and Kolter, 1996; Missiakas et al., 1996; Rouvière and Gross, 1996). Lack of SurA interferes with an early folding step in LamB maturation, the conversion of unfolded to folded monomer, and negatively affects expression of the major trimeric porins OmpC, OmpF and LamB, as well as of OmpA (Rouvière and Gross, 1996). surA– cells exhibit the pleiotropic phenotypes characteristic of a defective cell envelope, such as lysis on plates and increased sensitivity to hydrophobic agents (e.g. SDS/EDTA, crystal violet and rifampicin). In addition, SurA influences intercompartmental communication. surA– cells are constitutively induced for the σE-dependent extracytoplasmic stress response (Missiakas et al., 1996; Rouvière and Gross, 1996), one of two signal transduction pathways known to communicate the folding state in the periplasm to the cytoplasm (Mecsas et al., 1993; Connolly et al., 1997; Danese and Silhavy, 1997).
The PPIase activity of SurA resides in only one of two iterative parvulin-like domains in the C-terminal half of the protein. In addition, SurA has a substantial N-terminal domain of previously unknown function, and a C-terminal tail. Here we demonstrate that this N-terminal region coupled to the C-terminal tail (i) has a PPIase-independent chaperone-like activity; (ii) is required for the selective recognition of OMPs by SurA; and (iii) almost completely complements the activities of intact SurA in vivo. We suggest that the chaperone-like activity of SurA may be used preferentially to assist OMP maturation.
Results
Complementation of the surA mutant phenotypes does not require wild-type PPIase activity
The only known enzymatic activity of SurA is a PPIase activity, which is carried in the second parvulin-like domain of the protein. Thus, to dissect the activities of SurA required for function, we first disabled this PPIase activity and then assessed whether cells still exhibited SurA function in vivo. Alanine substitution of three highly conserved amino acid residues (G, H and I) in the parvulin-like domain of Pin1, the human homolog of SurA, has been reported to result in the loss of detectable PPIase activity (Lu et al., 1996); the histidine residue is believed to be part of the active site (Ranganathan et al., 1997). The residues proposed to form the hydrophobic pocket for binding the cyclic side chain of the substrate proline in Pin1 are well conserved in domain II of SurA, and the predicted secondary structure of SurA using PHD PredictProtein and Swiss-Model closely matches that observed for Pin1 (data not shown). Accordingly, we chose to substitute the homologous H and I residues of SurA (His376 and Ile378) with alanine, called here SurA-2A. The presence of glycine often signals either very tight packing of residues or a turn in secondary structure. Therefore, we did not substitute the glycine with alanine as we were concerned that this would destabilize the protein.
The SurA-2A protein properly fractionated into the periplasm (Figure 1), and complemented the two defects exhibited by surA– cells in vivo: induction of the extracytoplasmic stress response mediated by σE (Figure 2) and sensitivity to membrane perturbants (Table I). As expected from restoration of SDS/EDTA resistance, SurA-2A shows normal amounts of OMPs in the outer membrane, rather than the low OMP content of surA– cells (data not shown). As little or no PPIase activity was detected for SurA-2A (Table II), these data suggest that wild-type levels of PPIase activity are not required for function of SurA in vivo.

Fig. 1. Schematic representation (A) and periplasmic location (B) of SurA variants used in this study. (A) The mature proteins without signal peptide sequence (amino acids 1–20 in SurA) are shown and their relative predicted molecular weights (Mr) are given. Numbers refer to amino acid positions in the non-processed peptide sequence. Black box, parvulin-like inactive domain I; white box, parvulin-like active domain II. The N-terminal region of SurA is shaded light gray, the very C-terminus (Ct) of SurA (amino acids 385–428) is shown as a zig-zag line. The positions of alanine substitutions in SurA-2A and SurAI+II-2A are indicated. For all in vivo experiments, these fragments were preceded by the SurA signal peptide sequence so that they would be secreted into the periplasm (see Materials and methods). (B) Western blot detection of SurA variants in periplasmic fractions of surA– cells producing plasmid-encoded: (a) SurA; (b) SurA-2A; (c) SurAΔC368–428; (d) SurAI+II; (e) SurAN+I; (f) SurAN+II; (g) SurAN+II-Ct; (h) SurAN+I-Ct; (i) SurAN-Ct; (j) SurAN; (k) SurAI; and (l) SurAII. Periplasmic fractions of wild-type cells (wt) and surA– cells (surA–) served respectively as positive and negative controls. Extracts from equal amounts of cells were loaded in each lane, and western blot analysis, loading controls and controls for contamination by cytoplasmic fraction (data not shown) were performed as described in Materials and methods. Arrows indicate specific signals. A non-specific cross-reaction with lysozyme (14 kDa signal in the right panel) was observed occasionally in western blots with SurA antibodies. The wild-type sized signal in lane c was not observed in other experiments and results from a spill-over from the neighboring lane.

Fig. 2. EσE activity in surA– cells producing periplasmic SurA variants. EσE activity was assayed by monitoring β-galactosidase activity originating from a single chromosomal copy of the reporter fusion φλ[rpoHP3::lacZ]. The differential rates of β-galactosidase synthesis in surA– cells were normalized to the activity of wild-type cells. Results represent the average of at least two experiments.
Table I. Complementation of the surA OM phenotype.
| Strain background | Relevant genotype | Plasmida | No. of colonies/ml on |
Plating efficiency | |
| |
|
|
LB |
LB + 0.5% SDS/1 mM EDTA |
|
| CAG16037 | wild type | pQE60 | 6.9 × 109 | 6.7 × 109 | 0.97 |
| CAG24029 | surA– | pQE60 | 4.0 × 109 | 6.2 × 104 | 1.6 × 10–5 |
| CAG24029 | surA– | pSurA | 6.9 × 109 | 6.7 × 109 | 0.97 |
| CAG24029 | surA– | pSurAI+II | 3.5 × 109 | 1.1 × 103 | 3.1 × 10–7 |
| CAG24029 | surA– | pSurAI | 3.5 × 109 | 5.9 × 103 | 1.7 × 10–6 |
| CAG24029 | surA– | pSurAII | 3.8 × 109 | 48 | 1.2 × 10–8 |
| CAG24029 | surA– | pSurAN+I | 4.6 × 109 | 6.6 × 104 | 1.4 × 10–5 |
| CAG24029 | surA– | pSurAN+II | 5.1 × 109 | 3.2 × 105 | 6.3 × 10–5 |
| CAG24029 | surA– | pSurAΔC368–428 | 3.8 × 109 | 1.4 × 105 | 3.7 × 10–5 |
|
CAG24029 |
surA– |
pSurA-2A |
6.8 × 109 |
6.1 × 109 |
0.90 |
| |
|
|
LB |
LB + 0.5% SDS/0.5 mM EDTA |
|
| CAG16037 | wild type | pQE60 | 6.1 × 109 | 7.0 × 109 | 1.15 |
| CAG24029 | surA– | pQE60 | 6.1 × 109 | 6.7 × 104 | 1.0 × 10–5 |
| CAG24029 | surA– | pSurAN-Ct | 6.9 × 109 | 7.3 × 109 | 1.1 |
| CAG24029 | surA– | pSurAN+I-Ct | 7.4 × 109 | 8.1 × 109 | 1.1 |
| CAG24029 | surA– | pSurAN+II-Ct | 6.4 × 109 | 7.8 × 109 | 1.2 |
All values are averages from at least three independent experiments.
aAll strains also contain pPLT13.
