

Abstract
We have conducted molecular dynamics simulations to gain insight into the atomic-scale properties of an isotropic system of cholesteryl oleate (CO) molecules. Cholesteryl esters are major constituents of low density lipoprotein particles, the key players in the formation of atherosclerosis, as well as the storage form of cholesterol. Here the aim is to clarify structural and dynamical properties of CO molecules under conditions, which are suggestive of those in the core of low density lipoprotein particles. The simulations in the fluid phase indicate that the system of CO molecules is characterized by an absence of translational order, as expected, while the orientational order between distinct CO molecules is significant at short distances, persisting over a molecular size. As for intramolecular properties, the bonds along the oleate chain are observed to be weakly ordered with respect to the sterol structure, unlike the bonds along the short hydrocarbon chain of cholesterol where the ordering is significant. The orientational distribution of the oleate chain as a whole with respect to the sterol moiety is of broad nature, having a major amount of extended and a less considerable proportion of bended structures. Distinct transient peaks at specific angles also appear. The diffusion of CO molecules is found to be a slow process and characterized by a diffusion coefficient of the order of 2 × 10−9 cm2/s. This is considerably slower than diffusion, e.g., in ordered domains of lipid membranes rich in sphingomyelin and cholesterol. Analysis of the rotational diffusion rates and trans-to-gauche transition rates yield results consistent with experiments.
INTRODUCTION
Cholesteryl esters (CEs) are lengthy molecules where a long fatty acyl chain is attached to the OH group of cholesterol. Practically, cholesteryl ester is the extracellular transport and intracellular storage form of cholesterol, containing mainly linoleate and oleate fatty acids in these functions, respectively (1). In the intravascular transport, cholesteryl esters are formed with the help of lecithin-cholesterol acyl transferase enzyme to carry cholesterol in a more hydrophobic form in the nonaqueous interior of lipoprotein particles, mostly low density lipoprotein (LDL) entities. LDL particles are known as key players in the formation of atherosclerotic lesions in the arterial intima because of the propensity of their modified forms to accumulate extracellularly in the form of small lipid droplets (2). Speculations have risen whether the composition of the LDL core would affect the pathological fate of the LDL particles through the differences in the physical state of the core and possibly the whole particle ((3–5), and references therein). In addition, the importance of CEs in the pathology of atherosclerosis is underlined by the fact that CEs are the major component of atherosclerotic plaques (6).
Due to the major role of cholesteryl esters in the formation of atherosclerosis, a lot of reports of the properties of cholesteryl esters have appeared (7). At low temperatures cholesteryl esters form liquid-crystalline smectic or cholesteric phases and, in proper conditions, crystals as well. In fact, atherosclerotic plaques have been suggested to contain crystalline, liquid-crystalline, and liquid domains (8,9). However, aside from the final pathological accumulation to the lesions, the isotropic liquid phase characteristic to high temperatures is abundant in the LDL core ((4), and references therein). The isotropic phase of cholesteryl esters has been studied experimentally at the molecular level mostly by 2H NMR, 13C NMR, x-ray, and neutron diffraction. The isotropic phase, at temperatures relatively close to the phase transition temperature, is suggested to be characterized by groups of parallel molecules and merely extended conformations (10,11). In addition, relatively strong intermolecular interactions between unsaturated CEs have been suggested to reduce the motions of the fatty acyl chains between the ester and the double bond, whereas the mobility has been found to increase toward the chain ends (12,13).
As for computational studies of lipoproteins, there are a few recent studies on the apolipoprotein A-1 surrounding a lipoparticle (14–16). In these cases, however, one did not focus on the core of the particles, which further was comprised of phosphatidylcholine molecules rather than cholesteryl esters. Hence, rather surprisingly, although the properties of cholesteryl ester systems have been widely explored through experimental studies, it seems evident that there are no prior studies of CEs through atomistic molecular dynamics (MD) simulations.
Yet, in recent years, the amount and quality, as well as the scope of different applications in the field of atomic-scale simulations of lipid systems has increased rapidly (17–20). The bilayers of saturated molecules such as dipalmitoylphosphatidylcholine are probably the most common objects of MD simulations of lipid systems, because the development of the methodology and the force fields has required solid experimental data to be available to allow comparison. Fortunately, the importance of MD simulations as a tool to enhance our insight of physiologically relevant molecular systems is emerging and many important lipid species have already been incorporated to simulations. Studies of naturally unsaturated phosphatidylcholine and sphingomyelin bilayers, cholesterol-phospholipid mixtures, and charged membranes are topical examples of the recent progress in the field (21–28). For comparison, the cholesteryl esters are a new type of lipid to be simulated, as they do not form bilayers but isotropic continuous environments.
Here, a molecular system of 128 cholesteryl oleate (CO) molecules has been constructed and simulated to gain insight into the properties of CO molecules in an isotropic environment, therefore gathering molecular information on their physiological role. Interestingly, the oleate is one of the most common fatty acyl chains in cholesteryl esters in the core of LDL (9), and it is the most prevailing fatty acid in the CEs of intracellular storage (1). The extensive analysis of this simulation data was focused on the structural as well as dynamical properties of the CO molecules. For future purposes, this type of model molecular system serves as a solid basis for the new type of simulation studies, where the important questions on the structural details of lipoproteins may be addressed.
METHODS
Modeling and computational details
The cholesteryl oleate molecule that was used to construct the cholesteryl ester system is shown in Fig. 1. In this work, the aim is to mimic the properties of biologically relevant fluid states of cholesteryl esters, such as the core of LDL particles, intracellular storages of CE and atherosclerotic plaques. In real LDL particles, the cholesteryl esters of the core contain a mixture of many different fatty acids, of which the diunsaturated linoleate is most abundant (9). The choice of oleate is supported by the availability of the validated force-field data, experimental data for comparison, and the fact that oleate is also an especially common fatty acid in LDL core. In addition, the cholesteryl oleate is the more common intracellular storage form of cholesterol (1), giving further motivation to study its properties. Just one type of fatty acid is chosen to allow comparison to experimental model systems. This is also justified by the fact that simulations of one-component systems are feasible, unlike many-component systems where the timescales to be simulated should be substantially larger due to the mixing of different molecular components; we discuss this issue briefly in Results and Discussion, Diffusion, below. Water or any other solvent is not included here since we model a homogeneous CO system in its assumed physiological conditions, where water is not present.
