

Abstract
Ceramides have been implicated in the initiation of apoptosis by permeabilizing the mitochondrial outer membrane to small proteins, including cytochrome c. In addition, ceramides were shown to form large metastable channels in planar membranes and liposomes, indicating that these lipids permeabilize membranes directly. Here we analyze molecular models of ceramide channels and test their stability in molecular dynamics simulations. The structural units are columns of four to six ceramides H-bonded via amide groups and arranged as staves in either a parallel or antiparallel manner. Two cylindrical assemblies of 14 columns (four or six molecules per column) were embedded in a fully hydrated palmitoyloleoyl-phosphatidylcholine phospholipid bilayer, and simulated for 24 ns in total. After equilibration, the water-filled pore adopted an hourglass-like shape as headgroups of ceramides and phospholipids formed a smooth continuous interface. The structure-stabilizing interactions were both hydrogen bonds between the headgroups (including water-mediated interactions) and packing of the hydrocarbon tails. Ceramide's essential double bond reduced the mobility of the hydrocarbon tails and stabilized their packing. The six-column assembly remained stable throughout a 10-ns simulation. During simulations of four-column assemblies, pairs of columns displayed the tendency of splitting out from the channels, consistent with the previously proposed mechanism of channel disassembly.
INTRODUCTION
In biology, polar lipids are best known for their self-assembly, in an aqueous environment, into lamellar phases that are the fundamental structures of membranes. However, the amphipathic nature of these lipids can result in a variety of structures. Recently, the sphingolipid, ceramide, has been shown to be able to self-assemble, within the environment of a phospholipid membrane, into a large channel (1) capable of allowing small proteins to cross membranes (2). These channels grow and shrink in size by the incorporation or loss of ceramide aggregates (3). The addition of ceramide to mitochondria results in channels in the outer membrane with an estimated radius of 3 nm.
The phenomenology of ceramide channel formation and dynamics observed in the biophysical experiments on mitochondria (2), planar membranes (3), and liposomes (4) can be strongly complemented by structural analysis and computational techniques. Molecular dynamics (MD) simulation can put proposed structures to qualitative and in certain cases quantitative testing to assess their overall stability, how the components interact with each other and their environment, and the importance of different structural features. The analysis can also yield new models for the structure of the channel.
There is no previous literature on MD of ceramide channels. Assemblies of sphingolipids have previously been studied using MD but not in the context of pore formation (5,6). Aqueous pores in phospholipid bilayers were also subjected to MD simulations (7,8); however, no sphingolipids were included in the system. Here we are trying to obtain computational insight into the possible molecular arrangements of ceramide channels that may explain their remarkable stability and conductive properties observed in experiments.
Channel-forming abilities of ceramide
Ceramide is a two-tailed lipid (Fig. 1 A) consisting of sphingosine with a 15-carbon aliphatic chain amide-linked to a fatty acid. Thus, similar to phospholipids, ceramides have two hydrocarbon chains. The fatty acid chains, however, vary in length and degree of unsaturation. One example is ceramide with a 16-carbon fatty acid chain (C16-ceramide) that is naturally present in mammalian cells. However, ceramides with fatty acid chains as short as two carbons (C2-ceramide) have been generated and are biologically active. When the difference in ability to translocate into and across membranes is compensated for, both short-chain and long-chain ceramides function similarly in their ability to induce apoptosis or form channels in membranes so the length of the acyl chain is not critical to ceramide channel model building.
FIGURE 1.

Model of the ceramide channel and patterns of hydrogen-bonding stabilizing the assembly. (A) Structure of C16 ceramide. For C2 ceramide, the fatty acid chain indicated by a bracket is just two carbons long. Headgroup atoms capable of H-bonding are labeled in green. Color coding is as on the next panel. (B) An initial model of the ceramide channel is composed of six layers and 14 columns of C16 molecules. Oxygens (red), carbons (cyan), and polar hydrogens (white) are in licorice representation. Nitrogen (blue) and oxygen of amide group are in van der Waals (VDW) spheres representation to visualize the antiparallel orientation of neighboring columns. (C) Circular hydrogen bonding occurs between the hydroxyls of neighboring ceramide molecules in the same layer. (C–E) Hydrogen bonds marked by hashing are colored according to the H-bond donor atom. (D,E) The hydrogen bonding between the amide groups is illustrated for two types of ceramide columns. An opposite orientation of the dipoles of amide groups makes these columns antiparallel to each other in the model on B. (F) Two types of arrangement of the ceramide columns in a barrel (parallel and antiparallel).
A critical feature is the trans double bond at the 4–5 position of the sphingosine base. A special enzyme inserts that bond into dihydroceramide to generate ceramide. Dihydroceramide is not able to form the channels or induce apoptosis.
Ceramide channels were proposed (1) to be barrel-stave arrangements with each stave consisting of a column of several ceramide monomers hydrogen-bonded together through the amide linkage. The tails were proposed to splay out in the plane of the membrane and the twin hydroxyl groups form a hydroxy-hydrogen-bonded network that interfaces with the water in the channel. The columns are organized in an antiparallel fashion to maximize the favorable interaction of the dipole moments formed by the polar H-bonded amide groups that link individual ceramides in each column (3).
Electrophysiological measurements in planar membranes indicate that ceramide channels are ordered cylindrical structures (3). The conductance induced by ceramide usually increases and decreases in a steplike manner, remaining on a somewhat stable plateau in between events. This is similar to channels formed by proteins or peptides suggesting a fairly rigid structure, unlike typical lipid pores. More interesting, large conductance decrements show a strong preference to be multiples of 4 nS (nanosiemens) in 1.0 M KCl (3). The large size of these channels means that the access resistance dominates (Methods and (14)), and the conductance varies proportionaly to the channel radius or circumference. Following the barrel-stave model of the ceramide channel, the decrements in conductance must correspond to changes in circumference that are multiples of the width of the stave. The multiplicity of 4 nS thus corresponds to the loss of an even number of columns implying the channel is a nearly perfect cylinder. The MD simulations reported here start with this model and explore various aspects including the number of ceramides per column, a parallel versus an antiparallel arrangement, and the degree of order in the cylinder.
METHODS
CHARMM parameters for ceramide
Although the CHARMM 22 force field used for simulations contains parameters for more than a dozen of the most popular lipid molecules, ceramides were not among them. Thus, we had to create the parameters for ceramides anew. CHARMM is an empiric force field, whose parameters are based on extensive experimental data. Parameterization of the ceramide molecule from scratch is a formidable task complicated by limited physicochemical data for ceramide molecules. To our advantage, ceramide molecules are composed of basic fragments that can be found in several molecules already parameterized for CHARMM. Based on the similarity and overlapping parts of the molecular fragments of lysine, serine, POPE, and acetylated N-terminus we have compiled the parameters for ceramide presented in Supplementary Material, Supplement A (Topology and Partial Atomic Charges of the C16 Ceramide Molecule) and Supplement B (Force-Field Parameters for Ceramides). Note that topologies are different for different ceramides due to the differences in acyl chains.