Table II. Catalysis of prolyl isomerization and proline-limited folding by variants of SurA.
| Protein | Prolyl isomerization kcat/Km (mM–1 s–1) | Proline-limited foldingc kcat/Km (mM–1 s–1) |
|---|---|---|
| SurA | 34a/53b | 35 |
| SurA-2A | not detectablea/not detectableb | not detectable/0.315 |
| SurAI | not detectablea | not detectable |
| SurAII | 22a/58b | 0.37 |
| SurAI+II | 30a | 12 |
| SurAI+II-2A | not detectablea/0.055b | 0.037 |
| SurAN-Ct | not determined | not detectable |
| SurAN+I-Ct | not detectablea | 0.42 |
| SurAN+II-Ct | 28a/36b | 62 |
Protease-coupled assay with the tetrapeptide succinyl-Ala-Leu-Pro-Phe-4-nitroanilide at 4°Ca/15°Cb.
cRefolding of RCM (S54G/P55N)-RNase T1. Average values of two independent protein preparations are shown for SurA, SurA-2A and SurAI+II-2A.
Regions of SurA required for in vivo function
The finding that wild-type levels of PPIase activity were not required for function raised the question of whether the two reiterated parvulin-like domains played any significant role in the function of SurA. One extreme possibility is that the large N-terminal region alone carries the core function of SurA. To elucidate the contribution of each region, we tested whether each region, either separately or in combination, restored SurA function.
The periplasmic location of SurA fragments (Figure 1A) generated by fusion to the surA signal peptide sequence (see Materials and methods) was confirmed by western blot analysis of periplasmic and spheroplast fractions (Figure 1B and data not shown) using polyclonal SurA antibodies. Duplicate membranes probed with α-β-lactamase indicated equal loading in all lanes (data not shown). All SurA fragments visualized were located predominantly in the periplasm and were present at levels equivalent to or greater than that of chromosomally encoded SurA (wild-type lane), permitting direct comparison of the functional capacity of fragments and the full-length protein. (SurAII is somewhat lower in this experiment, but its level is usually equivalent to that of SurA.) The N-terminal domain alone was not visualized on this blot (lanes i and j). Experiments conducted later indicated that this domain does not react with our SurA polyclonal antisera.
Although correctly localized, no individual portion of SurA complemented either surA– mutant phenotype. Cells were still induced for the extracytoplasmic stress response (Figure 2) and sensitive to membrane perturbants (Table I). Indeed, the parvulin domains either together or separately actually reduce the capacity to plate on SDS/EDTA (Table I), indicating that SurA requires the N-terminal domain for function. We were unable to assess meaningfully the ability of the N-terminal domain alone to complement as we could not quantify its level of expression. However, we did test constructs containing the N-terminal domain plus one of the two parvulin-like domains (either SurAN+I or SurAN+II). Such constructs showed slightly reduced σE activity (Figure 2), but still showed a >104-fold plating defect on SDS/EDTA (Table I), indicating a major perturbation in the barrier function of the outer membrane. Thus, these partial proteins are, at best, only slightly functional.
One caveat to the conclusion that all of SurA is required for function in vivo was our observation that the SurA fragments N+I and N+II showed double bands (Figure 1B, lanes e and f), which are likely to reflect degradation. We therefore considered the possibility that the low activity of these fragments resulted from incorrect folding because they lacked a stabilizing region of the protein. The extreme C-terminus of the protein (amino acids 385–428), which was absent from these constructs, was a candidate for a stabilizing region as it is very basic (predicted isoelectric point of 10.5) and might interact with the rather acidic N-terminal region (predicted isoelectric point of 5.3). Indeed, the C-terminal tail does stabilize N-terminal fragments, as they could now be purified and showed a single band in western blots (Figure 1B, lanes g and h). Moreover, when the C-terminal tail was present, the N-terminal fragment alone (SurAN-Ct) or in combination with domain I (SurAN+I-Ct) or domain II (SurAN+II-Ct) completely restored membrane functionality as judged by plating on SDS/EDTA (Table I), and almost completely eliminated the extracytoplasmic stress response as judged by the almost wild-type activity of σE (Figure 2). These fragments are no more than 2- to 3-fold overexpressed relative to the wild-type level of the protein (Figure 1B), indicating that gross overexpression is not necessary for complementation. Thus, these assays indicate that the major functions of SurA are carried within the N-terminal domain coupled to the C-terminal tail. The two reiterated parvulin-like domains do make a minor contribution to SurA function, as the three partial constructs are still slightly induced for σE activity (Figure 2).
Catalysis of proline-limited folding by SurA and variants of SurA
Recent work has indicated that active site mutations result in increased sensitivity of several cyclophilin- and FKBP-like PPIases to chymotrypsin digestion, the protease used in the tetrapeptide PPIase assay (Dolinski et al., 1997). Both SurA and the SurA with an alanine-substituted active site (SurA-2A) were degraded rapidly by this protease (data not shown). We therefore retested the PPIase activity of these proteins by examining the refolding of the reduced and carboxymethylated RNase T1 variant (S54G/P55N)-RNase T1 (RCM-T1), whose folding is controlled by the slow trans→cis isomerization about the Tyr38–Pro39 peptide bond (Mücke and Schmid, 1994).
The relative rate of SurA-catalyzed RCM-T1 folding increases strongly with SurA concentrations (Figure 3). This increase is linear at low SurA concentrations (0–100 nM; see inset) and becomes non-linear when enzyme (SurA) and substrate (RCM-T1) concentrations become similar. kcat/Km values were therefore calculated from SurA concentrations ≤100 nM. With a catalytic efficiency of kcat/Km = 30–40 × 103 M–1 s–1 (Table II), SurA exhibits a folding activity similar to that reported for parvulin in this assay (30 × 103 M–1 s–1; Scholz et al., 1997b). In contrast, the alanine double mutant SurA-2A has an activity ≤1% of the wild-type protein activity, indicating that PPIase activity is eliminated effectively by the double alanine substitution.

Fig. 3. Refolding of RCM-T1 by wild-type SurA (filled circles), the alanine double mutant SurA-2A (filled squares), the SurA fragments SurAI (domain I, filled triangles), SurAII (domain II, open circles), SurAI+II (domain I+II, open squares) and SurAI+II-2A (domain I+II-2A, open triangles) at 15°C. The dependence of the apparent rate of slow folding (Kapp) on the concentration of these SurA variants is shown. The concentration of unfolded RCM-T1 substrate was 0.5 µM for SurA and SurAI+II and 2.0 µM for SurA-2A and SurAI. The kcat/Km values (Table II) were obtained from the slopes of the indicated regression lines. The inset gives a more precise picture of the refolding of RCM-T1 by wild-type SurA and SurAI+II at concentrations between 0 and 1 µM. SurA proteins with an N-terminal region have the SurA signal sequence at their N-terminus and a C-terminal His6 tag; these proteins were purified from periplasmic fractions. All other fragments contained an N-terminal His6 tag and were purified from whole-cell extracts.