FIGURE 1.

(a) CO molecule used to construct the cholesteryl ester system, including the numbering used elsewhere in this work. (b) Schematic description of how the director, the angles φ and θ, and the distance r are defined. The director for a given CO molecule is defined as the vector from Ca to Cb, and r is the distance between the centers of two directors. The θ describes the orientation of the oleate chain with respect to the director of the same molecule, and φ is defined as the angle between the directors of two different CO molecules to characterize intermolecular orientational order (see Eq. 1).
Initial configuration for a single CO molecule was prepared from coordinate files for cholesterol and dipalmitoylphosphatidylcholine (DPPC) by working interactively with the molecular modeling program Cerius2. In practice, an acyl chain obtained from DPPC was attached to the OH group of cholesterol. Having done that, the double bond between C9 and C10 carbons (see Fig. 1) was constructed using the double-bond region of the oleoyl chain of palmitoyloleoylphosphatidylcholine (POPC).
Force fields for CO molecules were generated from existing force fields for cholesterol (22,29) and DPPC (the ester bond region and the singly bonded parts of the oleate chain), the latter being available at http://www.gromacs.org. To describe the double-bond region in the CO molecule, we employed two approaches. First, we used the force field designed for POPC available at http://moose.bio.ucalgary.ca/Downloads/. Second, we adapted the force field originally developed for palmitoyllinoleylphosphatidylcholine (PLPC) (30), see below. Our decision to test the above two descriptions for the double-bond region is based on recent quantum-mechanical studies (28,30), which have suggested skew states to be important in describing the structure of the hydrocarbon chain around the double bond. The description by Bachar et al. (30) takes this feature into account.
Partial charges were obtained from corresponding studies. They are available in Höltje et al. (29) for cholesterol, at http://www.gromacs.org for the ester region and the singly bonded parts of the oleate chain, and in Bachar et al. (30) for the double-bond region. The long-range electrostatic interactions were computed using the particle-mesh Ewald summation method (31,32), which has been shown to be a reliable means to handle long-range interactions in lipid membrane systems (33,34). The Lennard-Jones potential was cut off at 1.0 nm. For the time step, we employed a value of 2.0 fs.
The model system was simulated under conditions of constant particle number, pressure, and temperature. This was accomplished by the Nose-Hoover thermostat (35,36) with a time constant τ = 0.1 ps, and the Parrinello-Rahman barostat (37,38) with τ = 1.0 ps. These are both coupling schemes that are based on an extended ensemble approach, meaning that the system is coupled to an external heat (or pressure) bath, whose equations of motion become part of the problem that the MD integrator must solve. The advantage is that this way the correct physical ensemble is maintained for the simulation system. The simulation temperature was chosen to be 60°C, to ensure the formation of a liquid isotropic phase, as the transition temperature of 46–51°C for a CO molecular system between liquid-crystalline and liquid phases has been reported (12). A higher than physiological temperature is needed to compensate for the fact that the homogeneous cholesteryl oleate system has a higher phase transition temperature than in realistic mixtures of cholesteryl esters of many different fatty acids (including multipally unsaturated ones). At the end of this work, we discuss how our results can be translated to physiological conditions at ∼37°C.
To create a disordered starting point for production simulations, the system of 128 CO molecules was stepwise built-up, with a short MD simulation between every construction phase to allow molecules that were copied from the same initial configuration to attain different conformations. First, a simulation of an isolated molecule was done, and four different conformations were extracted from it. These were brought together by hand using Cerius2, subjected to energy minimization, and then simulated for 16 ns using periodic boundary conditions in all directions. The final conformation of this system was copied to double the system size in all directions, randomly orienting each copy. The random orientation required that the copies were spread further away from each other than the box size of the four-molecule configuration. This resulted in a low-density system of 32 CO molecules that was simulated for ∼4 ns. The last configuration of this simulation had a shape that allowed it to be copied in only two directions resulting in a roughly cubic box of 128 CO molecules that was finally used in the simulations using periodic boundary conditions in all directions.
The system of 128 CO molecules was first preequilibrated for a reasonably long time to allow the system size and molecular conformations to relax toward their equilibrium behavior. Next, we equilibrated the system by carrying out a simulation lasting for 100 ns using the double-bond description available at http://moose.bio.ucalgary.ca/Downloads/ (see above). This was followed by the actual production simulation of 100 ns using the double-bond parameterization of Bachar et al. (30). Although the results based on the two double-bond descriptions were found to be essentially similar, the results discussed in this work are based on the final simulation of 100 ns employing a description of Bachar et al. (30), as the results of Feller et al. (28) and Bachar et al. (30) suggest that it possibly describes the specific nature of the double-bond region in a more reliable manner.
GROMACS (39) was used to carry out all the simulations. A snapshot of the system illustrating the structure and ordering of a few specific CO molecules is shown in Fig. 2.
FIGURE 2.

A snapshot of the MD simulations, illustrating also the structure and ordering of three tagged cholesteryl oleate molecules in more detail. The molecule on the right is in a straight conformation (θ ≈ 180°), the one on the left is partly bent (θ ≈ 90°), and the molecule in the middle is highly bent (θ ≈ 30°).
Data analysis
The temporal behavior of the density of the system is followed to determine whether the system has reached equilibrium. For the same purpose, we gauge the time evolution of local quantities such as radial distribution functions (see below).
Intermolecular translational order is characterized by radial distribution functions (RDFs). The RDF of the center-of-mass (CM) positions of the molecules provides insight into the phase behavior of the system, while more detailed insight into the atomic-scale structure is gained through, e.g., studies of intermolecular RDFs between C3 carbons (see Fig. 1).
To characterize intermolecular orientational order, we first define the director of CO as a vector from Ca to Cb. Then we define the ring-ring correlation function as
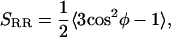 |
(1) |
where φ is the angle between the directors of two different CO molecules (see Fig. 1). The order parameter SRR is computed as a function of the distance between the centers of the directors. Hence, SRR can be employed to consider the decay of intermolecular orientational order.