Previously, CHARMM permitted accurate calculations of lateral pressure/tension profiles in phospholipid bilayers (9), and the results obtained with this force field, including the ceramide channels studied here, can be easily compared with other simulations performed with this force field. The same system potentially can be used for further MD studies with proteins, which are thoroughly parameterized in CHARMM. Protein-ceramide pore interactions can be critical for studies of suggested regulation of cytochrome c release through the channel.
Assembly of the simulation system
The channel models were assembled of individual C16 ceramide molecules using WebLab viewerPro 4.0 (Molecular Simulations, Cambridge, UK), VMD (10), and PSFGEN, part of the NAMD package (11). Cylindrical assemblies were oriented to have the symmetry axis coinciding with the z axis of the simulation cell. The length of ceramide columns was either four or six molecules (representing circular layers). The channel arrangement composed of 14 antiparallel columns was used for all-atom simulations in an explicit fully hydrated palmitoyloleoyl-phosphatidylcholine (POPC) bilayer. Models were superimposed on the segment of preequilibrated POPC bilayer and all phospholipids overlapping with the channel-conducting pathway or ceramide headgroup region were removed. Lipids overlapping with the peripheral regions of the hydrophobic ceramide tails (starting from the seventh carbon) were preserved. In total, 56 ceramide molecules composed the four-layer (4L) channel, and 252 POPC molecules formed the bilayer for 4L simulation (84 and 275 molecules respectively for 6L simulation). The system was hydrated with 13,320 water molecules (13,831 for 6L), and 49 Potassium and 49 Chloride ions were added (50 and 50 for 6L) making 200 mM KCl, and bringing the total to 79,706 atoms (87,263 for 6L).
All the simulations were performed using NAMD 2.5 (Theoretical Biophysics Group and the National Institutes of Health Resource for Macromolecular Modeling and Bioinformatics, Beckman Institute, University at Illinois, Urbana-Champaign, Urbana, IL (11)) with CHARMM22 force field (12). Simulations were performed in a flexible hexagonal periodic box as an NPT ensemble at 310 K (Langevin method) and 1 atm pressure (Langevin piston method). The flexible TIP3P water model (13) was used. Electrostatic interactions were calculated using the particle-mesh Ewald method with a grid spacing of ∼1 Å and full direct electrostatics calculation within 10 Å of the atom. Results were analyzed with PDBAN software custom written in MatLab (www.life.umd.edu/biology/sukharevlab/download.htm#PDBAN) and TCL scripts for VMD, and visualized using VMD 1.8.3 (10). Time-dependent trajectories were plotted using MS Excel.
The overlaps between ceramide and POPC were gradually removed using a brief energy minimization (100 steps) first with fixed ceramide atoms allowing phospholipids to relax and avoid most drastic clashes. After that, only headgroups of the ceramide were fixed and the system was minimized for 1000 steps, allowing both C16 tails and POPC molecules to remove the remaining overlaps, and finally the whole system was minimized for 1000 steps without any restraints. The system was then heated to 310 K and simulated for 2 ns with the headgroup and aliphatic double-bond carbons (C1–C5, and C21; see Supplementary Material, Supplement A, Ceramide Topology) harmonically restrained near the modeled positions with a soft spring constant (kf = 1 kcal/mol/Å2). After this restrained simulation, allowing C16 tails and phospholipid membrane to adjust to each other, all the restraints were lifted and each of the systems was simulated for 10 ns.
Mean cross-sectional area and estimated conductivity of the pore
Solvent-accessible surfaces were created for 25 conformations of the ceramide channel simulated in the membrane (taken every 500 ps). The van der Waals (VDW) radii from the CHARMM22 force field and a spherical probe of a 1.4 Å radius were used. A set of cross sections normal to the pore axis was created for solvent-accessible surfaces with a step of 1 Å. Average pore radii were calculated as described in the next section. All calculations were done using custom-written program PDBAN.
Conductance of the channel (G) was calculated taking into account the access resistances (14) and the resistance of the pore itself in a solution of given bulk conductivity g (78.08 mS/cm in our calculations):
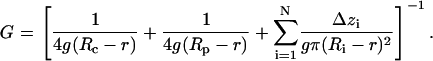 |
The effective radius of the pore (Ri) in a given location along the channel axis (zi) was defined as a radius of the circle equal in area to the corresponding cross section of the solvent-accessible pore surface. Channel pore was represented as a set of N cylindrical slices of thickness Δzi = 1 Å with radii Ri. The effective ion-accessible pore area was determined with a probe of r = 3.42 Å in radius, which approximates a generic ion (2.02 Å, the average between radii of K+ and Cl−) coated with half of its hydration shell (1.4 Å). Access resistances were determined from the effective radii of the upper (Rc) and lower (Rp) pore entrances.
Electrostatic potential inside the channel
To estimate the electrostatic potential in the ceramide channel relative to the bulk of the simulation cell, particle-mesh Ewald potential maps were calculated using the PMEPot Plug-in (Vers. 1.0) for VMD (10). Potential was estimated for every 0.1-ns step over the whole duration of the unrestrained simulations with an Ewald factor of 0.25 A−1. The smallest equipotential surface covering the whole cross section of the water-filled interior of the channel was detected and the value of the potential was used as a characteristic value for the potential inside the ceramide channel.
Computation of numbers of contacts between ceramide, POPC, and water
Contacts between the ceramide, POPC, and water atoms were detected by the average distance criterion −2.3 Å between the water oxygen and any of POPC or C16 atoms, and 2.7 Å for ceramide-POPC contacts. These values for the threshold distance were selected based on the position of the first minimum in the radial correlation functions of all the atoms of water, ceramide, and POPC. Nonpolar ceramide or POPC atoms were defined as atoms possessing a partial charge in the range −0.3 to +0.2. To count water molecules that contact ceramide or POPC only near nonpolar atoms, we estimated the number of water molecules that are located within the threshold distance from any nonpolar atom and do not contain any polar atoms within this distance. The converse was used for waters contacting C16 or POPC only near polar atoms. The formulated criteria are approximate, as they exclude water molecules in contact with both polar and nonpolar groups at the same time. Nevertheless, this approach was fast and effective and proved to be useful in quantifying and tracking time dependences for hydrophilic and hydrophobic surfaces exposed to water.