We also used the RCM-T1 folding assay to reinvestigate the PPIase activity of the isolated parvulin-like domains I and II. Parvulin-like domain I of SurA (SurAI) is folded as judged from its native-like CD spectrum (data not shown), but inactive in the folding assay (Figure 3; Table II). This confirms that domain I has no PPIase activity on its own and indicates that the lack of activity previously observed with the tetrapeptide assay does not arise from protease sensitivity. Parvulin-like domain II (SurAII), which shows essentially wild-type prolyl isomerase activity when assayed with a peptide (Rouvière and Gross, 1996; Table II), exhibits only ∼1% of the activity of wild-type SurA in the protein folding assay (Figure 3; Table II). Again, CD spectroscopy indicates that SurAII is folded properly (data not shown). Interestingly, SurAI+II, which contains both parvulin-like domains, shows a kcat/Km of 12 × 103 M–1 s–1 and thus almost 50% of the activity of the intact wild-type protein. This suggests that domain I, though inactive as a prolyl isomerase on its own, provides determinants that allow domain II to interact with RCM-T1 such that it becomes a better substrate protein for catalysis at the active site of domain II. Again, the double alanine substitution in this fragment (SurAI+II-2A) effectively eliminates PPIase activity (Figure 3; Table II).
We also assayed the N-terminal domain alone or in combination with either PPIase domain I or domain II, using variants that were coupled to the C-terminus of SurA (Table II). As expected, the N-terminal domain alone (SurAN-Ct) exhibited no activity and the N-terminal domain in combination with the second parvulin-like domain (SurAN+II-Ct) exhibited high activity in the proline-limited folding assay, comparable to or slightly better than that of native SurA. Unexpectedly, SurAN+I-Ct exhibited a very slight amount of PPIase activity (≤1% of wild type). Thus, in the context of the N-terminal domain of SurA, but not on its own, parvulin-like domain I exhibits very weak PPIase activity.
SurA protects citrate synthase from aggregation during thermal stress
In vivo studies suggest that SurA promotes folding of OMPs. Given the fact that PPIase activity does not appear central to the activity of SurA, we considered the possibility that SurA has chaperone function. To test this, we asked whether SurA prevented aggregation of thermally denatured citrate synthase. A wide range of chaperones including GroEL, Hsp70, Hsp90, small heat shock proteins and several periplasmic binding proteins have been shown to possess this activity (Buchner et al., 1991; Wiech et al., 1992; Jakob et al., 1993; Ehrnsperger et al., 1997; Richarme and Caldas, 1997). When SurA and thermally denatured citrate synthase are present at equimolar concentrations (based on citrate synthase monomer), aggregation is reduced (Figure 4A). A 64-fold excess of SurA eliminates aggregation. In contrast, bovine serum albumin (BSA) is much less effective, requiring a 64-fold excess over citrate synthase monomer to have the same preventative effect as equimolar concentrations of SurA. Several other control proteins (lysozyme, ovalbumin and cytochrome c) show no or only minor effects at comparable concentrations (data not shown). Protease contamination can not explain the chaperone effects of SurA, as citrate synthase remained intact throughout a 1200 s incubation with an 8-fold excess of SurA (as judged by SDS–PAGE and silver stain; data not shown).

Fig. 4. Aggregation of citrate synthase during thermal stress (A) and during renaturation (B) in the absence of additional protein (buffer control) or in the presence of BSA or SurA in increasing concentrations. Light-scattering measurements of citrate synthase (0.15 µM monomer) were performed in 40 mM HEPES–KOH pH 7.5 at 43°C for thermal aggregation and in 50 mM Tris–HCl pH 8.0 at 25°C for renaturation experiments, respectively. To exclude effects that may be caused by the protein buffer (10 mM Tris–HCl pH 8.0, 300 mM NaCl, 5% glycerol), all samples were added to the assay in constant volume.
SurA also reduces the extent of aggregation during the renaturation of citrate synthase. When unfolded citrate synthase (in 6 M guanidinium hydrochloride) was renatured at 25°C by a 200-fold dilution, a 2-fold molar excess of SurA (0.3 µM SurA versus 0.15 µM citrate synthase monomer) reduced the final light scattering signal by ∼30%. A further increase in SurA concentration provided only slightly more protection (Figure 4B). Complete protection could not be achieved. BSA was ineffective in this assay. It is possible that SurA does not recognize the early folding intermediates of citrate synthase efficiently and thus its effect in this assay would be limited.
The chaperone-like activity in SurA resides in the N-terminal domain coupled to the C-terminal tail
As the N-terminal domain of SurA coupled to the C-terminal tail (SurAN-Ct) complemented function in vivo, we asked whether this variant has chaperone function. Indeed, SurAN-Ct protects citrate synthase from thermal aggregation even more effectively than intact SurA. At an equimolar concentration with citrate synthase monomer, SurAN-Ct significantly inhibits aggregation (Figure 5A). Addition of either domain I or domain II to the N-terminus (SurAN+I-Ct or SurAN+II-Ct) slightly enhanced anti-aggregation activity, consistent with the idea that these domains play a minor but important role in vivo. As expected, neither the domain I (SurAI) nor the domain I+II (SurAI+II) fragments of SurA exhibited chaperone activity, even when present at high concentration (Figure 5B). Thus, the N-terminal region when coupled to the C-terminal tail is necessary and sufficient for chaperone activity.

Fig. 5. Influence of SurA fragments with (A) and without (B) the N-terminal region of the protein on thermal aggregation of citrate synthase. Light-scattering measurements of citrate synthase (0.15 µM monomer) were performed without additional protein (buffer control) or in the presence of the indicated concentrations of BSA, SurA, SurAN-Ct, SurAN+I-Ct, SurAN+II-Ct, SurAI and SurAI+II. Buffer compositions and assay conditions were as in Figure 4.
SurA preferentially binds to in vitro synthesized non-native porin
SurA is thought to influence an early folding step in porin maturation in vivo. We therefore asked whether SurA possessed the requisite substrate recognition properties by asking whether selected proteins co-immunoprecipitated with SurA. Both prePhoE and mPhoE were co-immunoprecipitated with SurA ∼50-fold more efficiently than other similarly sized proteins, indicating that SurA preferentially recognized PhoE (Figure 6; Table III). Moreover, SurA has broad specificity for porin proteins as the co-immunoprecipitation of preLamB and preOmpF with SurA was significantly more efficient than that observed for PhoE. We were unable to assess directly the ability of the N-terminal domain of the protein coupled to the C-terminal tail to bind OMPs because it is not recognized by the SurA antibodies that we have generated (Figure 1B, lane i). However, we have shown that the N-terminal region of SurA in combination with either domain I or domain II and coupled to the C-terminal tail can selectively recognize OMPs, and that domain I+II alone can not (H.de Cock, unpublished results). Thus, these studies directly show that the N-terminal domain of SurA coupled to the C-terminal tail is required for OMP recognition.