To get a firm idea of typical coarse-grained conformations of the molecules, we consider an angle θ between the director of a given molecule's sterol structure and the oleate chain (vector from Cb to Cc). The angle θ thus describes the average orientation of the oleate chain with respect to the ring structure, and its distribution P(θ) allows us to gain insight into the structure and orientational properties of individual CO molecules. Using the same approach, we consider the average orientation of the short hydrocarbon chain by a vector from Ca to Cd at the other end of the cholesterol ring structure.
To characterize intramolecular orientational order, we use the directors of the CO molecules. Then, the average orientation of the fatty acyl chain segments with respect to the steroid structure of the same cholesteryl ester molecule is described by an order parameter
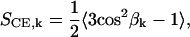 |
(2) |
where βk is the angle between the director and a C–H bond at the kth carbon atom in the oleate chain. For this purpose, as the apolar hydrogens are not explicitly present in united-atom simulations, we reconstructed the corresponding C–H vectors using backbone chain configuration. In a similar manner, we characterize the orientation of the acyl chain segments of the short hydrocarbon chain at the end of the cholesterol ring structure. The above order parameter is like the deuterium order parameter commonly measured from lipid membrane systems (22), except that here we cannot consider the membrane normal direction as the reference direction.
The dihedral term in the bonded interaction potential for saturated carbon chains has local minimums at three values of the dihedral angle. The global minimum is found at the trans conformation. The two other local minimum conformations, gauche+ and gauche−, are found with dihedral angle values of ∼−60 and +60°, respectively. For every group of four consecutive carbons in the oleate chain, the average fraction of trans and gauche dihedrals is determined at that position of the chain. The single bonds C8–C9 and C10–C11 next to the double bond, however, reproduce a so-called skew type isomerism (30), which means that the bond has two main low energy conformations at ∼−120 and +120°. Therefore, the relative amounts of these conformations were determined, too. Transition rates between the conformations of each bond were also calculated.
As for intramolecular dynamics we determine the autocorrelation functions of the C–H bonds (as in Eq. 5) from trajectories that are postprocessed to include hydrogen atoms. In simulation studies, the C–H bond autocorrelation functions are often used to characterize motions in different parts of molecules by looking at the half-time of the autocorrelation function. Another complementary means is to calculate the autocorrelation time by integrating the autocorrelation function, subtracted by its equilibrium value (26). Due to the isotropic nature of our system, the long time limit of the autocorrelation functions would be zero. However, due to slow dynamics characterized by autocorrelation times larger than the simulation time used in this work, many of them do not decay to zero during the course of the simulations. Therefore, we prefer to compute only the half-times of the autocorrelation functions, which characterize the short time motions of the hydrocarbon chains.
As for dynamics, we also calculate the diffusion coefficient of CO molecules using the expression
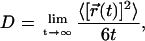 |
(3) |
where the mean-squared displacement is defined as
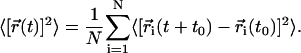 |
(4) |
The average is over a given set of N = 128 molecules and time origins t0 at 1-ns intervals, and 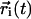 is the center-of-mass position of molecule i at time t.
is the center-of-mass position of molecule i at time t.
We further look into the rotational motion of CO molecules through the rotational correlation function
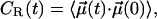 |
(5) |
where 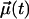 is a vector of unit length at time t. For that, we consider two cases as defined in Fig. 1: one where
is a vector of unit length at time t. For that, we consider two cases as defined in Fig. 1: one where 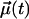 is in parallel to the principal axis (director) of the molecule, and another where
is in parallel to the principal axis (director) of the molecule, and another where  is perpendicular to it. The decay of the respective correlation functions,
is perpendicular to it. The decay of the respective correlation functions, 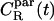 and
and  allows comparison to 13C NMR measurements (7,12). The CR(t) was calculated by averaging over molecules and all possible time origins (configurations of the system were saved every 10 ps).
allows comparison to 13C NMR measurements (7,12). The CR(t) was calculated by averaging over molecules and all possible time origins (configurations of the system were saved every 10 ps).
RESULTS AND DISCUSSION
Density
After preequilibration, we conducted a simulation of 100 ns to fully equilibrate the system, followed by a 100-ns production simulation to collect the data for analysis. As shown in Fig. 3, the density remains very stable, implying that the system has reached equilibrium. Analysis of further quantities such as RDFs confirmed this (no noticeable drift). The size of the simulated system was ∼(5.2 nm)3. As the production simulation of 100 ns provides more than enough data for most purposes, averages of most quantities are computed over 10-ns time-slices from 100 to 200 ns, allowing us to estimate errors in the quantities calculated.
FIGURE 3.

Time-dependence of the density of the CO system after preequilibration. At t = 100 ns, the double-bond description of CO was changed to the one described in Bachar et al. (30). The latter period from 100 ns to 200 ns was used for analysis.
For the average density, we find 989.8 ± 0.6 kg/m3. Rough estimates can be made whether this agrees with experiments, as we are unaware of direct experimental studies for the density of cholesteryl oleates in an isotropic phase. Low-density lipoproteins are defined as having a density in the range from 1019 kg/m3 to 1063 kg/m3, containing ∼20% protein. In chylomicrons and very low density lipoproteins, where the lipid proportion approaches 100%, the density of the particles goes below 1000 kg/m3. In this light, a density of slightly below 1000 kg/m3 is more than reasonable for a system with the composition mimicking that of an LDL core. In addition, standard densities for cholesterol and oleic acid are 1052 and 895 kg/m3, respectively, giving the upper and lower limits for the density of the CO system.
Radial distribution functions
On the basis of Fig. 2 showing a snapshot of the system at the end of the simulation, there is no prevalent ordering, which readily suggests the system to be in a liquidlike state. This is confirmed by the intermolecular radial distribution functions shown in Fig. 4 for pairs of cholesteryl carbons Cb (for labels, see Fig. 1), the centers of masses of each molecule, and the centers of the directors. In general, the RDFs indicate a liquidlike phase, as there is no distinct translational long-range order. Rather, the RDFs decay to unity at distances of the order of 1.5–2.0 nm, which is of the order of molecular size.
FIGURE 4.

Intermolecular radial distribution functions g(r) versus distance r between (a) pairs of Cb atoms at the ester bond region, (b) center-of-mass positions of the CO molecules, and (c) the centers of the directors. Two suggestive error bars are given in each figure.