Number of ions in the channel
Ions occupying the channel were counted within 20 Å from the pore axis, in the range of z coordinate of ±15 Å for 6L and ±10 Å for 4L channel models. A cylinder of this size encompasses the pore itself and the vestibules of the channels, both in the initial state and in the course of simulated transformations of channel shape. Estimations were performed for every 1-ps timestep.
Number of hydrogen bonds and H-bonded ceramide-water-ceramide bridges
Hydrogen bonds maintaining the stability of the simulated ceramide channels were estimated for all the bonding combinations involving hydroxyl and amide groups of C16, and also between these groups and water. H-bonds were defined by the geometric criteria (a donor-acceptor distance of <3.5 Å, and the angle formed by the donor-hydrogen and hydrogen-acceptor lines <35°). Water molecules participating in hydrogen bonding with two and more ceramide molecules were considered to be H-bonded water bridges between the ceramide molecules. If such water molecules possessed more than two H-bonds to ceramide, each possible interceramide bridge was counted, e.g., a water molecule having three H-bonds: one bond with first ceramide and two bonds with second ceramide was considered to bring two H-bridges. Most of the water bridges were single; a small fraction formed double bridges, and practically no water molecules formed three or more bridges simultaneously. H-bridges were counted every 1-ps timestep.
Ordered packing of the ceramide columns
Packing of the individual ceramide molecules in a column was observed to be cooperative, so that typically in the simulations of the channels in POPC bilayer the whole column was either ordered (shorter and uniform distances between the amide groups, tails packed in parallel, as illustrated by the black columns on Fig. 7 A) or disordered (nonuniform distances between the amide groups, randomly spread tails, illustrated by the white column on Fig. 7 A). The column was considered ordered if more than 60% of it preserved the ordered packing. Ordered and disordered columns were counted by visual inspection of the structures every 0.5 ns of the simulation.
FIGURE 7.

Ordered packing of ceramide tails correlates with stability of the columns, and contact of double bond regions stabilizes packing of columns. Snapshot from the end of 6L14C C16 simulation (A) shows that in the most stable columns (black) hydrophobic tails of ceramide are packed in highly ordered crystal-like manner, whereas damaged columns (white) occur where tails are disordered. Stable association of the pair of columns is often maintained by the tight packing of the double-bond regions (black) of the hydrophobic tails (B) and interlayer H-bonding of the hydroxyl groups. Direct intercolumn H-bonding is more extensive over the double-bond–double-bond columns packing than over the amide-amide one (marked by the double arrows d-d and a-a, respectively).
Distances between the nitrogen atoms of the neighboring ceramide columns
These distances were measured every 1-ps step. Only atoms of the closest neighboring molecules that were located within the same layer were considered. Nitrogen pairs for the amide-amide and double bond-double bond interfaces (see Results) were treated separately.
An averaged shape of the ceramide molecules
Coordinates of the ceramide molecules were extracted from the last six nanoseconds of the 6L14C channel simulation in the fully hydrated POPC bilayer. Snapshots were taken every one-nanosecond, producing 588 C16 molecules in total. All the molecules were spatially aligned using the minimal RMSD for all the ceramide atoms as a criterion. Space occupied by a single molecule was defined as a space within its VDW volume (radii from the CHARMM22 force field). Occupied volumes from all the aligned molecules were superimposed and the averaged shape of the molecule was considered to be a space occupied by the molecule with probability >50%.
RESULTS AND DISCUSSION
Ceramide channel models, structure, and bonding
In the original ceramide channel model (Fig. 1 B) (1), the barrel-stave assembly is maintained by vertical hydrogen bonds between the amide groups within the columns and by circular hydroxyl-hydroxyl bonds linking adjacent columns. (Fig. 1, C–E). Since experiments show that both C2 and C16 ceramides can form channels in phospholipid membranes, any of these types of ceramide molecules should be considered as building blocks for the models.
The columns are inherently asymmetrical, and thus, with n columns, the number of possible structures would be 2n (disregarding symmetry considerations) simply by varying the orientation of each column. However, each column should have a strong dipole moment arising from the aligned amide groups forming vertical H-bonds (Fig. 1, D and E). These are located on only one of the two hydrocarbon chains. By pairing columns in an antiparallel fashion, one would gain additional structural stabilization. Thus, two fundamental structures to consider would be an entirely parallel arrangement and a paired antiparallel arrangement (Fig. 1 F). Recordings from the ceramide channels favor channel disassembly in multiples of two columns and thus favor the antiparallel model (3).
Each of the conformations of the ceramide molecule shown in Fig. 1 results in two different curvatures orthogonal to one another. The lengths of interhydroxyl hydrogen bonds and the ceramide headgroup versus tail packing favors negative curvature inside the pore. The width of the ceramide molecule near the circular OH…OH bonds is small (∼5.8 Å) relative to the characteristic size of two packed aliphatic tails (∼8.6 Å); thus, in the x,y plane the assembly would promote a negative spontaneous curvature. In the z-direction, H-bonded amide groups N-H…O=C are longer than the tail thickness; thus, the effective size of the headgroup in this direction is slightly larger than the size of two aliphatic tails oriented in the plane of the membrane. This could force the pore surface to adopt a positive curvature (in x,z plane) creating a continuous boundary with both surfaces of the bilayer. This ability of ceramides to support dual curvature, thus perfectly fitting the headgroup-lined pore in the flat membrane, revealed itself spontaneously in the simulations below.
Another model of the ceramide channel satisfying the barrel-stave principle can be built with different packing of the ceramides. Rotation of every ceramide molecule by 90° around the head-tail axis would result in amide-amide hydrogen bonds forming circles around the pore axis and H-bonds between the hydroxyls maintaining the columns. To satisfy the hydrogen-bond length requirements, that packing would produce narrower and taller channels and the number of layers to span the membrane would be approximately two times smaller (2–3 layers). This type of assembly is expected to be less stable both on geometric grounds and because it has a larger number of hydrophobic ceramide atoms exposed to the aqueous pore. The preferred curvature of individual ceramides assembled this way (positive in x,y plane, negative in x,z plane) is in conflict with the dual curvature of the pore lining (negative and positive, respectively). We will not consider this alternative model further.
The number of ceramide molecules in the column could reasonably range between four and six and still interface well with the phospholipid bilayer. More than six layers would introduce apolar ceramide tails into the polar headgroup region, while less than four layers (∼18 Å) seem to be insufficient to cover most of the hydrophobic core of the membrane resulting in strong bending of the membrane rim. Thus, tests for spatial compatibility of the channel assembly with the lipid bilayer were performed with four- and six-layer models in the explicit membrane.