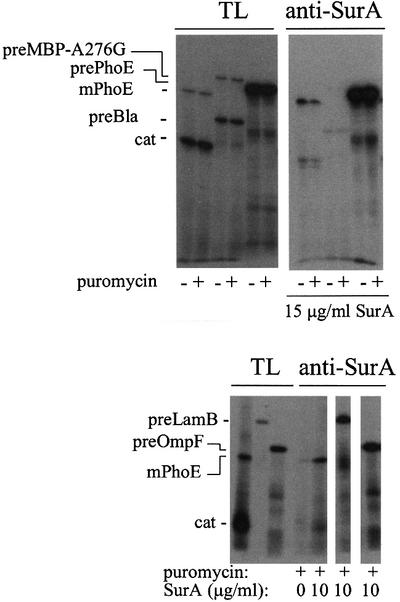
Fig. 6. Co-immunoprecipitation of in vitro synthezised proteins and SurA. Precursor and mature PhoE proteins (prePhoE and mPhoE), chloramphenicol acetyltransferase (cat) and the precursor proteins of LamB (preLamB), OmpF (preOmpF), the slow folding mutant maltose-binding protein (preMBP-A276G; Chun et al., 1993) and β-lactamase (preBla) were synthesized in vitro in the absence of SurA or with 10 or 15 µg/ml SurA added to the translation mixture and co-precipitated with anti-SurA antibodies as described in Materials and methods. The lanes with total translation products (TL) contain 10% of the total amount of translation mixtures used in the co-immunoprecipitations (anti-SurA). The amount of PhoE protein associating with SurA is similar in the absence or presence of puromycin (10 µM), which, where indicated, was added after protein synthesis to release the peptide chains from ribosomes.
Table III. Substrate selectivity of SurA.
| Protein | % precipitated protein of total protein synthesizeda |
|
|---|---|---|
| 10 µg/ml SurA | 15 µg/ml SurA | |
| prePhoE | ND | 4.4 |
| mPhoE | 4.2 | 3.4 |
| preLamB | 39 | ND |
| preOmpF | 13.2 | ND |
| preMBP-A276G | ND | 0.07 |
| preBla | ND | 0.07 |
| CAT | ND | 0.09 |
aAs quantified from + puromycin lanes ‘TL’ and ‘anti-SurA’ in Figure 6. ND, not determined. A background of 0.06 ± 0.04 resulting from endogenous SurA in the S135 extracts was observed in these experiments.
Discussion
Among the four known periplasmic PPIases, SurA appears to play a pivotal role in the biogenesis of trimeric outer membrane porins, outer membrane function and proper protein folding in the periplasm. We have investigated the biochemical basis of SurA function. SurA consists of a large N-terminal region, two successive parvulin-like domains with only the second one active as a PPIase, and a C-terminal tail. Our studies show that the PPIase activity of SurA is dispensable for function and that the N-terminal region coupled to the C-terminal tail has a chaperone-like activity and is almost completely functional for SurA activity in vivo. We show that SurA preferentially recognizes porins and that this activity requires the N-terminus of the protein coupled to the C-terminal tail. Together, these studies suggest that the role of SurA in porin biogenesis is to selectively recognize OMPs and aid in the folding and assembly pathway, with the PPIase activity of SurA playing a minor role, if any, in the overall function of SurA. Our work lends credence to the emerging notion that a web of proteins initially identified as folding catalysts may predominantly provide the chaperone activities that are needed for protein remodeling in the periplasm.
SurA as a folding catalyst
The sensitivity of SurA and its variants to the proteolysis step inherent in the peptide-based assay necessitated re-examination of its PPIase activity with an assay that avoids proteolysis. The proline-limited folding of the RNase T1 variant RCM-T1 (Mücke and Schmid, 1994) provided such an assay. The RNase refolding assay also reports on the affinity of a PPIase for a folding protein, such as RCM-T1. Single-domain PPIases such as parvulin often show much lower activity in the RNase refolding assay than in the peptide assay, which has been attributed to their inability to bind well to the folding protein substrate (Scholz et al., 1997a). In support of this idea, larger PPIases such as trigger factor show equivalent high activity in both assays, whereas the isolated FKBP-like domain of trigger factor is ∼1000-fold less active in the folding assay than in the peptide-based assay (Scholz et al., 1997b). SurA resembles trigger factor in this respect. The PPIase domain II of SurA in isolation (SurAII) is 50-fold less active in the folding assay than in the peptide assay. Domain I (which has no PPIase activity on its own) restores PPIase activity to 30% of that of the intact protein, indicating that both parvulin domains in combination (in SurAI+II) interact more productively with RNase T1 than does domain II alone. The N-terminal portion of SurA seems to play a minor role and increases the activity in RCM-T1 folding only ∼3-fold. However, given the fact that the rate of SurA-catalyzed folding does not increase linearly with SurA concentration, small differences in rates may not be significant. We have confirmed that this non-linearity does not result from substrate concentrations being near the Km value (data not shown). Possibly, SurA and its derivatives may multimerize or otherwise lose activity at higher concentration.
The PPIase activity of SurA was decreased ≥100-fold by substituting two highly conserved amino acid residues in domain II with alanine. Unexpectedly, we found that domain I, which is inactive as a PPIase alone, has a small amount of isomerase activity when combined with the N-terminal domain of the protein. As this amount of activity is comparable to that exhibited by SurA-2A (Table II), we suggest that the residual activity of SurA-2A results from parvulin domain I.
Chaperone-like activity of SurA
We have used three different assays to assess the chaperone activity. SurA exhibits robust activity in preventing thermal aggregation of citrate synthase, a classic assay for chaperone function (Buchner et al., 1998). A 50-fold molar excess of intact SurA prevents aggregation and an equimolar amount of SurA slows aggregation significantly. SurA is more effective in preventing aggregation than several control proteins but is less effective than the classic cytoplasmic chaperones (Buchner et al., 1998). Its anti-aggregation potential is most similar to that of the periplasmic binding proteins MglB and OppA (Richarme and Caldas, 1997). SurA is less robust at preventing renaturing citrate synthase from aggregating, decreasing the extent of aggregation but not the initial rate of aggregation. However, the effect is significant, as control proteins could not prevent aggregation in this assay. The reduced light scattering signal may result from a reduced total number of aggregating molecules or from alterations in the form and size of the aggregates. Finally, SurA was unable to restore citrate synthase activity after oxalic acid-induced reactivation of thermally denatured citrate synthase (as described in Buchner et al., 1998) (data not shown). The weak or absent chaperone activity in the latter two assays may reflect poor binding of SurA to particular partially folded forms of citrate synthase. This would be consistent with our finding, discussed below, that SurA has strong substrate specificity.
Our results establish that the chaperone and PPIase activities of SurA are in distinct domains of the protein. The chaperone activity of SurA is contained solely in the N-terminal region coupled to the C-terminal tail (SurAN-Ct): SurAN-Ct actually exhibits higher anti-aggregation activity than does intact SurA, whereas the PPIase domains alone (SurAI+II) exhibit no anti-aggregation activity, even at very high concentration. In contrast, the PPIase activity of SurA is contained solely in the parvulin-homology domains I + II. The segregation of chaperone and PPIase function into separate domains of SurA stands in contrast to recent findings for the FkpA PPIase, whose chaperone activity resides in the PPIase domain (A.Plückthun and K.Ramm, personal communication).