A more careful examination of Fig. 4 reveals that the RDF for carbon Cb shows a larger first coordination shell associated with the leading peak than the RDFs for the centers of masses. This suggests that ring structures are often packed next to each other. The pronounced peak at r ≈ 0.5 nm in Fig. 4 c and the results below for the ring-ring order parameter (SRR) support this idea. As for the RDF of the CM positions in Fig. 4 b, we find that there is some structure at very small distances, r < 0.5 nm. This results from the fact that we are dealing with CM positions of lengthy molecules, that is, neighboring molecules may entangle around one another such that the center of mass of a given molecule may occasionally (almost) merge with the CM of a neighboring one.
Intermolecular orientational ordering
The ring-ring order parameter SRR describes the distance-dependent correlation between the orientations of the cholesteryl rings in CO molecules and is shown in Fig. 5. The corresponding intermolecular RDF between the centers of the directors is shown in Fig. 4 c.
FIGURE 5.

The order parameter SRR characterizing the orientational ordering between the ring structures of two cholesteryl ester molecules. Here, r is the distance between the centers of their directors. Two suggestive error bars are given to illustrate typical fluctuations in the data.
We find that the order parameter has essentially three distinct regimes. At very short distances, at ∼0.25 nm, we find a minor dip in SRR. This dip is backed up by the RDF in Fig. 4 c and is real, though likely due to a rare event: analysis of the data revealed an event where two CO molecules were in close contact, their steroid rings being against each other but such that the angle between the directors was ∼90°. Next, for larger distances there is a broad and pronounced double peak characterizing COs standing next to each other, peak maxima being located at ∼0.40 and 0.53 nm. A similar structure with peaks at exactly the same positions is observed in the corresponding RDF (see Fig. 4 c). The peaks in SRR highlight orientational ordering of nearby sterol structures, yet they do not differentiate between pairs of COs in parallel and antiparallel configurations. A closer analysis of this regime revealed that neighboring COs preferred an antiparallel arrangement: ∼58% of cholesteryl oleate pairs characterized by the peak at 0.40 nm were in an antiparallel configuration, and in the more pronounced peak at 0.53 nm the corresponding number was 70%. This is in accord with x-ray diffraction studies of Wendorff and Price (11), who suggested on the basis of packing properties that the arrangement of adjacent cholesteryl ester molecules is antiparallel rather than parallel. Finally, beyond the double-peak region, the correlation quite completely dies away at twice the nearest-neighbor distance, and it is clear that the phase can be characterized as liquid, rather than liquid-crystalline.
Summarizing, the features shown in Fig. 5 imply that the orientations of neighboring cholesteryl rings are strongly correlated, but this order persists over a short distance only. This is in line with the findings of Wendorf and Price (11), who suggested that the isotropic phase of saturated cholesteryl esters is characterized by small groups of molecules, whose main axes are more or less parallel (φ ∼0 or 180°) (11).
Intramolecular orientational ordering
The distribution of the angle θ, describing the average orientation of the oleate chain with respect to the cholesteryl ring structure is depicted in Fig. 6 a. Also shown here is the corresponding distribution for the angle between the short hydrocarbon chain and the ring structure of cholesterol. We can notice that the short chains are to a large degree extended with respect to the orientation of the ring.
FIGURE 6.

(a) The distribution of the angle θ, which describes the average orientation of the oleate chain of CO with respect to its director (solid line), and the corresponding distribution for the short acyl chain of cholesterol (dashed line). As for the oleate chain, the three snapshots from Fig. 2 are repeated here to better illustrate typical structures for θ ≈ 180° (snapshot on the right), θ ≈ 90° (middle), and θ ≈ 30° (left). (b) Distinct peaks observed at limited time intervals for a single tagged molecule.
As for the orientation of the oleate chain, the zero intensity region at very small angles essentially indicates that the ring and the chain cannot overlap. In the limit of large angles, close to 180°, the distribution decays to zero approximately in a linear fashion due to entropic reasons. For intermediate angles, we find a peak at ∼35°, corresponding to kinked conformations where the oleate chain lies next to the sterol ring structure of the same molecule. At larger angles, we find a broad distribution of conformations, with extended ones slightly favored. A closer look at individual molecules reveals, however, that the broad distribution in Fig. 6 a is comprised of several distinct regions in which the molecules reside over time intervals of <10 ns. To demonstrate this issue, distributions over a few distinct 10-ns periods for a selected molecule are shown in Fig. 6 b. These short-time distributions highlight a process where the conformations of a tagged molecule move from one regime to another during the given period. Specifically, here the conformational change takes place from the most kinked conformation (θ ≈ 30°) to a more elongated one (θ ≈ 90°). It also turns out that during the present transition another cholesteryl oleate molecule moves between the ring and the oleate chain parts of the tagged molecule, suggesting a possibility that concerted motions play a significant role here. However, due to a small number of transitions, we have not addressed this question in detail.
Occasionally, major transitions between conformations separated by a large angle difference occur, too, but these were found to be rare processes. In these cases, they took place through a set of smaller transitions, such as those shown in Fig. 6 b. Consequently, the timescales associated with such major transitions were larger than 10 ns.
Three of the most typical conformations giving rise to the distribution in Fig. 6 are shown in Fig. 2. They clearly demonstrate the role of carbons C3–C5 of the oleate chain in the formation of these different classes of molecules: if the molecule is not straight, it is bent around C3–C5. Fig. 6 further illustrates that a majority of the CO molecules are in merely extended (θ > 90°) conformations during the simulation, in line with the conclusions both based on 13C NMR experiments (12) of unsaturated CEs and scattering experiments of saturated CEs in isotropic fluid phase (11). Yet, we also find a significant minority of the molecules to be in a kinked conformation, characterized by θ < 60°. It is interesting to speculate whether the kinked conformation accounts for the shape of cholesteryl ester molecules in surface layers of lipoproteins as suggested in several experimental studies (40,41). This conformation would be beneficial for the hydrolysis of CEs that should happen at the surface of lipoproteins (42).