Another parameter, which can be varied significantly in the models, is the number of columns in the barrel, which defines the size of the channel pore (in the assumption that the pore is round or of some regular shape). From bilayer experiments, ceramide channels range in conductance from <1 nS to ∼200 nS in 1 M KCl. That corresponds to a channel composed of just a few columns to over 70 columns. In mitochondria, the observed cutoff for the size of proteins passing through the ceramide channel was 60 kDa, corresponding to a pore diameter of ∼6 nm or ∼40 columns. As the channel increases in size, the effective packing angle between columns changes, as does the radius of curvature. Thus, inevitably, the detailed structure must change. Although limited by computation time to simulating relatively small channels, for the full-scale computation we chose a structure of 14 columns and 2.4 nm in internal diameter that is comparable to the size of cytochrome c.
All-atom simulations of ceramide channels in the fully hydrated POPC bilayer
A ceramide channel consisting of 14 antiparallel columns was chosen because its inner diameter is comparable with the size of cytochrome c, but the size of the system still permits multi-nanosecond all-atom simulations in an explicit fully hydrated bilayer. We selected a channel formed by C16 ceramide because naturally occurring ceramide exists with two long chains. Both four- and six-layer variants of the channel were simulated.
Upon embedding the ceramide ring into lipids, bad molecular contacts were resolved within 1000 minimization steps. The initial two nanoseconds of simulations were conducted with ceramide headgroups restrained, allowing formation of the ceramide-POPC boundary. This was followed by 10-ns unrestrained simulations to observe the channel dynamics. Both four- and six-layer channels remained relatively stable during 10 nanoseconds of simulation. Since most of the estimated parameters stabilized during the first three nanoseconds of the simulation, this indicates likely stability on a longer timescale.
Dynamics of the phospholipid pore: a control without ceramide
To have a negative control for stability we used a lipid system of comparable configuration and size, which could give us a lower boundary of the collapse timescale. A POPC bilayer having a cylindrical perforation (hydrophobic pore) instead of a ceramide channel was simulated under similar conditions. All the lipids within a cylinder of 24 Å in diameter were removed and the perforation was filled with water (Fig. 2 C). The size of the pore was comparable to the size of the water pathway in the 14-column ceramide channel models. Rapid (hundreds of picoseconds) constriction of the pore was observed, mostly due to protruding POPC acyl chains from the middle of the membrane into the pore lumen, and (to a smaller extent) due to the bending of the choline groups of POPC. The expansion profile of the bilayer edge was similar to what would be expected considering the lateral pressure profile for a POPC bilayer (9). After ∼1.5 ns, the hydrated pathway constricted down to a single column of water molecules at the level of the middle of the membrane, and near 1.8 ns the water pathway was interrupted, thus resealing the perforation completely (Fig. 2 D). During the following 1–1.5 ns, full contact between the headgroups was also restored. The system was simulated for an entire four nanoseconds.
FIGURE 2.

Adaptation of the lipid bilayer with the incorporated ceramide channel versus quick collapse of the introduced bilayer puncture. (A) Cross section of the initial all-atom simulation cell at the beginning of the heating process. Columns of fatty tails of the C16 ceramide channel (yellow) interdigitate with the tails (cyan) of the POPC bilayer. There are initially hydrophobic tail atoms exposed to water (thin blue lines) in a rectangular gap between the polar headgroups of POPC and C16. Chloride and potassium ions (orange and green VDW spheres, respectively) were distributed randomly in the bulk water. Snapshots at the end of 10-ns simulation of the six-layer (B) and four-layer (E) 14-column models show the water pathway that adopted an hourglass shape. A characteristic segregation of aliphatic tails and the continuous lipid-water boundary lined with polar groups are observed. The bilayer accommodated the channel by bending and conformational adjustment of the phospholipids (see also Fig. 4). When, instead of the ceramide channel, a round water-filled puncture of comparable size (20 Å in diameter) was introduced into the POPC membrane (C), after ∼1.8 ns of simulation under the same conditions lipid tails of the opposite walls of the puncture fused together, interrupting the water pathway (D).
For systems with a less pronounced instability, the characteristic time of degradation would be significantly longer. Collapse of inverted phospholipid pores of a similar size, where the water pathway is lined by hydrophilic headgroups, takes an order-of-magnitude longer (∼20 ns (15)), but significant transformations and the general tendency are already visible within 10 ns. Thus, the ceramide channel structure does not represent a very unstable structure, because no significant tendencies to collapse were observed within the timescale of our simulations.
Channel-bilayer contact
In the preparation of the simulation cell, an important and difficult issue was building the interface between the ceramide channel and phospholipid membrane. As was described in Methods, phospholipid molecules were removed from the region overlapping with the water pathway and headgroups of the ceramide channel but were preserved in the region overlapping with the peripheral two-thirds of the hydrophobic C16 tails. To remove the overlap between the POPC and C16 atoms, the system was subjected to energy minimization. During this process only the ceramide headgroups were restrained in their initial positions, allowing both C16 tails and whole POPC molecules to relax. During the 1000 steps of energy minimization most of the VDW conflicts were resolved with an average ∼3 Å displacement of atoms. Interdigitation of POPC tails and C16 columns remained after the minimization.
After the minimization stage, two-nanosecond simulations with partial restraints were performed where six carbons of the ceramide headgroup (atoms C1–C5 and C21) were harmonically restrained near their initial positions, so that hydroxyl groups and apolar tails could rearrange. During the first 500 ps of the restrained simulations most of the POPC tails segregated from the C16 ring, in this manner removing the interdigitation of tails (Fig. 2 A), and shaping the external surface of the ceramide channel into an oblate shape (Fig. 3; in cross section shown in Fig. 2 B). This resulted in a sharp interface between ceramide and POPC tails as indicated. The time course of the segregation can be illustrated by the plot of the number of tight contacts between the POPC and ceramide tails (Fig. 4 A) for both six- and four-layer ceramide channels. This arrangement of the interface appears to be rather stable since neither the general external shape of the channels nor the number of hydrophobic POPC-C16 contacts changed during the following unrestrained simulation.
FIGURE 3.

Conformational adjustments in the POPC molecules surrounding the 6L14C C16 channel. Hydrocarbon tails of POPC (cyan) are mostly expelled from the ceramide tails (yellow). Following the lenslike shape of the ceramide channels, the outer half of the fatty chains of annular POPC reaches an angle of ∼60° to bilayer normal, whereas polar POPC headgroups (blue is nitrogen, red is oxygen, tan is phosphorus) more often orient normal to bilayer and maintain the contact with the rest of the membrane headgroup region. Their attachment to the ceramide hydroxyl and amide groups is maintained through the H-bonding with oxygens belonging to glycerol or carbonyls groups (frame 2), and/or phosphate of POPC (frame 3). A smaller fraction (∼30%) of phospholipids have choline groups in tight contact with ceramide (frame 1).