The biological role of SurA
Gram-negative bacteria are highly impermeable to most small molecules, protecting cells against a variety of insults in their environment, including antibiotics. This barrier function is performed by the outer membrane, in a collaboration between lipopolysaccharides (LPS), the OMPs and divalent cations. Negative charges in LPS and OMPs are likely to participate in a strong and tight network with divalent cations in the outer monolayer of the outer membrane to create the barrier; porins allow entry of selected small molecules through this barrier. Production of these two outer membrane constituents is co-regulated to give the appropriate barrier (W.C.Suh, V.Rhodius and C.Gross, in preparation). This barrier function is destroyed when the amount of either component is drastically altered. For example, cells that have few porins in the outer membrane, due to mutational alteration, exhibit increased sensitivity to a variety of external agents. Studies have shown that this results from addition of normal phospholipids to the membrane and its outer leaflet to compensate for lack of porins, thus breaching the barrier function (Smit et al., 1975; Kamio and Nikaido, 1976). Thus it is crucial to cells to maintain their normal complement of porins in the outer membrane.
All indications are that the biological role of SurA is to facilitate maturation of outer membrane porins. Cells lacking SurA are defective in inserting porins in the outer membrane, as indicated both by direct assay and by their extreme sensitivity to membrane perturbants such as SDS/EDTA. Our studies showing that SurA has chaperone function and selectively recognizes OMPs suggest that SurA is likely to be a chaperone that is involved specifically in OMP maturation. Although the only clearly documented role of SurA thus far is promoting formation of the native-like folded porin monomer, it is possible that SurA works at several steps in porin maturation. The major functions of SurA are located in the N-terminal region coupled to the C-terminal tail, as this region of the protein provides complete protection against SDS/EDTA in vivo, has chaperone function and is required for selective recognition of OMPs by SurA. Although we were unable to show directly that the N-terminal region stabilized by the C-terminal tail is sufficient for OMP recognition, this is likely to be the case as this region restores resistance to SDS/EDTA in vivo, which requires OMP insertion in the outer membrane. Further studies will be required to establish whether the C-terminal tail simply stabilizes the N-terminal region or plays additional roles in carrying out SurA function.
It is likely that several other proteins collaborate with SurA. Skp, a small periplasmic protein implicated in OMP maturation, is likely to collaborate with SurA, as SurA and Skp deletions are synthetically lethal (S.Behrens, unpublished data). Additionally, deletion of either PpiD or FkpA, two other periplasmic PPIases, gives phenotypes that are similar to but less severe than those exhibited by cells that lack SurA (Horne and Young, 1995; Missiakas et al., 1996; Dartigalongue and Raina, 1998). This suggests that all three periplasmic PPIases participate in the same processes and are partially redundant in function. Direct evidence for this proposition comes from the finding that SurA and PpiD deletions are synthetically lethal (Dartigalongue and Raina, 1998). As FkpA has recently been shown to have chaperone activity, it is biochemically capable of performing some SurA functions (Ramm and Plückthun, 2000). It would not be surprising if PpiD were found to be a chaperone as well. We suggest that these three multidomain periplasmic PPIases play a major role in mediating porin maturation and possibly other protein folding activities in the periplasmic compartment of the cell.
The cytoplasmic chaperones exhibit rather broad specificity (Houry et al., 1999; Mogk et al., 1999). In contrast, PapD, the best characterized periplasmic chaperone, is specialized to interact with pili subunits to ensure their specific assembly at the cell surface. Interestingly, SurA, like PapD, exhibits strong selectivity, recognizing three different outer membrane porins much more efficiently than several other similarly sized proteins. The chaperone function of SurA may be directed predominantly at maturation of bacterial porins. Resolution of this point requires detailed studies on the interaction of SurA with its biologically relevant porin substrates.
Materials and methods
Growth media
Luria–Bertani (LB) and M9 minimal media were prepared as described (Sambrook et al., 1989). Ampicillin (Ap), kanamycin (Km), chloramphenicol (Cm), spectinomycin (Spec) and tetracycline (Tc) were used at final concentrations of 100, 30, 20, 50 and 10 µg/ml, respectively. SDS/EDTA plates were prepared freshly 1 day in advance.
Strains and plasmids
Strains and plasmids are listed in Table IV. All β-galactosidase assays were performed in cells deleted for the chromosomal copy of lacZ.
Table IV. Bacterial strains and plasmids.
| Strain/plasmid | Relevant genotype | Source/reference |
|---|---|---|
| Strain | ||
| CAG16037 | MC1061 φλ[rpoH P3::lacZ] | Mecsas et al. (1993) |
| CAG24029 | CAG16037 surA::Tn10dCm, CmR | Rouvière and Gross (1996) |
| CAG44102 | MC4100 surA::Tn10dCm slyD1 zhg::Tn10, CmR TcR | this work |
| Plasmids | ||
| pQE60 | C-His6 expression vector, PT5/Olac, ColEI ori, ApR | Qiagen |
| pQE30 | N-His6 expression vector, PT5/Olac, ColEI ori, ApR | Qiagen |
| pASK75 | expression vector, P/Otet, tetR, ColEI ori, ApR | Skerra (1994) |
| pASKSurA | surA in pASK75, ApR | this work |
| pASKSurA-2A | surA H376A I378A in pASK75, ApR | this work |
| pPER93a | surA in pQE60, ApR | Rouvière and Gross (1996) |
| pPER94 | surA domain I in pQE30, ApR | Rouvière and Gross (1996) |
| pPER95 | surA domain II in pQE30, ApR | Rouvière and Gross (1996) |
| pPER96 | surA domain I+II in pQE30, ApR | Rouvière and Gross (1996) |
| pPER96-2A | surA domain I+II H376A I378A in pQE30, ApR | this work |
| pSurA-2A | surA H376A I378A in pQE60, ApR | this work |
| pSurAN | surA ssb/domainN in pQE30, ApR | this work |
| pSurAN-Ct | surA ssb/domainN/ctc in pQE30, ApR | this work |
| pSurAN+I | surA ssb/domainN+I in pQE30, ApR | this work |
| pSurAN+I-Ct | surA ssb/domainN/ctc in pQE30, ApR | this work |
| pSurAI | surA ssb/domain I in pQE30, ApR | this work |
| pSurAI+II | surA ssb/domain I+II in pQE30, ApR | this work |
| pSurAII | surA ssb/domain II in pQE30, ApR | this work |
| pSurAN+II | surA ssb/domainN+II in pQE30, ApR | this work |
| pSurAN+II-Ct | surA ssb/domainN+II/ctc in pQE30, ApR | this work |
| pSurAΔC368–428 | surAΔC368–428 in pQE60, ApR | this work |
| pJP29 | mature PhoE-encoding sequence in pACYC184, CmR | Bosch et al. (1986) |
| pJP370 | precursor of PhoE-encoding sequence in pACYC184, CmR | de Cock et al. (1990) |
| pMalE276G | carries the preMBP-A276G-encoding sequence, ApR | Chun et al. (1993) |
| pPER98 | precursor of OmpF-encoding sequence in pKK223-3 | P.Rouvière, laboratory collection |
| pPER99 | precursor of LamB-encoding sequence in pKK223-3 | Rouvière and Gross (1996) |
| pPLT13 | mini-F carrying lacIq, KmR | Tavormina et al. (1996) |
| pML121 | lacIq Ω::spec in pACYC184, SpecR | M.Lonetto, laboratory collection |
aReferred to as pSurA in the text.
bss = signal peptide sequence including the two adjacent 3′ codons of surA (codons 1–22).
cct = 43 C-terminal codons of surA.
pSurA-2A was constructed by altering codons 376 and 378 of surA to alanine using primers SurA52-2A (5′-GCAGTTCGGCTAAAGCCCAGCC-3′) and SurA53-2A (5′-GGCTGGGCTTTAGCCGAACTGC-3′) with primers SurA35 and SurA27 (Rouvière and Gross, 1996); the resulting EcoRV–BglII fragment was subcloned back to yield pSurA-2A. pPER96-2A was constructed similarly, except that primer SurA37 (Rouvière and Gross, 1996) was used instead of SurA27.