Ordering of hydrocarbon chains
The ordering of the oleate hydrocarbon chain as well as individual acyl-chain segments with respect to the ring structure are studied via the order parameter profile, SCE. The average order parameters are shown in Fig. 7. Although the first carbons of the oleate chain are found to be relatively ordered, overall the chain is highly disordered. For carbons C3 and above, the order parameters form a wide plateau characterized by low order parameter values (−SCE ≈ 0.05). For comparison, the values of SCD, comparable to the SCE calculated here, found in neat lipid bilayer systems comprised, for example, of saturated acyl chains in fluidlike glycerophospholipid bilayers are ∼0.20 (see (22) and references therein).
FIGURE 7.

The order parameter SCE describing the orientational order of the fatty acyl chain segments of the oleate with respect to the cholesteryl ring structure of the given molecule (solid line), and order of the short chain of cholesterol also with respect to the sterol rings (dashed line). Error bars are smaller than the symbol size.
The results in Fig. 7 indicate that there is certain, though weak preference to conformations where the acyl-chain segments along a chain are aligned in parallel to the cholesteryl ring moiety. This overall behavior is slightly complemented by a reduction of order at a few well specified sites. In particular, we find a dip at a region from the carbon C4 to C5, and a significant increase around C9–C10.
The dip in the vicinity of C4 and C5 indicates that cholesteryl oleates tend to bend at this particular site, thus in part favoring conformations where the acyl chain of a given molecule turns in the opposite direction and lies perpendicular to, or along its cholesteryl ring moiety. This is in agreement with our findings above for the intramolecular order in terms of the distribution of θ (see Fig. 6; see also conformations of the three molecules in Fig. 2).
The second major change at carbons C9 and C10 is presumably related to the double bond in the oleate chain. Interestingly, it resembles the one observed in the 2H NMR order parameter profile of the oleate chains in liquid-crystalline lipid bilayers (43). Further, similar drops of order in the vicinity of double bonds have been found in more recent studies for unsaturated lipid membranes (25,30,44–46). This underlines the common nature of the effects of double bonds on the ordering of fatty acyl chains, as the isotropic environment considered here would in principle allow free orientations of the double bonds. Yet, the effect of the cis double bond at carbons C9 and C10 is notable (see Fig. 7). 13C NMR relaxation time measurements of Ginsburg et al. (12) support this interpretation, since in their data one can easily identify the position of the double bond.
After the double bond, the order parameter values in Fig. 7 rise back to the same level as before the double bond, decreasing again toward zero as one approaches the end of the oleate chain. The decrease of the order parameter profile in the end of the chain has also been observed in numerous lipid bilayer systems ((22,25,30), and references therein). It is likely that they have a similar origin, namely increased thermal fluctuations in the last carbon segments allowing for more flexible conformations. This is also suggested by 13C NMR studies of isotropic CO systems, where enhanced motions toward the chain ends are indicated by relaxation time gradients (12). Hence, the decrease in the order parameter around carbon C4 and in the methyl end of the oleate chain are clearly of different origin.
As compared to oleate, the short chain of the cholesterol is highly ordered with respect to the ring. This is in line with previous observations for the chain orientation depicted in Fig. 6 a. As one approaches the end of the chain, the order decreases due to thermal fluctuations.
Bond isomerization in hydrocarbon chains
To further characterize the conformational flexibility of the oleate, the number of different states per bond were determined along the chain, together with the isomerization rates per each bond. The relative amount of trans states along the chain was mostly 70–75%, which is consistent with the findings in the fatty acyl chains of phosphatidylcholines, where the relative amount of trans state bonds per carbon segment vary, roughly, between 70 and 80% (47). We found a slight decrease in the amount of trans states in the C3–C4 positions, possibly related to the dip in the order parameter profile at the same region. At the same time, the dihedral isomerization rates were found to vary along the oleate chain between 10 and 30 transitions per molecule per nanosecond, the rates being the lowest at the region of carbons C4–C6. The dihedrals close to the end of the chain are the most flexible ones as is to be expected. As for dihedrals in skew states close to the double bond, we found 19 transitions per molecule per nanosecond for the dihedral closer to the steroid ring structure, and ∼23 transitions for the other case closer to the methyl end.
Interestingly, a plateau in the relaxation time profile was earlier found in the same region by 13C NMR experiments (12), which indicates restricted motions at that part of the chain. Ginsburg et al. (12) suggested that this could be due to the relatively strong intermolecular interactions within this region, especially between the hydrocarbon chains. However, the slightly increased amount of gauche states would oppose this idea by possibly interfering with the close contacts. On the other hand, the increase observed here is minor and may not have enough effect.
Intramolecular dynamics
Autocorrelation of C–H bonds along the hydrocarbon chains of CO molecules was monitored to determine the half-times of their autocorrelation functions, shown in Fig. 8. The half-time describes the short time dynamics of a given bond and can be qualitatively related to the experimentally measurable NMR spin-lattice relaxation time T1; the longer the relaxation times, the shorter the effective correlation times (12).
FIGURE 8.

Half-times of the autocorrelation functions for C–H bonds along the hydrocarbon chains of oleate and cholesterol. Error bars for small values of t1/2 are ∼20%, and smaller elsewhere. However, since the results are given on a logarithmic scale, this is hardly a problem.
Except for the double-bond region in the oleate chain, we find the half-times to decrease as we go along the hydrocarbon chain toward its end. This is consistent with the general conclusions for the fatty acyl chains of cholesteryl esters in isotropic fluids (12). Similar findings have been made in deuterium NMR studies of both saturated (48) and polyunsaturated (49) phospholipid bilayers. The decrease is rather modest up to carbon C8 in the oleate chain, whereas after the double bond, for carbons C10 and above, the half-times decrease significantly, in full agreement with the findings of Ginsburg et al. (12) for a corresponding system.
The double bond gives rise to a major but local increase in the half-time values. Similar findings have been made in experiments (12), and in other simulation studies containing cis unsaturated double bonds ((50) and S. Ollila, M. T. Hyvönen, and I. Vattulainen, unpublished). This is most likely due to the hindrance of motion that the fixed cis bond causes. The half-times at the region of C3–C8 are comparable, or only slightly longer than the values obtained for the saturated lipid chains in dipalmitoylphosphatidylcholine bilayers over a similar region (26). These results further suggest that the ends of the oleate chains are the most flexible parts of the system.