FIGURE 4.

The time course of the POPC-ceramide-water system development. (A) Number of contacts between the hydrophobic tails of C16 and POPC. For all the plots, estimations were performed every 10 ps of simulation, except for F where pore conductivity was calculated every 500 ps. All the parameters in plots A, D, G–L are recalculated per one ceramide molecule, and in plots B and C per one ceramide in the outer layer (28 molecules per channel). Solid and shaded lines represent simulations of 6L14C and 4L14C C16 channel, respectively. Time from −2 to 0 ns represents the bilayer equilibration stage with carbons of ceramide headgroups (atoms C1–C5 and C21) restrained harmonically in the positions of the energy-minimized structure. Black and shaded circles at the time −2 ns represent the initial value of the parameter after the energy minimization. (B,C) Number of water contacts with the nonpolar atoms of ceramide (B) and annular POPC (C). (D) Number of contacts of the hydrophilic ceramide atoms with water. (E) Number of ions in the channel. (F) Estimated ion conductance of the water pathway in the channel. (G) Number of ceramide-water hydrogen bonds. (H) Number of ceramide-water-ceramide H-bond bridges. (I,J) Number of ceramide H-bonds between the amide groups (I) and between the hydroxyl groups (J). (K) Total number of ceramide-ceramide H-bonds. (L) Total number of ceramide-ceramide H-bonded connections, including direct hydrogen bonds and H-bonded C16-water-C16 bridges. See Methods for parameter calculation details.
Instead of being normal to the membrane plane and interdigitating with aliphatic C16 chains, as in the starting configuration, the expelled tails of POPC tilted and bent around the newly formed lenslike ceramide assembly. Methyl ends of fatty acid chains were close to the normal orientation, but near the carbonyl group the chains were tilted by ∼60° from the original orientation (Fig. 3). During this mutual adjustment process, tight contacts between the ceramide and POPC tails developed, and nearly all hydrophobic regions contacting with water, for both 6L and 4L assemblies, were eliminated. The number of contacts of the hydrophobic tails with water gradually decreased by ∼40% for ceramide (Fig. 4 D) and by ∼20% for POPC near the channel (Fig. 4 C). A comparison between the 6L and 4L models shows that the degree of hydrophobic exposure in ceramide molecules is similar for both models whereas the exposure of annular POPC molecules is smaller for 6L model, suggesting a better match of the channel height to the membrane thickness and less curvature perturbation for each leaflet of the bilayer. Nevertheless, the timescale of the decrease of hydrophobic surface exposure is on the order of several nanoseconds, which is approximately one order-of-magnitude slower than the segregation of the ceramide tails. This timescale, and the timescale of the hourglass channel shaping (see below), suggests that the driving force for the fast chain demixing and formation of C16-POPC interface in the simulations is not mainly due to the hydrophobic interactions, but rather the entropic characteristics of tails packing and the viscoelastic properties of the membrane.
Headgroups of POPC contacting the channel were mostly found in two different configurations. In one of them (approximately one-third of the molecules) the whole headgroup including the choline part is tightly attached to C16 polar atoms (Fig. 3, frame 1). In the other configuration (approximately two-thirds) the headgroup is oriented normal to the bilayer and maintains strong bonding to the rest of the phospholipids, whereas contacts with ceramides are mediated mostly by oxygens of carbonyl (Fig. 3, frame 2) and phosphate groups of POPC (Fig. 3, frame 3). Among the annular lipids, ∼47% (in both 4L and 6L simulations) had polar atoms of the headgroup in direct contact (within 5 Å) with polar atoms of C16 capable of H-bonding, and the rest of POPC molecules had water mediating this contact. After equilibration, the choline, phosphate, and carbonyl groups each provided approximately one-third of the direct polar contacts between the headgroups in both simulations. In 58% of molecules, only one of these three contact types was present in the direct interaction; in 26%, two types simultaneously; and in 17%, all three POPC groups were involved for the 4L model (50, 33, and 17% for 6L).
In the course of the initial restrained simulation, water-exposed surfaces of the ceramide channel smoothened, eliminating the orthogonal orientation of the C16 ceramide and lipids of the initial assembly (Fig. 2 A). The resultant inner surface of the pore is lined primarily by ceramide hydroxyls, leaving space for only a few apolar atoms exposed to water (Fig. 2, B and E). Both surfaces of the membrane converge in the midplane of the ceramide pore, forming one smooth and continuous boundary with a radius of curvature of ∼20 Å. This curvature was the same for both the four- or six-layer channels. The only difference was that for the 4L channel the boundary covered by phospholipids continued further into the vestibules, so that the thinner channel ring could be accommodated. The lenslike shape of the ceramide channel maintained a tight and highly ordered crystal-like packing of the ceramide hydrophobic tails in 43% of the molecules near the headgroup, while the rest (57%) were disordered. As will be shown below, the aliphatic chain packing correlates with the column stability.
The hourglass-shaped conductive pathway
The simulation transformed the headgroup region of the ceramide channel from cylindrical to hourglass-like shape, in both 6L and 4L simulations (Figs. 2 and 6). This was the result of a slight expansion of the vestibules, mostly during the first three nanoseconds of unrestrained simulation. An hourglass-like shape is essential for the generation of a continuous smooth membrane-water boundary between the pore and the lipid bilayer. The surface of the ceramide channels is characterized by a dual curvature, and this duality arises from the fundamental structure of the ceramide molecule described above. To illustrate the average shape that ceramides adopt when forming the channel, we extracted C16 coordinates every one-nanosecond from the last six nanoseconds of the 6L14C simulation, when the geometry of the pore was already stable. After aligning the molecules with each other (see Methods), the relative spatial distribution of the ceramide atoms was calculated. An averaged shape of the molecule can be roughly characterized by the surface that encloses the space occupied with a probability of 50% or more (Fig. 5 A). The shape has double-wedge characteristics with the headgroup being narrower but taller than the tails. The stiff core of the molecule (probability >80%) has a similar shape, revealing the higher stability of the headgroup and frequent contacts between the two hydrocarbon tails at the position behind the double bond and the amide group. This averaged shape can be fitted easily both into the circular layer with negative curvature (Fig. 6 B) and the bent column with positive curvature (Fig. 6 C).
FIGURE 6.