Fragments of SurA to be secreted to the periplasm were generated by fusing the respective gene fragments to the signal sequence of surA. pSurAI, pSurAII and pSurAI+II were created by replacing the N-His6 tag-encoding EcoRI–SacI vector fragment in pPER94, pPER95 and pPER96 with an EcoRI–SacI PCR fragment encoding the first 22 amino acids of SurA (the signal peptide sequence and its two adjacent 3′ codons). This fragment was amplified from pSurA using the primers TIII/IV (Qiagen) and SurA50 (5′-GTCGACGAGCTCGGGGGCAGCGAAAC-3′; the SacI site is underlined). To create pSurAN and pSurAN+I, the domain N and domain N+I gene fragments were PCR amplified from pSurA using the primer TIII/IV in combination with SurA51 (5′-CAGTCTAAAGCTTGCGTCGTTTTGGTTAC-3′) and SurA36 (Rouvière and Gross, 1996), digested with EcoRI and HindIII and cloned into pQE30. Insertion of a fragment encoding the C-terminal 43 amino acids of SurA (Ct) and the His6 tag into the HindIII site of these plasmids yielded pSurAN-Ct and pSurAN+I-Ct. The Ct fragment was amplified using the primer pair CT2 (5′-CTGGATACCCGAAGCTTCGATAAAACCGAC-3′)/QErev (Qiagen). For pSurAN+II, the domain N fragment was amplified by PCR using the primers TIII/IV and DNSacI (5′-GGCTCAGGTTGAGCTCAGTGCTGG-3′), cleaved with EcoRI and SacI, and inserted into the corresponding sites of pPER95. To yield pSurAN+II-Ct, the Ct-encoding fragment was amplified from pSurA with the primer pair CT1 (5′-CTGGATACCCGTAAGCTTGATAAAACCGAC-3′)/QErev (Qiagen) (HindIII site underlined) and cloned into the HindIII site of pSurAN+II.
For pSurAΔC368–428, pSurA was cleaved at AgeI and BglII, blunt-ended and religated. For pASKSurA and pASKSurA-2A, the corresponding genes from pSurA and pSurA-2A were cloned into pASK75 (Skerra, 1994) using HindIII and blunted NcoI (surA fragments) or XbaI (pASK75) restriction sites. All sequences were confirmed by DNA sequencing. pQE-based plasmids were maintained in strains expressing the LacIq repressor from either pPLT13 or pML121.
Protein purification
C-terminally His6-tagged SurA proteins (from pASKSurA and pASKSurA-2A) were purified from the periplasmic fraction of slyD– surA– (CAG44102) cells following growth in LB medium at 30°C and induction with 10 µg/ml tetracycline at an OD600 of 0.2–0.25 for 4 h. N-terminally His6-tagged fragments of SurA (from pPER94, pPER95, pPER96 and pPER96-2A) were purified from whole-cell extracts (Rouvière and Gross, 1996).
Peptide-based PPIase assay
PPIase activity was assayed and calculated as described (Fischer et al., 1992; Rahfeld et al., 1994; Rouvière and Gross, 1996). The reaction was monitored at 395 nm on a Kontron Uvikon 933 spectrophotometer.
Refolding of RCM (S54G/P55N)-RNase T1 (RCM-T1)
Reduced and carboxymethylated (S54G, P55N)-RNase T1 was prepared according to Mücke and Schmid (1994). Unfolding of the enzyme and refolding reactions were performed essentially as described (Scholz et al., 1997a). As RCM-T1 refolding did not follow monoexponential kinetics at high concentrations of SurA and SurAI+II, the specificity constant kcat/Km for these proteins was determined from low enzyme concentrations as indicated.
Analysis of chaperone function
SurA effects on thermal aggregation and on aggregation during renaturation of citrate synthase were determined according to Buchner et al. (1998). For refolding, citrate synthase was denatured in 6 M guanidinium hydrochloride, 50 mM Tris–HCl pH 8.0, 20 mM dithiothreitol for at least 2 h at room temperature. Aggregation was monitored on a Hitachi F-4500 spectrofluorometer with both excitation and emission wavelengths set to 500 nm at a spectral bandwidth of 2.5 nm. Data points were recorded every 0.5 s.
Determination of protein concentrations
Protein concentrations were determined spectrophotometrically with absorption coefficients of 21 060 M–1 cm–1 at 278 nm for RCM-RNase T1 (Takahashi et al., 1970) and of 1.78 cm–1 for a 1 mg/ml solution at 280 nm for citrate synthase (West et al., 1990). Absorption coefficients for SurA, SurA-2A and SurA fragments were calculated according to Pace et al. (1995).
σE activity assays
σE activity was assayed by monitoring β-galactosidase activity from the chromosomal σE-dependent reporter fusion in rpoHP3::lacZ (Mecsas et al., 1993) in cells growing exponentially in M9 glucose medium at 30°C. Starting at an OD450 of 0.1–0.15, duplicate samples were taken at fixed time intervals for calculation of the differential rates of β-galactosidase synthesis.
Plating efficiencies
Fresh overnight cultures grown at 30°C in LB plus 1 mM isopropyl- β-d-thiogalactopyranoside (IPTG) and appropriate antibiotics were diluted and plated in duplicate on LB, LB plus 0.5% (w/v) SDS/0.5 or 1 mM EDTA plus antibiotics. Plating efficiency was calculated from the colony count after incubation at 30°C for 24–30 h.
Preparation of soluble cell extracts and analysis by western blotting
Cells growing in LB medium at 30°C were induced with 1 mM IPTG at OD600 = 0.25 for 2 h. Pellets from 1 ml samples were resuspended in 1 ml of 100 mM Tris–HCl pH 8.0, 20% sucrose (buffer A) and equilibrated for 10 min on ice. To make spheroplasts, pelleted cells were resuspended in 0.5 ml of buffer A plus 10 mM EDTA, incubated with lysozyme (10 µg/ml) for 15 min on ice, adjusted to 20 mM MgSO4 and sedimented at low speed. The periplasmic fractions were transferred into fresh Eppendorf tubes. Spheroplasts were washed in 1 ml of buffer A + 10 mM EDTA and resuspended in protein sample buffer. Extracts from equal numbers of cells were run on SDS–polyacrylamide gels, transferred to nitrocellulose membranes and subjected to western blot analysis (Connolly et al., 1997). Polyclonal antibodies against SurA, σE and β-lactamase (5Prime-3Prime, Inc.) were used at 1:10 000 concentrations. Anti-rabbit alkaline phosphatase (Sigma-Aldrich, Corp.; 10 000 dilution) was the secondary antibody. The blots were developed by incubation in reaction buffer (100 mM Tris–HCl pH 8.8, 100 mM NaCl, 5 mM MgCl2, 37.5 µg/ml NBT, 150 µg/ml BCIP). To confirm equal loading and test for cytoplasmic contamination, duplicate membranes were probed with β-lactamase and σE antibodies, respectively.
