

Abstract
The retroviruses including the human pathogens HIV-1 and HIV-2 are diploid inasmuch as they encapsidate two copies of their RNA genome. Prior to or during encapsidation, two copies of full-length genomic RNA recognize and stably bind each other in a process called dimerization. RNA structures within the viral genome promote dimerization in both HIV-1 and HIV-2 and are located in the 5′ untranslated leader region. Inhibition of dimerization by mutation of these RNA signals has been demonstrated to drastically reduce virus infectivity and replication kinetics, and thus represents a potential target for antiretroviral therapy. In this study, we identified sites in HIV-2 leader region RNA that are functionally accessible to hybridization with oligonucleotides by reverse transcription with random oligonucleotide libraries, or RT-ROL (Allawi et al., 2001). We then tested specific oligonucleotides directed against these regions for their efficacy in inhibiting RNA dimerization in vitro. We determined that of several hybridization-competent oligonucleotides, only two were very effective in inhibiting RNA dimerization. Both of these oligonucleotides were complementary to viral RNA at the primer binding site (PBS). These results identify regions with high accessibility to oligonucleotide binding on HIV-2 RNA and help to map the region(s) essential for dimerization within the viral RNA.
Introduction:
An essential step in the replication cycle of all retroviruses is the dimerization of two copies of full length genomic RNA prior to or concomitant with encapsidation and budding. Interference of the dimerization process through mutagenesis of a conserved motif within the 5′ leader region of HIV-1 RNA called the dimerization initiation site (DIS) has been shown to be detrimental to viral infectivity and replication in cell culture studies (Berkhout and van Wamel, 1996; Haddrick et al., 1996; Paillart et al., 1996; Clever and Parslow, 1997; Hirota et al., 1997; Laughrea et al., 1997; St. Louis et al., 1998; Laughrea et al., 1999). These studies demonstrated multiple effects of deletion or mutation of the dimerization domain, including diminished dimerization, encapsidation, and proviral DNA production. Because the dimerization step is crucial, and the fact that there is considerable conservation of the sequence(s) involved, it represents a potential antiretroviral therapeutic target.
Human immunodeficiency virus type 2 (HIV-2), one of the two causative agents of AIDS in humans, is prevalent in parts of West Africa and Asia, but is still relatively rare in Europe and North America. HIV-2 infection is typically characterized by a less aggressive pathology and longer latency period compared to HIV-1 infection, and a greater proportion of HIV-2 infected individuals do not progress to full-blown AIDS (Reeves and Doms, 2002). However, those patients who develop HIV-2 AIDS have symptoms and prognoses similar to those with HIV-1 AIDS. In part because of its scarcity in western nations, HIV-2 has been less extensively studied than HIV-1 despite the fact that it represents a significant worldwide health threat.
HIV-2 shares significant sequence and genomic organization similarity with HIV-1, but the viruses are less than 50% identical at the nucleotide level. Whereas dimerization of HIV-1 RNA has been studied extensively by several laboratories, less is known about the structures and mechanisms involved in dimerization of HIV-2 RNA. A structure homologous to the HIV-1 DIS called stem loop 1 (SL-1) exists in the HIV-2 5′ leader region, but it was initially shown not to be required for dimerization under the experimental conditions classically used to study dimerization in HIV-1 (Jossinet et al., 2001). Instead, a region overlapping the primer binding site (PBS) was implicated in promoting dimerization. Subsequently, it was demonstrated using certain truncated RNA transcripts and buffer and electrophoretic conditions lacking magnesium and involving a heating step to greater than 50°C during incubation, that SL-1 could also promote dimer formation (Dirac et al., 2001; Lanchy and Lodmell, 2002).