The short chain of cholesterol turns out to be more rigid than the oleate (see Fig. 8). Its motion in the beginning of the chain is quite restricted, which is understandable due to the presence of a methyl group attached to the first carbon of this chain. The flexibility in the end of the short chain is, however, already comparable to the segment C11 in the oleate chain, next to the double bond.
Diffusion
The mean-squared displacement of cholesteryl oleate molecules is shown in Fig. 9. At long times, after 20 ns, the slope of the mean-squared displacement is by and large constant, and allows us to determine the diffusion coefficient. We find a value of D = (2.2 ± 0.3) × 10−9 cm2/s. For comparison, the diffusion coefficients of almost all solute molecules in three-dimensional fluids are of the order of 10−5 cm2/s. In pure lipid bilayers of glycerophospholipids in the fluid phase, the diffusion coefficient is ∼1 × 10−7 cm2/s (51), whereas in strongly ordered sphingomyelin bilayers it is ∼5 × 10−8 cm2/s (26) and in gel-phase bilayers <10−10 cm2/s (51). It is evident that, despite its fluid nature, the diffusion in the CO system is slowed down considerably because of entanglement effects, for example.
FIGURE 9.

Mean-squared displacement of CO diffusion versus time t. The fit corresponding to the diffusion coefficient of D = (2.2 ± 0.3) × 10−9 cm2/s is shown by a dashed line. Here the error estimate has been determined from separate plots for the three distinct diffusion components (x, y, z).
The found result implies that the diffusion of COs is a slow process, which evidently plays a role in the timescales of mixing of different molecular components in the core of lipoproteins as well as in intracellular storages. For high density lipoproteins such as the smallest lipoparticles, the relevant size is their radius, which is ∼5 nm. Considering 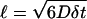 as the diffusion length during a period of δt, one finds that CO molecules can, on average, migrate a distance of ℓ = 5 nm during a timescale of δt = 19 μs.
as the diffusion length during a period of δt, one finds that CO molecules can, on average, migrate a distance of ℓ = 5 nm during a timescale of δt = 19 μs.
To explore rotational degrees of freedom, we considered the rotational motion of CO molecules through the correlation functions 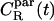 and
and 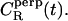 Unfortunately, it turned out that the decay of these correlation functions is a very slow process. This is in line with experiments, where the characteristic times of rotational motions have been found to be of the order of 1–100 ns (12). This implies that we cannot gauge the true long-time behavior as in experiments, but only provide results based on the short-time decay which obviously should be taken as suggestive only. Thus, by analyzing the decay of
Unfortunately, it turned out that the decay of these correlation functions is a very slow process. This is in line with experiments, where the characteristic times of rotational motions have been found to be of the order of 1–100 ns (12). This implies that we cannot gauge the true long-time behavior as in experiments, but only provide results based on the short-time decay which obviously should be taken as suggestive only. Thus, by analyzing the decay of 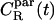 and
and 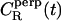 at short times, and assuming the decay to be exponential (CR ∼ exp(– t/τR)), we extracted the respective characteristic times
at short times, and assuming the decay to be exponential (CR ∼ exp(– t/τR)), we extracted the respective characteristic times 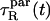 and
and  We found
We found 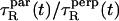 ≈ 25 at short times. For comparison, 10° above the liquid-crystalline transition temperature (as is essentially the case in our work), 13C NMR measurements (12) have yielded a ratio of 46.
≈ 25 at short times. For comparison, 10° above the liquid-crystalline transition temperature (as is essentially the case in our work), 13C NMR measurements (12) have yielded a ratio of 46.
CONCLUDING REMARKS
Atherosclerotic plaques contain cholesterol in the form of liquid, liquid-crystalline, and crystalline cholesteryl esters, which are known to originate mainly from the low density lipoproteins. The molecular level characteristics of cholesteryl esters are thought to affect the propensity of the LDL particles to end up in atherosclerotic plaques, as well as the likelihood of cholesterol to escape from plaques to high density lipoproteins by reverse cholesterol transport. To gather detailed information on the properties of cholesteryl esters, we have conducted lengthy atomic-scale simulations for an isotropic system of cholesteryl oleates.
Although the radial distribution functions support the idea that the system is in the fluid state, some short-scale orientational ordering is still evident. In particular, the ordering of the ring structures of cholesteryl oleate molecules relative to each other appears to be prevailing at short distances up to ∼8 Å. Thereafter intermolecular orientational ordering gradually vanishes. The oleate chains are mainly in extended conformations relative to ring structures, in accordance with the suggestions from experimental studies. However, a smaller population of bended conformations also exists. The oleate chains are also highly mobile as expected in the fluid state. The mobility increases moderately between the carbons C3 and C8, slows down considerably at the double-bond region, and increases quite strongly again toward the methyl end of the oleate chain. The short acyl chain of cholesterol is very extended and mobile, although the methyl group attached to the first carbon of the chain apparently reduces the degrees of freedom for the first two carbons. Finally, the diffusion of cholesteryl oleate molecules is found to be of the order of 2 × 10−9 cm2/s, which is very slow compared to the diffusion of phospholipids in fluid or liquid-ordered bilayers.
Because these studies were carried out in the disordered (fluid) phase at a relatively high temperature of 60°C, one may ask what happens at physiological conditions close to 37°C. For a neat CO system a melting temperature of 46–51°C has been reported (12), while for cholesteryl linoleate, which is another commonly found cholesteryl ester in LDLs, the corresponding melting temperature is between 36 and 42°C. At 37°C, pure cholesteryl oleate and cholesteryl linoleate systems are expected to be in a smectic and cholesteric phase, in respective order (12). However, in human plasma LDLs there are also small amounts of triglycerides and unesterified cholesterol, and the chains of lipids in the surface layer penetrate to the core region of LDL (4). Consequently, the local order of cholesteryl ester domains in the core is likely perturbed implying that the structure inside LDL particles is less pronounced than in neat cholesteryl ester systems. This is supported by experimental findings, since the isotropic liquid phase characteristic to high temperatures is indeed abundant in the LDL core ((4), and references therein). This supports the idea that the results discussed in this article are biologically relevant and provide insight into the structural and dynamical properties of cholesteryl esters inside LDLs, under conditions suggestive of the physiologically important fluid phase.