In vitro synthesis and co-immunoprecipitation
In vitro transcription and translation reactions were performed as described (de Vrije et al., 1987). pJP370 and pJP29 directed synthesis of [35S]methionine-labeled mature and precursor forms of the PhoE protein, respectively, and of chloramphenicol acetyltransferase; pMalEA276G of preMBPA276G and β-lactamase precursor; pPER98 of preOmpF; and pPER99 of preLamB. In vitro translation reactions contained 0, 10 or 15 µg/ml SurA in 10 mM Tris–HCl pH 8.0, 300 mM NaCl, 5% glycerol. Complexes with SurA were immunoprecipitated with SurA antibodies as described (de Cock et al., 1992). Samples were analyzed by gel electrophoresis and the data quantified with a PhosphorImager (Molecular Dynamics).
Acknowledgments
Acknowledgements
We thank R.Young for the gift of slyD– strains, J.Weissman for helpful discussion and B.Alba for comments. We are grateful to A.Plückthun and K.Ramm for sharing unpublished results. This work was supported by the US Public Health Service Grant GM36278 from the National Institutes of Health and by a postdoctoral research grant of the DFG to S.B.
References
- Bardwell J.C. and Beckwith,J. (1993) The bonds that tie: catalyzed disulfide bond formation. Cell, 74, 769–771. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C., McGovern,K. and Beckwith,J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell, 67, 581–589. [DOI] [PubMed] [Google Scholar]
- Bosch D., Leunissen,J., Verbakel,J., de Jong,M., van Erp,H. and Tommassen,J. (1986) Periplasmic accumulation of truncated forms of outer-membrane PhoE protein of Escherichia coli K-12. J. Mol. Biol., 189, 449–455. [DOI] [PubMed] [Google Scholar]
- Bothmann H. and Plückthun,A. (1998) Selection for a periplasmic factor improving phage display and functional periplasmic expression. Nature Biotechnol., 16, 376–380. [DOI] [PubMed] [Google Scholar]
- Buchner J., Schmidt,M., Fuchs,M., Jaenicke,R., Rudolph,R., Schmid,F.X. and Kiefhaber,T. (1991) GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry, 30, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Buchner J., Grallert,H. and Jakob,U. (1998) Analysis of chaperone function using citrate synthase as nonnative substrate protein. Methods Enzymol., 290, 323–338. [DOI] [PubMed] [Google Scholar]
- Bukau B. and Horwich,A.L. (1998) The Hsp70 and Hsp60 chaperone machines. Cell, 92, 351–366. [DOI] [PubMed] [Google Scholar]
- Chen J., Song,J.L., Zhang,S., Wang,Y., Cui,D.F. and Wang,C.C. (1999) Chaperone activity of DsbC. J. Biol. Chem., 274, 19601–19605. [DOI] [PubMed] [Google Scholar]
- Chen R. and Henning,U. (1996) A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol. Microbiol., 19, 1287–1294. [DOI] [PubMed] [Google Scholar]
- Chun S.Y., Strobel,S., Bassford,P.,Jr and Randall,L.L. (1993) Folding of maltose-binding protein. Evidence for the identity of the rate-determining step in vivo and in vitro. J. Biol. Chem., 268, 20855–20862. [PubMed] [Google Scholar]
- Connolly L., De Las Peñas,A., Alba,B.M. and Gross,C.A. (1997) The response to extracytoplasmic stress in Escherichia coli is controlled by partially overlapping pathways. Genes Dev., 11, 2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese P.N. and Silhavy,T.J. (1997) The σ(E) and the Cpx signal transduction systems control the synthesis of periplasmic protein-folding enzymes in Escherichia coli. Genes Dev., 11, 1183–1193. [DOI] [PubMed] [Google Scholar]
- Dartigalongue C. and Raina,S. (1998) A new heat-shock gene, ppiD, encodes a peptidyl-prolyl isomerase required for folding of outer membrane proteins in Escherichia coli. EMBO J., 17, 3968–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cock H., Hekstra,D. and Tommassen,J. (1990) In vitro trimerization of outer membrane protein PhoE. Biochimie, 72, 177–182. [DOI] [PubMed] [Google Scholar]
- de Cock H., Overeem,W. and Tommassen,J. (1992) Biogenesis of outer membrane protein PhoE of Escherichia coli. Evidence for multiple SecB-binding sites in the mature portion of the PhoE protein. J. Mol. Biol., 224, 369–379. [DOI] [PubMed] [Google Scholar]
- de Cock H., Schäfer,U., Potgeter,M., Demel,R., Müller,M. and Tommassen,J. (1999) Affinity of the periplasmic chaperone Skp of Escherichia coli for phospholipids, lipopolysaccharides and non-native outer membrane proteins. Role of Skp in the biogenesis of outer membrane protein. Eur. J. Biochem., 259, 96–103. [DOI] [PubMed] [Google Scholar]
- de Vrije T., Tommassen,J. and De Kruijff,B. (1987) Optimal posttranslational translocation of the precursor of PhoE protein across Escherichia coli membrane vesicles requires both ATP and the protonmotive force. Biochim. Biophys. Acta, 900, 63–72. [DOI] [PubMed] [Google Scholar]
- Dolinski K., Scholz,C., Muir,R.S., Rospert,S., Schmid,F.X., Cardenas, M.E. and Heitman,J. (1997) Functions of FKBP12 and mitochondrial cyclophilin active site residues in vitro and in vivo in Saccharomyces cerevisiae. Mol. Biol. Cell, 8, 2267–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnsperger M., Graber,S., Gaestel,M. and Buchner,J. (1997) Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J., 16, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G., Bang,H., Ludwig,B., Mann,K. and Hacker,J. (1992) Mip protein of Legionella pneumophila exhibits peptidyl-prolyl-cis/trans isomerase (PPlase) activity. Mol. Microbiol., 6, 1375–1383. [DOI] [PubMed] [Google Scholar]
- Hayano T., Takahashi,N., Kato,S., Maki,N. and Suzuki,M. (1991) Two distinct forms of peptidylprolyl-cis-trans-isomerase are expressed separately in periplasmic and cytoplasmic compartments of Escherichia coli cells. Biochemistry, 30, 3041–3048. [DOI] [PubMed] [Google Scholar]
- Hayhurst A. and Harris,W.J. (1999) Escherichia coli skp chaperone coexpression improves solubility and phage display of single-chain antibody fragments. Protein Expr. Purif., 15, 336–343. [DOI] [PubMed] [Google Scholar]
- Horne S.M. and Young,K.D. (1995) Escherichia coli and other species of the Enterobacteriaceae encode a protein similar to the family of Mip-like FK506-binding proteins. Arch. Microbiol., 163, 357–365. [DOI] [PubMed] [Google Scholar]