Dipole-dipole interactions and hydrogen bonding in the simulated ceramide channels. Snapshots of the 4L14C (A) and 6L14C (B) C16 simulations in the membrane show that in the stationary phase approximately one-half of the columns preserve the antiparallel orientation of column dipoles arising from the H-bonded amide groups. The original regular hydrogen-bonding pattern (C) randomizes quickly, and on average, in the 6L14C C16 simulation approximately one-third of the bonds are dynamically maintained (D). A new pattern of hydroxyl bonding often appears, linking both layers and columns. Water (green) participates in the interceramide hydrogen bonding, acting as a bridge, and partially compensating for the lost bonds. Amide-amide and double-bond–double-bond contact interfaces between the neighboring columns are marked by the double arrows a-a and d-d, respectively.
FIGURE 5.

An averaged shape of the ceramide molecule and tendency toward surfaces with dual curvature. Ceramide molecules from the last six nanoseconds of 6L14C simulation were aligned and overlapped as described in Methods. The averaged shape of the molecule (A) is illustrated by the cyan surface that encloses the space where C16 atoms are found with a probability of >50%. The dark blue core visible inside shows the most rigid zone occupied with probability >80%. Fourteen alternating contours of the top projection (in the plane of the membrane) of the averaged surface are arranged into a ring (B) with negative monolayer curvature. Six side-projection contours are assembled as a bent column with positive curvature lining an hourglass-shaped conductive pathway (C).
In addition to the persisting hourglass-like shape, the stability of the conducting pathway is also reflected by the pore occupancy with water and ions. Both channels had very similar conductance and this conductance was stable with time (Fig. 4 F), showing only periodic fluctuations.
The number of potassium and chloride ions in the simulation cell corresponded to a 200 mM salt concentration. Ions were initially placed randomly into the bulk of water outside of the pore. They gradually diffused into the water pathway of the channel (Fig. 4 E), reaching an ionic strength comparable to the bulk. According to experimental observations, a small ceramide channel displays a weak preference for cations (−30 mV reversal potential, six cations per one anion, per 10-fold KCl gradient for ∼1 nS conductance, corresponding to a 6–8 column channel). The cation selectivity (reversal potential) drops sixfold when the channel diameter increases twofold, reaching an estimated size comparable with the models in this simulation. Thus one should expect a virtually nonselective channel. However, the simulated channels of this size preferred anions to cations (Fig. 4 E). The abundance of anions in the channel can be explained by the positive potential (0.6 V and 0.9 V for 4L and 6L simulations, respectively, at the beginning of the simulation) generated in the channel due to the specific orientation of the ceramide molecules and attenuation by oriented water dipoles. Once the ions diffused into the pore, the positive potential was compensated down to 0.3 V and 0.6 V for 4L and 6L, respectively (an average over the last five nanoseconds).
The electrostatic potential of the ceramide molecule itself shows the most positive region near the hydrogen of the amide group and the negative region is distributed mostly near the oxygens of hydroxyl groups and the amide oxygen. Smaller fractions of the negative potential are spread between the methyl ends and the middle of the aliphatic tails but ceramides arrange in the channel in such a manner that their individual potentials superimpose and mostly cancel out. This still results in a net positive potential in the pore. Our preliminary studies with different modifications of the barrel-stave arrangement indicate that the sign and magnitude of this potential depends on the conformations of the ceramides, and more critically on the orientation of the hydroxyl dipoles. Minor rearrangements without changing the general architecture were able to produce the channels with negative potential inside. Thus, a slight change in the headgroup conformation may explain the difference in selectivity.
Hydrogen bonding and interactions with water
A significant increase in the amount of water intercalated between ceramide headgroups was observed in both models of the ceramide channel. On average, the hydration shell of the 4L and 6L channels stabilized after approximately two nanoseconds at the level of three water molecules per one C16 headgroup (Fig. 4 D), but since some water molecules are located between two ceramides the number of water contacts is actually larger and reaches ∼4.5 per one C16 molecule.
On average, 67% of the water molecules contacting a ceramide headgroup form hydrogen bonds with it (Fig. 4 G). Each headgroup forms approximately three H-bonds with water and participates in two hydrogen bonds with other ceramide molecules (Fig. 4 K). Since each C16 can potentially have up to nine H-bonds, ∼56% of its bonding capacity is satisfied. (This actual number is slightly higher since we do not consider H-bonding of the two boundary layers with phospholipids.)
Thirteen percent of water molecules H-bonded to the ceramide have more than one hydrogen bond (90% of such molecules have two, and ∼10% have three bonds). Of the water molecules with multiple H-bonds, 8% are bonded with the same ceramide molecule and 92% bridge two or more ceramide molecules. At the beginning of the simulations, ceramide headgroups were more densely packed and the extent of hydration was low; most of the observed H-bonded water bridges had a structure HC16-Owater-HC16. However, during the stationary phase of simulation, most of the bridges had a longer structure HC16-OHwater-OC16. Fig. 6 D illustrates the participation of water bridges in the ceramide bonding network. One can see that more bridges occur near the vestibules (five on the top and five on the bottom) than in the narrower middle of the channel (one bridge). Switching toward the longer conformation of water bridges reflects an increase of the width of channel vestibules during the simulation and a corresponding water adjustment such that longer water bridges can fit better in the increased gaps between the headgroups. In this case, water acts as a structural element in the channel wall, partially compensating for the intercolumn H-bonding loss in the channel rims (vestibules) and maintaining the channel stability. The decrease in the number of the direct interceramide hydrogen bonds during the first three nanoseconds of the simulations (Fig. 4 K) is paralleled by the increase in the number of H-bonded water bridges (Fig. 4 H), so that the total amount of hydrogen-bonded connections between ceramide molecules remains nearly constant (Fig. 4 L). Involvement of water in the formation of interceramide contacts correlates with the significantly restrained water dynamics in ceramide tetrasaccharide vesicles, shown to be dependent on the character of lipid packing (16).
The amount of direct hydrogen bonding between both the hydroxyl (Fig. 4 I) and the amide (Fig. 4 J) groups of ceramide decreases with time. By the end of the simulation, it was approximately one-half that of the restrained simulation or approximately one-third of the number of H-bonds in the initial model. The fast drop in the number of hydrogen bonds during the first 100 ps of the restrained simulation reflects the impact of the local thermal disorder compared to the frozen model structure, whereas a slower decrease after the beginning of unrestrained stage clearly reflects rearrangements in the ceramide packing. Interhydroxyl H-bonds decrease faster and stabilize after one nanosecond of unrestrained simulation, whereas interamide bonds decrease more slowly, stabilizing only after five nanoseconds. A small apparent fraction of amide-hydroxyl ceramide H-bonds slightly compensates for this decrease, so that the total number of direct interceramide bonds decreased ∼1.4 times relative to the restrained simulation and 2.4 times compared to the model. In the course of unrestrained simulation, the extent of hydrogen bonding stabilized after ∼1.5–2 ns.