In the present work we have used RNAs encompassing all or part of HIV-2 5′ leader region RNA to characterize and map sequences in the vicinity of the PBS that are accessible to oligonucleotide hybridization and that may be important for dimerization of viral RNA in vitro. First, we used a combinatorial technique called reverse transcription with random oligonucleotide libraries, or RT-ROL (Allawi et al., 2001), to identify several oligonucleotide-accessible sites in the HIV-2 leader region RNA. We then tested the sites identified by RT-ROL for their capacity to bind complementary oligodeoxynucleotides as assayed by digestion of complexes with RNase H or by electrophoresis mobility shift assay. Finally, we tested the oligonucleotides for their ability to inhibit dimerization of HIV-2 RNAs under native dimerization and electrophoresis conditions. Of the several oligonucleotides tested, only those that were complementary to parts of the PBS were capable of efficiently inhibiting dimerization. The data presented here implicate some potentially vulnerable sites in the 5′ leader region of HIV-2 RNA that were not obvious in the secondary structure model for this RNA. In addition, the fact that only a subset of the high-affinity oligonucleotides identified by RT-ROL affected dimerization enabled us to identify the parts of leader region RNA that are most important for promoting dimerization in vitro without any a priori supposition of dimerization signals in this region.
Materials and Methods:
RNA synthesis and purification.
RNA comprised of nucleotides 1-381 or 1-561 of the natural HIV-2 ROD isolate of HIV-2 was transcribed using phage T7 RNA polymerase from a DNA template derived from pROD10 (provided by Drs. J.-M. Bechet and A. M. L. Lever through the Centralised Facility for AIDS Reagents (UK Medical Research Council)). The 1-381 or 1-561 region of pROD10 was PCR amplified using a 5′ primer containing a BamHI site and a T7 promoter and a 3′ primer containing an EcoRI site (primers are described in Table 1). The PCR products flanked by BamHI and EcoRI were cloned into a pUC18 vector and sequenced. RNA was transcribed from the EcoRI digested plasmid using the AmpliScribe T7 transcription kit (Epicentre). DNA was removed from the RNA by DNase I treatment. RNA was purified by ammonium acetate precipitation, exclusion chromatography (Bio-Gel P-4, Bio-Rad), and ethanol precipitation.
Table 1.
Oligodeoxynucleotides used in this study.
| Name | Sequence | Description |
|---|---|---|
| RNDTAG | 5′-TAA GGT AGG ACT ACG NNN NNN NN | RT-ROL random tagged oligonucleotide |
| TAG | 5′-TAA GGT AGG ACT ACG | RT-ROL amplification oligonucleotide |
| sRODRNA | 5′-GGT CGC TCT GCG GAG AGG CTG | detection oligonucleotide for 5′ end of viral RNA |
| sROD145 | 5′-CCT CTT AAT AAA GCT GCC AG | detection oligonucleotide for the 145 region of viral RNA |
| sROD 228 | 5′-CCT GAG TAA CAA GAC CCT GG | detection oligonucleotide for 228 region of viral RNA |
| asROD32 | 5′-GCT CAA TCT GCC AG | antisense oligonucleotide complementary to 32 region of viral RNA |
| asROD99 | 5′-GTG CTG GTG AGA CTC TAG CAG | antisense oligonucleotide complementary to 99 region of viral RNA |
| asROD172 | 5′-GCT TCT AAC TGG C | antisense oligonucleotide complementary to 172 region of viral RNA |
| asROD258 | 5′-GTC CTA ACA GAC CAG G | antisense oligonucleotide complementary to 258 region of viral RNA |
| asROD309 | 5′-GGC GCC AAC CTG CTA GGG ATT | antisense oligonucleotide complementary to 309 region of viral RNA |
| asRODPBS | 5′-GTC CCT GTT CAG GCG CCA | antisense oligonucleotide complementary to the 18nt PBS sequence |
| asROD344 | 5′-CCA AGA CTT CTC AGT CTT C | antisense oligonucleotide complementary to 344 region of viral RNA |
| asROD418 | 5′-CGG CCC GCG CCT TTC TAG | control oligonucleotide not complementary to any region of 1-381 RNA |
Reverse transcription using random oligonucleotide libraries (RT-ROL).