- Houry W.A., Frishman,D., Eckerskorn,C., Lottspeich,F. and Hartl,F.U. (1999) Identification of in vivo substrates of the chaperonin GroEL. Nature, 402, 147–154. [DOI] [PubMed] [Google Scholar]
- Hultgren S.J., Jacob-Dubuisson,F., Jones,C.H. and Branden,C.I. (1993) PapD and superfamily of periplasmic immunoglobulin-like pilus chaperones. Adv. Protein Chem., 44, 99–123. [DOI] [PubMed] [Google Scholar]
- Jacob-Dubuisson F., Pinkner,J., Xu,Z., Striker,R., Padmanhaban,A. and Hultgren,S.J. (1994) PapD chaperone function in pilus biogenesis depends on oxidant and chaperone-like activities of DsbA. Proc. Natl Acad. Sci. USA, 91, 11552–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U., Gaestel,M., Engel,K. and Buchner,J. (1993) Small heat shock proteins are molecular chaperones. Biol. Chem., 268, 1517–1520. [PubMed] [Google Scholar]
- Kamio Y. and Nikaido,H. (1976) Outer membrane of Salmonella typhimurium: accessibility of phospholipid head groups to phospholipase C and cyanogen bromide activated dextran in the external medium. Biochemistry, 15, 2561–2570. [DOI] [PubMed] [Google Scholar]
- Lazar S.W. and Kolter,R. (1996) SurA assists the folding of Escherichia coli outer membrane proteins. J. Bacteriol., 178, 1770–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. and Walsh,C.T. (1990) Peptidyl-prolyl cis-trans-isomerase from Escherichia coli: a periplasmic homolog of cyclophilin that is not inhibited by cyclosporin A. Proc. Natl Acad. Sci. USA, 87, 4028–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K.P., Hanes,S.D. and Hunter,T. (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature, 380, 544–547. [DOI] [PubMed] [Google Scholar]
- Mecsas J., Rouvière,P.E., Erickson,J.W., Donohue,T.J. and Gross,C.A. (1993) The activity of σE, an Escherichia coli heat-inducible σ-factor, is modulated by expression of outer membrane proteins. Genes Dev., 7, 2618–2628. [DOI] [PubMed] [Google Scholar]
- Missiakas D., Georgopoulos,C. and Raina,S. (1994) The Escherichia coli dsbC (xprA) gene encodes a periplasmic protein involved in disulfide bond formation. EMBO J., 13, 2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D., Betton,J.M. and Raina,S. (1996) New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol. Microbiol., 21, 871–884. [DOI] [PubMed] [Google Scholar]
- Mogk A., Tomoyasu,T., Goloubinoff,P., Rudiger,S., Roder,D., Langen,H. and Bukau,B. (1999) Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J., 18, 6934–6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mücke M. and Schmid,F.X. (1994) Folding mechanism of ribonuclease T1 in the absence of the disulfide bonds. Biochemistry, 33, 14608–14619. [DOI] [PubMed] [Google Scholar]
- Nikaido H. (1994) Porins and specific diffusion channels in bacterial outer membranes. J. Biol. Chem., 269, 3905–3908. [PubMed] [Google Scholar]
- Pace C.N., Vajdos,F., Fee,L., Grimsley,G. and Gray,T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci., 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahfeld J.U., Schierhorn,A., Mann,K. and Fischer,G. (1994) A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett., 343, 65–69. [DOI] [PubMed] [Google Scholar]
- Ramm K. and Plückthun,A. (2000) The periplasmic E.coli peptidyl-prolyl-isomerase FkpA (II): isomerase-independent chaperone activity in vitro. J. Biol. Chem., 275, 17106–17113. [DOI] [PubMed] [Google Scholar]
- Ranganathan R., Lu,K.P., Hunter,T. and Noel,J.P. (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell, 89, 875–886. [DOI] [PubMed] [Google Scholar]
- Richarme G. and Caldas,T.D. (1997) Chaperone properties of the bacterial periplasmic substrate-binding proteins. J. Biol. Chem., 272, 15607–15612. [DOI] [PubMed] [Google Scholar]
- Rietsch A., Belin,D., Martin,N. and Beckwith,J. (1996) An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl Acad. Sci. USA, 93, 13048–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouvière P.E. and Gross,C.A. (1996) SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev., 10, 3170–3182. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schäfer U., Beck,K. and Müller,M. (1999) Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J. Biol. Chem., 274, 24567–24574. [DOI] [PubMed] [Google Scholar]
- Scholz C., Rahfeld,J., Fischer,G. and Schmid,F.X. (1997a) Catalysis of protein folding by parvulin. J. Mol. Biol., 273, 752–762. [DOI] [PubMed] [Google Scholar]
- Scholz C., Stoller,G., Zarnt,T., Fischer,G. and Schmid,F.X. (1997b) Cooperation of enzymatic and chaperone functions of trigger factor in the catalysis of protein folding. EMBO J., 16, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerra A. (1994) Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene, 151, 131–135. [DOI] [PubMed] [Google Scholar]
- Smit J., Yoshiyuki,K. and Nikaido,H. (1975) Outer membrane of Salmonella typhimurium: chemical analysis and freeze-fracture studies with lipopolysaccharide mutants. J. Bacteriol., 124, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto G.E., Dodson,K.W., Ogg,D., Liu,C., Heuser,J., Knight,S., Kihlberg,J., Jones,C.H. and Hultgren,S.J. (1998) Periplasmic chaperone recognition motif of subunits mediates quaternary interactions in the pilus. EMBO J., 17, 6155–6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Uchida,T. and Egami,F. (1970) Ribonuclease T1, structure and function. Adv. Biophys., 1, 53–98. [PubMed] [Google Scholar]
- Tavormina P.L., Reznikoff,W.S. and Gross,C.A. (1996) Identifying interacting regions in the β subunit of Escherichia coli RNA polymerase. J. Mol. Biol., 258, 213–223. [DOI] [PubMed] [Google Scholar]
- West S.M., Kelly,S.M. and Price,N.C. (1990) The unfolding and attempted refolding of citrate synthase from pig heart. Biochim. Biophys. Acta, 1037, 332–336. [DOI] [PubMed] [Google Scholar]
- Wiech H., Buchner,J., Zimmermann,R. and Jakob,U. (1992) Hsp90 chaperones protein folding in vitro. Nature, 358, 169–170. [DOI] [PubMed] [Google Scholar]
- Wülfing C. and Plückthun,A. (1994) Protein folding in the periplasm of Escherichia coli. Mol. Microbiol., 12, 685–692. [DOI] [PubMed] [Google Scholar]
- Zheng W.D., Quan,H., Song,J.L., Yang,S.L. and Wang,C.C. (1997) Does DsbA have chaperone-like activity? Arch. Biochem. Biophys., 337, 326–331. [DOI] [PubMed] [Google Scholar]