Hydrogen bonds between the OH groups within the same ceramide molecule were initially present in one-half of the molecules in the original 14-column antiparallel model (and in all molecules in the models with parallel columns). During the 4L and 6L simulations, the fraction of ceramides possessing an intramolecular H-bond decreased 2.5 times, i.e., to ∼20%. Usually only more isolated and disordered molecules on the periphery of the channel had the internal bond, while the molecules closer to the middle of the channel used all their bonds in the interactions with other ceramides or water (Fig. 6 D).
The estimated numbers of interceramide and ceramide-water hydrogen contacts are very similar in 6L and 4L channels, suggesting that both assemblies reached similar local structures. Nevertheless there were differences in the distribution of hydrogen bonding. Packing of the molecules was more stable in the 6L channel, providing a more stable core with a higher density of H-bonds. However, due to the hourglass shape adopted by the longer 6L channel, the columns split further at the rims and lost part of their intercolumn H-bonding there. Therefore the net number of hydrogen bonds per one ceramide in the 6L channel became approximately equal to those in the 4L channel.
Stability of ceramide columns
The stability of the ceramide column arrangement is determined both by the hydrogen bonding between the amide groups and by cooperative dipole-dipole interactions. Visual inspection of the snapshots at the end of simulation (Fig. 6 B) shows that ∼50% of dipoles in 6L channel preserve their consistent cooperative orientation or preserved this orientation in fragments of columns.
Among the disordered molecules, a small fraction restored pairwise amide-amide contacts in the horizontal direction (parallel to the membrane plane). This can be observed more often in the 4L assembly. Nevertheless, these structures were unstable and there were no circular in-plane assemblies formed larger than a pair. At the same time, it was also frequently observed that the peripheral ceramide molecules switched from one column to another, instead of simply dissociating from the channel rim. This confirms the theoretically expected stability of the amide-amide type of column assembly (vertical column rather that horizontal circle) as a structural element of the channel wall. Preferential amide dipole-dipole orientation and columnar packing are clearly seen in these simulations. The persistence of the antiparallel columns was higher in the 6L ceramide channel, compared to the 4L channel.
During the 10 nanoseconds of simulation, some of the interhydroxyl H-bonds, originally connecting adjacent columns within one layer of the channel, formed H-bonds between layers (Fig. 6 D). However, the whole network of hydrogen bonds is dynamic, maintaining approximately one-half of the originally modeled bonds at any time (Fig. 4 L).
A strong correlation exists between the ordering of the ceramide tails and column stability. The columns that were preserved throughout the 10-ns simulation of the 6L ceramide channel all maintained tails in a crystal-like pattern (Fig. 7 A, black), excluding intermixing with POPC tails. Columns showing greater disorder had tails that were spread randomly and noticeably intermixed with the acyl chains of phospholipids (Fig. 7 A, white). At this stage it is speculative to suggest cause and effect. A comparison of the dynamics of individual molecules of ceramide versus POPC shows that on average hydrocarbon chains of the ceramide stay closer to each other (as defined by the distance between the first carbons of the tails), in accordance to experimental observations of the restrained dynamics of ceramide acyl chain in glycolipid-phospholipid bilayers (17). The tight packing can enhance interactions between the chains within the same column. An essential feature of the ceramide molecule, which may allow for the ordered aggregation of molecules and stable column formation, is that the amide group mediating the polar intermolecular contacts is located directly at the hydrocarbon chain that participates in the tail packing, thus making these two types of intermolecular interactions highly cooperative and codirectional. Unlike the ceramide, most of the polar atoms of the POPC headgroup are more distant from the tails and are connected to them in a very flexible manner, thus allowing the direction of the polar interaction to be uncoupled from the packing of its accompanying tail with adjacent hydrocarbon chains.
An additional factor in stabilizing intercolumn packing is the nature of the interface. The antiparallel arrangement produces two types of interfaces: one in which the double bonds interact and the other where the dipoles of the amide linkages interact (marked by d-d and a-a arrows, respectively, on Figs. 6 and 7). In the simulation the most stable packing of the hydrophobic tails was observed on the double-bond–double-bond type of the interface (Fig. 7 B, black rods). In the course of thermal fluctuations, pairs of columns contacting over this interface moved as a single domain. As a result, an average distance between the nitrogen atoms of the neighboring columns across the double-bond interface was stable over the course of the entire simulation; over the last five nanoseconds, it equaled 9.7 Å and 9.5 Å for 4L and 6L simulations, respectively (close to the value of 9.3 Å at the beginning of unrestrained simulations). Individual pairwise distances varied in time with standard deviations of 1.05 Å and 1.15 Å, respectively. Stability of this type of the interface may suggest why the double bond restraining the chain mobility is essential for channel formation. This interface was also stabilized by more extensive hydrogen bonding (Fig. 7, B and D). In contrast, the amide-amide interface displayed more rounded contact surfaces, appeared to be more flexible, and exhibited a lower degree of tail packing as compared to the double bond interface. Most of the slippage of the neighboring pairs of columns over the time occurred at the amide interface. An average distance between the nitrogen atoms of the neighboring columns here increased from 5.4 Å to 9.8 Å for 4L simulation (5.3–8.9 for 6L) during the first five nanoseconds, and then reached plateau. Comparing to the double-bond interface, the amide contact displayed a larger standard deviation (34% and 14% for 4L and 6L, respectively) of individual pairwise distances over the last five nanoseconds. At the same time, the amide interface may be stabilized by longer-range electrostatic interactions between the column dipoles as opposed to the shorter-range VDW interactions and hydrogen bonding of the double-bond interface. This long-range and low angular dependence of the amide interface is consistent with its higher flexibility and may be necessary for the process of insertion/removal of columns from the channel barrel. This higher flexibility could allow for energetically favorable barrel-stave assembly over a wide range of channel diameters. The presence of two types of interfaces with different structural characteristics correlates with the preference for an even number of columns in channel disassembly.
Self-organization of channels, assembly, and disassembly
Despite the general stability of the simulated channel assemblies as judged by the geometry of the water-conducting pathway and estimated channel conductance (Fig. 4 F), we observed certain tendencies for channel disassembly in both all-atom simulations, mostly in the more fragile 4L structure. The first tendency was a simple separation of several of the most external ceramide molecules from the body of the channel, usually followed by their reattachment to the same or the neighboring column (in the last case, the dent in the channel wall was filled by a POPC molecule). Since the simulated pure POPC bilayer did not have dissolved ceramides, the normal dynamic equilibrium between monomers in the channel and on each monolayer should result in a tendency for disassembly under the conditions used. In real experiments the monolayers have an estimated 4% content of ceramide (1). (This means that 11 out of 275 POPC lipids should be replaced by ceramides.) Thus, these separation events may simply represent a tendency toward dynamic equilibrium.