RT-ROL was carried out essentially as described (Allawi et al., 2001). For the reverse transcription step, 0.3 pmol 1-561 RNA was mixed with 6 pmol of random-tagged oligonucleotide (RNDTAG; see Table 1) in a total volume of 20 μl. The mixture was heated to 95°C for 2 minutes, quenched on ice for 2 minutes, then incubated at room temperature for 40 minutes to hybridize the random oligonucleotides. Then 4.5 μl H2O, 3 μl 10X buffer (100mM Tris HCl/500mM KCl), 1.5 μl dNTP mix (10mM each dNTP), 1.5 μl MgCl2 (30mM), and 1.5 μl AMV-RT (Seikagaku America/Associates of Cape Cod) solution (stock AMV-RT diluted 10X in above Tris/KCl at 1X) were added and mixed. This mixture was incubated at 22°C for one hour, then 90°C for 10 minutes to inactive the RT.
To amplify the cDNA products, a PCR routine was performed as follows. A mixture containing 7μl H2O, 4 μl RT-ROL product, 2 μl 10X Thermopol buffer, 1 μl dNTP mix (10 mM each dNTP), 1 μl TAG oligonucleotide (10 pmol/μl), 1 μl sense detection oligonucleotide (10 pmol/μl; see Table 1), and 1 μl Vent exo- polymerase (New England Biolabs) was subjected to 30 cycles of 95°C (1 minute), 55°C (1 minute), then 72°C (1 minute). PCR products were visualized on a 1% agarose gel in 1X TAE with ethidium bromide staining.
Agarose gel analysis gave an approximate location of the hybridization sites on the HIV-2 RNA based upon the size of the resultant PCR products. However, to determine an exact location of the binding site of the oligonucleotide, sequencing reactions were required. First, the detection oligonucleotide was 5′ end labeled with 32P using T4 polynucleotide kinase. The RT-ROL products were amplified using this labeled oligonucleotide and the TAG oligonucleotide. The size of the radioactive PCR product was determined by running normal DNA sequencing reactions in adjacent lanes and could be used to determine the exact site of the random oligonucleotide hybridization by subtracting the nucleotides comprising the TAG sequence on the original random oligomers. For example, a band on a sequencing gel at position 180 would be interpreted as a random oligonucleotide hybridization site of approximately 157-165, since 15 nucleotides of the PCR product are comprised of the TAG sequence, and 8 nucleotides comprise the random oligonucleotide sequence. Furthermore, all of the hybridization sites yielded multiple bands on the sequencing gel, indicating a ‘window’ of RNA sequence that was available for hybridization with the random 8 nucleotide segment of the RNDTAG oligonucleotides.
The extension reactions were carried out as follows. 10 μl H2O were mixed with 5 μl RT-ROL product, 2 μl 10X Thermopol buffer, 1 μl dNTP mix (10mM), 1 μl TAG oligonucleotide (10 pmol/μl), 1 μl Vent exo- polymerase, and 200,000 CPM of the detection primer. The extension reactions were carried out with 30 cycles of 95°C (1 minute), 55°C (1 minute), then 72°C (1 minute). Extension products were loaded onto a 6% denaturing polyacrylamide gel alongside normal G,A,T,C sequencing lanes (plasmid DNA was used as the sequencing template), and the bands were revealed by autoradiography.
Oligonucleotide:RNA binding specificity assay using RNase H.