In all, the properties of cholesteryl oleate molecules found in this work were well in line with the knowledge based on experimental studies. Our data complements this picture in many ways and introduces new topics to be explored. Perhaps the most topical and feasible one is to explore the structure within the surface layer of lipoparticles. Namely, these results concerning the packing and ordering of cholesteryl oleate molecules show, interestingly, that there is a wide variety of both elongated and kinked conformations. When cholesteryl esters within the core come into contact with the surface layer of a lipoprotein particle, it hence seems plausible that there would be a significant amount of interdigitation. What conformations, then, would be most abundant? The extended ones seem to be prevailing in the core, while the kinked ones would be beneficial for the hydrolysis of cholesteryl esters taking place at the surface of lipoproteins (42). Work is in progress to resolve related issues.
Acknowledgments
The Wihuri Research Institute is maintained by the Jenny and Antti Wihuri Foundation. This work has, in part, been supported by grants from the Federation of Finnish Insurance Companies, by the Academy of Finland through its Center of Excellence Program (to I.V.), and the Academy of Finland grant No. 80851 (M.H.) and grant No. 80246 (to I.V.). The Finnish IT Center for Science and the HorseShoe (DCSC) Supercluster Computing Facility at the University of Southern Denmark are thanked for computer resources.
References
- 1.Smith, E. B. 1974. The relationship between plasma and tissue lipids in human atherosclerosis. Adv. Lipid Res. 12:1–49. [DOI] [PubMed] [Google Scholar]
- 2.Öörni, K., M. O. Pentikäinen, M. Ala-Korpela, and P. T. Kovanen. 2000. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J. Lipid Res. 41:1703–1714. [PubMed] [Google Scholar]
- 3.Babiak, J., and L. Rudel. 1987. Lipoproteins and atherosclerosis. Baillieres Clin. Endocrinol. Metab. 1:515–550. [DOI] [PubMed] [Google Scholar]
- 4.Hevonoja, T., M. O. Pentikäinen, M. T. Hyvönen, P. T. Kovanen, and M. Ala-Korpela. 2000. Structure of low density lipoprotein (LDL) particles. Basis for understanding molecular changes in modified LDL. Biochim. Biophys. Acta. 1488:189–210. [DOI] [PubMed] [Google Scholar]
- 5.Segrest, J. P., M. K. Jones, H. D. Loof, and N. Dashti. 2001. Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res. 42:1346–1367. [PubMed] [Google Scholar]
- 6.Small, D. M. 1988. Progression and regression of atherosclerotic lesions. Insights from lipid physical biochemistry. Arteriosclerosis. 8:103–129. [DOI] [PubMed] [Google Scholar]
- 7.Ginsburg, G. S., D. Atkinson, and D. M. Small. 1984. Physical properties of cholesteryl esters. Prog. Lipid Res. 23:135–167. [DOI] [PubMed] [Google Scholar]
- 8.Guo, W., and J. A. Hamilton. 1996. 13C MAS NMR studies of crystalline cholesterol and lipid mixtures modeling atherosclerotic plaques. Biophys. J. 71:2857–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton, J. A., E. H. Cordes, and C. J. Glueck. 1979. Lipid dynamics in human low density lipoproteins and human aortic tissue with fibrous plaques. J. Biol. Chem. 254:5435–5441. [PubMed] [Google Scholar]
- 10.Burks, C., and D. M. Engelman. 1981. Cholesteryl myristate conformation in liquid crystalline mesophases determined by neutron scattering. Proc. Natl. Acad. Sci. USA. 78:6863–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendorff, J. H., and F. P. Price. 1973. The structure of mesophases of cholesteryl esters. Mol. Cryst. Liq. Cryst. 24:120–144. [Google Scholar]
- 12.Ginsburg, G. S., D. M. Small, and J. A. Hamilton. 1982. Temperature-dependent molecular motions of cholesterol esters: a carbon-13 nuclear magnetic resonance study. Biochemistry. 21:6857–6866. [DOI] [PubMed] [Google Scholar]
- 13.Treleaven, W. D., Y. I. Parmar, H. Gorrissen, and R. J. Cushley. 1986. Orientational order of cholesteryl oleate in low-density lipoprotein observed by 2H-NMR. Biochim. Biophys. Acta. 877:198–210. [DOI] [PubMed] [Google Scholar]
- 14.Klon, A. E., J. P. Segrest, and S. C. Harvey. 2002. Molecular dynamics simulations on discoidal HDL particles suggest a mechanism for rotation in the Apo A-I belt model. J. Mol. Biol. 324:703–721. [DOI] [PubMed] [Google Scholar]
- 15.Phillips, J. C., W. Wriggers, Z. Li, A. Jonas, and K. Schulten. 1997. Predicting the structure of apolipoprotein A-I in reconstituted high-density lipoprotein disks. Biophys. J. 73:2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih, A. Y., I. G. Denisov, J. C. Phillips, S. G. Sligar, and K. Schulten. 2005. Molecular dynamics simulations of discoidal bilayers assembled from truncated human lipoproteins. Biophys. J. 88:548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feller, S. E. 2000. Molecular dynamics simulations of lipid bilayers. Curr. Opin. Colloid Interface Sci. 5:217–223. [Google Scholar]
- 18.Saiz, L., and M. L. Klein. 2002. Computer simulation studies of model biological membranes. Acc. Chem. Res. 35:482–489. [DOI] [PubMed] [Google Scholar]
- 19.Scott, H. L. 2002. Modeling the lipid component of membranes. Curr. Opin. Struct. Biol. 12:495–502. [DOI] [PubMed] [Google Scholar]
- 20.Vattulainen, I., and M. Karttunen. 2005. Modeling of biologically motivated soft matter systems. In Handbook of Theoretical and Computational Nanotechnology. M. Rieth and W. Schommers, editors. American Scientific Publishers, Stevenson Ranch, CA; in press.