The second and more prevalent mechanism of disassembly was observed in the simulation of the 4L structure. It was a gradual sliding (splitting) of a partially ordered pair of ceramide columns. This supports the mechanism of channel disassembly suggested by experiments. The breach in the channel structure was filled by POPC molecules, thus preserving the pore geometry. Since this slow process was not finished within the simulation time it is not clear if the contact between the remaining ceramide columns would have reformed, or if POPC molecules would have continued to fill the gap, or if this sliding of columns would occasionally lead to the collapse of the remaining part of the 4L channel. We did not observe a clear tendency to splitting or sliding out of pairs of columns in simulations of the more stable 6L assemblies in POPC. Only the very beginning of splitting of one column was registered. Longer simulations would be required to characterize the equilibrium and dynamics of assembly/disassembly processes.
CONCLUSIONS
Molecular dynamics simulations described above revealed several important issues related to the models of ceramide channels and their stability. The simulations have emphasized the key structural features of individual ceramide molecules allowing them to interact specifically with other ceramides and in a less specific fashion with phospholipids that accommodate ringlike ceramide assemblies in the bilayer structure. Both 4L and 6L ceramide channels in the explicit membrane were relatively stable on a 10-ns timescale. Their properties stabilized in the first three nanoseconds, indicating the formation of a persistent structure. In the course of 10-ns simulations, the six-layer (6L) channel assemblies displayed higher stability, whereas the 4L channel displayed a tendency to disassemble in a manner consistent with the previously proposed barrel-stave mechanism.
The factors contributing to the stability of the simulated channel models can be summarized as follows:
Amide-hydrogen bonding and cooperative tails' packing keep the molecules in columns.
Double-bond–double-bond packing and the interlayer-intercolumn pattern of hydrogen bonds keeps the pairs of antiparallel columns together.
Long-range electrostatic interaction between the column dipoles, circular hydrogen bonding between hydroxyls near the middle of channel, and water bridges in the funnel-like channel vestibules keep pairs of columns assembled in the channel.
Long-range dipole-dipole interaction can also slightly stabilize pairs of columns. After initial equilibration, the width of the water-conducting pathway was stable throughout the simulation, and the structure of the channel was secured by a dynamic network of hydrogen bonds, maintaining a stable number of H-bonds at approximately one-half the maximal number.
Simulations revealed the overall oblate shape of ceramide inclusions in planar phospholipid bilayers and suggested the nature of the contact zone and the character of intermolecular interactions between the surrounding phospholipid and ceramides. Tilted ceramides formed boundaries with tilted phospholipids connecting the channel with each of the leaflets of the membrane. The mutual fit of the two components produced a smooth rounded surface of the transmembrane pore, which is continuous with the rest of the bilayer. After mutual adjustment, no significant intermixing or interdigitation of the ceramide and POPC acyl chains was observed. Packing of the separated ceramide tails into the lenslike structure and an hourglass shape of the aqueous pathway were typical characteristics of the equilibrated channel model. The dual curvature of the assembled channels, negative in the x,y plane and positive in the z-direction, reflect the fundamental underlying properties of the ceramide monomers and its hydrogen bonding with adjacent monomers.
The observed anionic preference of the simulated ceramide pores is the only marked deviation from the experimentally observed weak cationic selectivity. Our preliminary data shows that small adjustments of the headgroup orientation can change the electric potential distribution inside the pore without changing the general architecture of the channel. The search for other possible headgroup conformations is in progress.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant No. NS42025.
Abbreviations used: C2, N-acetyl-D-erythro-sphingosine; C16, N-hexadecyl-D-erythro-sphingosine; 6L14C, ceramide channel containing six layers and 14 antiparallel columns of C16 ceramide molecules; 4L14C, ceramide channel containing four layers and 14 antiparallel columns of C16 ceramide molecules.
References
- 1.Siskind, L. J., and M. Colombini. 2000. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J. Biol. Chem. 275:38640–38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siskind, L. J., R. N. Kolesnick, and M. Colombini. 2002. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 277:26796–26803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siskind, L. J., A. Davoody, N. Lewin, S. Marshall, and M. Colombini. 2003. Enlargement and contracture of C2-ceramide channels. Biophys. J. 85:1560–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montes, L. R., M. B. Ruiz-Arguello, F. M. Goni, and A. Alonso. 2002. Membrane restructuring via ceramide results in enhanced solute efflux. J. Biol. Chem. 277:11788–11794. [DOI] [PubMed] [Google Scholar]
- 5.Chiu, S. W., S. Vasudevan, E. Jakobsson, R. J. Mashl, and H. L. Scott. 2003. Structure of sphingomyelin bilayers: a simulation study. Biophys. J. 85:3624–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niemela, P., M. T. Hyvonen, and I. Vattulainen. 2004. Structure and dynamics of sphingomyelin bilayer: insight gained through systematic comparison to phosphatidylcholine. Biophys. J. 87:2976–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tieleman, D. P., H. Leontiadou, A. E. Mark, and S. J. Marrink. 2003. Simulation of pore formation in lipid bilayers by mechanical stress and electric fields. J. Am. Chem. Soc. 125:6382–6383. [DOI] [PubMed] [Google Scholar]
- 8.Tieleman, D. P. 2004. The molecular basis of electroporation. BMC Biochem. 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gullingsrud, J., and K. Schulten. 2004. Lipid bilayer pressure profiles and mechanosensitive channel gating. Biophys. J. 86:3496–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 11.Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- 12.MacKerell, A. D., D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 14.Hall, J. E. 1975. Access resistance of a small circular hole. J. Gen. Physiol. 66:531–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marrink, S. J., E. Lindahl, O. Edholm, and A. E. Mark. 2001. Simulation of the spontaneous aggregation of phospholipids into bilayers. J. Am. Chem. Soc. 123:8638–8639. [DOI] [PubMed] [Google Scholar]
- 16.Arnulphi, C., C. A. Martin, and G. D. Fidelio. 2003. Mixed lipid aggregates containing gangliosides impose different 2H-NMR dynamical parameters on water environment depending on their lipid composition. Mol. Membr. Biol. 20:319–327. [DOI] [PubMed] [Google Scholar]
- 17.Fenske, D. B., K. Hamilton, H. C. Jarrell, E. Florio, K. R. Barber, and C. W. Grant. 1991. Glycosphingolipids: 2H NMR study of the influence of carbohydrate headgroup structure on ceramide acyl chain behavior in glycolipid-phospholipid bilayers. Biochemistry. 30:4503–4509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.