- 21.Böckmann, R. A., A. Hac, T. Heimburg, and H. Grubmüller. 2003. Effect of sodium chloride on a lipid bilayer. Biophys. J. 85:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falck, E., M. Patra, M. Karttunen, M. T. Hyvönen, and I. Vattulainen. 2004. Lessons of slicing membranes: interplay of packing, free area, and lateral diffusion in phospholipid/cholesterol bilayers. Biophys. J. 87:1076–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurtovenko, A., M. Patra, M. Karttunen, and I. Vattulainen. 2004. Cationic DMPC/DMTAP lipid bilayers: molecular dynamics study. Biophys. J. 86:3461–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofsäß, C., E. Lindahl, and O. Edholm. 2003. Molecular dynamics simulations of phospholipid bilayers with cholesterol. Biophys. J. 84:2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyvönen, M. T., and P. T. Kovanen. 2005. Molecular dynamics simulations of unsaturated lipid bilayers: effects of varying the numbers of double bonds. Eur. Biophys. J. 34:294–305. [DOI] [PubMed] [Google Scholar]
- 26.Niemelä, P., M. T. Hyvönen, and I. Vattulainen. 2004. Structure and dynamics of sphingomyelin bilayer: insight gained through systematic comparison to phosphatidylcholine. Biophys. J. 87:2976–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pandit, S. A., S. Vasudevan, R. J. M. S. W. Chiu, E. Jakobsson, and H. L. Scott. 2004. Sphingomyelin-cholesterol domains in phospholipid membranes: atomistic simulation. Biophys. J. 87:1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feller, S. E., K. Gawrisch, and A. D. MacKerell, Jr. 2002. Polyunsaturated fatty acids in lipid bilayers: intrinsic and environmental contributions to their unique physical properties. J. Am. Chem. Soc. 124:318–326. [DOI] [PubMed] [Google Scholar]
- 29.Höltje, M., T. Förster, B. Brandt, T. Engels, W. von Rybinski, and H.-D. Höltje. 2001. Molecular dynamics simulations of stratum corneum lipid models: fatty acids and cholesterol. Biochim. Biophys. Acta. 1511:156–167. [DOI] [PubMed] [Google Scholar]
- 30.Bachar, M., P. Brunelle, D. P. Tieleman, and A. Rauk. 2004. Molecular dynamics simulation of a polyunsaturated lipid bilayer susceptible to lipid peroxidation. J. Phys. Chem. B. 108:7170–7179. [Google Scholar]
- 31.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an n log(n) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 32.Essman, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 33.Patra, M., M. Karttunen, M. T. Hyvönen, E. Falck, P. Lindqvist, and I. Vattulainen. 2003. Molecular dynamics simulations of lipid bilayers: major artifacts due to truncating electrostatic interactions. Biophys. J. 84:3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patra, M., M. Karttunen, M. T. Hyvönen, E. Falck, and I. Vattulainen. 2004. Lipid bilayers driven to a wrong lane in molecular dynamics simulations by subtle changes in long-range electrostatic interactions. J. Phys. Chem. B. 108:4485–4494. [Google Scholar]
- 35.Hoover, W. G. 1985. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 36.Nosé, S. 1984. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52:255–268. [Google Scholar]
- 37.Nosé, S., and M. L. Klein. 1983. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50:1055–1076. [Google Scholar]
- 38.Parrinello, M., and A. Rahman. 1981. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52:7182–7190. [Google Scholar]
- 39.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- 40.Grover, A. K., B. J. Forrest, R. G. Buchinski, and R. J. Cushley. 1979. ESR studies on the orientation of cholesteryl ester in phosphatidylcholine multilayers. Biochim. Biophys. Acta. 550:212–221. [DOI] [PubMed] [Google Scholar]
- 41.Janiak, M. J., C. R. Loomis, G. G. Shipley, and D. M. Small. 1974. The ternary phase diagram of lecithin, cholesteryl linolinate and water: phase behavior and structure. J. Mol. Biol. 86:325–339. [DOI] [PubMed] [Google Scholar]
- 42.Smaby, J. M., and H. L. Brockman. 1987. Acyl unsaturation and cholesteryl ester miscibility in surfaces. Formation of lecithin-cholesterol ester complexes. J. Lipid Res. 28:1078–1087. [PubMed] [Google Scholar]
- 43.Seelig, J., and N. Waespe-Sarcevic. 1978. Molecular order in cis and trans unsaturated phospholipid bilayer. Biochemistry. 17:3311–3315. [DOI] [PubMed] [Google Scholar]
- 44.Huber, T., K. Rajamoorthi, V. F. Kurze, K. Beyer, and M. F. Brown. 2002. Structure of docosahexaenoic acid-containing phospholipid bilayers as studied by 2H NMR and molecular dynamics simulations. J. Am. Chem. Soc. 124:298–309. [DOI] [PubMed] [Google Scholar]
- 45.Hyvönen, M. T., T. T. Rantala, and M. Ala-Korpela. 1997. Structure and dynamic properties of diunsaturated 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphatidylcholine lipid bilayer from molecular dynamics simulation. Biophys. J. 73:2907–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitchell, D. C., and B. J. Litman. 1998. Molecular order and dynamics in bilayers consisting of highly polyunsaturated phospholipids. Biophys. J. 74:879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mendelsohn, R., M. A. Davies, H. F. Schuster, Z. G. Zu, and R. Bittman. 1991. CD2 rocking modes as quantitative infrared probes of one-, two-, and three-bond conformational disorder in dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylcholine/cholesterol mixtures. Biochemistry. 30:8558–8563. [DOI] [PubMed] [Google Scholar]
- 48.Brown, M. F., J. Seelig, and U. Häberlen. 1979. Structural dynamics in phospholipid bilayers from deuterium spin-lattice relaxation time measurements. J. Chem. Phys. 70:5045–5053. [Google Scholar]
- 49.Eldho, N. V., S. E. Feller, S. Tristam-Nagle, I. V. Polozov, and K. Gawrisch. 2003. Polyunsaturated docosahexaenoic vs. docosapentaenoic acid—differences in lipid matrix properties from the loss of one double bond. J. Am. Chem. Soc. 125:6409–6421. [DOI] [PubMed] [Google Scholar]
- 50.Niemelä, P., M. T. Hyvönen, and I. Vattulainen. 2005. Influence of chain length and unsaturation on sphingomyelin bilayers. Biophys. J. 90:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vattulainen, I., and O. G. Mouritsen. 2005. Diffusion in membranes. In Diffusion in Condensed Matter: Methods, Materials, Models, 2nd Ed. P. Heitjans and J. Kärger, editors. Springer-Verlag, Berlin, Germany.