After determining the oligonucleotide-accessible sites by RT-ROL, specific complementary oligonucleotides were synthesized (Integrated DNA Technologies). To determine whether they bound to the RNA at a single site and at the expected location, the oligonucleotides were mixed at a 2:1 ratio with HIV-2 1-381 or 1-561 RNA and denatured at 90°C, snap-cooled, then incubated in monomer buffer for 15 minutes at 37°C. Then RNase H (1U; Takara) was added and allowed to incubate at 24°C for 5 minutes. The RNAs were then loaded in formamide buffer onto a 5% acrylamide, 7M urea, 1XTBE gel. Bands were visualized by ethidium bromide staining. The fragments resulting from the cleavage of the RNA at the site(s) of interaction of the oligonucleotide with the RNA gave an accurate indication of the hybridization site(s).
Oligonucleotide recruitment onto the viral RNA determined by EMSA.
To determine whether the oligonucleotide bound efficiently to the viral RNA, an electorphoretic mobility shift assay (EMSA) was performed. First, the oligonucleotides were 5′ end labeled using 32P-ATP and T4 polynucleotide kinase. After purification, labeled oligonucleotide was added to viral RNA during a dimerization protocol (see below). The resulting incubation mixtures were loaded onto non-denaturing agarose gels (1% agarose, 0.5X Tris-Borate, 0.1 mM MgCl2 run at 4°C) to fractionate the free oligonucleotide, the monomeric RNA, and the dimeric RNA. The bands were then visualized both by ethidium bromide staining and by autoradiography. In this manner, it was possible to determine not only whether the oligonucleotide was bound to the RNA on the gel, but also whether it bound preferentially to either the monomeric or dimeric RNA.
Dimerization of 1-381 viral RNA.
Five pmol of RNA were diluted in 8 μl H2O and heated to 90°C for 2 minutes then snap-cooled on ice. Two microliters of 5X concentrated dimerization buffer were added (final concentrations: 50 mM Tris-HCl pH 7.5, 300 mM KCl, 5 mM MgCl2) followed by incubation at 37°C for 15 minutes. The samples were then placed on ice. Finally, 2μl of glycerol loading dye were added just prior to loading onto a 0.8% agarose/0.5X Tris-Borate/0.1 mM MgCl2 gel chilled to 4°C. The electrophoresis was carried out at 7V/cm (to avoid heating of the gel) for one hour in the cold. Bands were visualized by ethidium bromide staining.
Inhibition of dimerization using antisense oligonucleotides.
Two methods were used to determine a given oligonucleotide's capacity to inhibit viral RNA dimerization. In the first type of assay, the oligonucleotide was added at the outset of the dimerization protocol in increasing concentrations relative to a fixed amount of RNA (5 pmol). Thus, the RNA, oligonucleotide, and water mixture was heated to 90°C for 2 minutes, followed by incubation on ice, then 37°C after addition of buffer, as described for the dimerization protocol above. In the alternate procedure, the RNA was allowed to dimerize at 37°C for 15 minutes in the absence of oligonucleotide, then increasing amounts of oligonucleotide were added to replicate tubes of pre-dimerized RNA. The oligonucleotides were allowed to ‘challenge’ the dimeric RNA for a further 15 minutes at 37°C, then the samples were placed on ice and electrophoresed as usual. The dimeric and monomeric RNA was quantified by fluorescence scanning of the ethidium bromide stained sample on a Fuji FLA-3000 fluorescence imager. The fraction of dimeric RNA was plotted as a function of oligonucleotide concentration using Kaleidagraph software.
Results:
Identification of oligonucleotide-accessible sites by RT-ROL.
We used the reverse transcription with random oligonucleotide libraries (RT-ROL) method (Allawi et al., 2001) to probe the leader region of HIV-2 RNA for sites that were amenable to oligonucleotide hybridization and subsequent extension by AMV reverse transcriptase. Several sites were identified in the 5′ leader region of HIV-2 RNA, as summarized in Figure 1.
Figure 1.

Summary of locations of hybridization-competent sites of HIV-2 leader region RNA as determined by reverse transcription with random oligonucleotide libraries (Allawi et al., 2001). The regions of RNA determined to be accessibly by RT-ROL are indicated with lines drawn on a secondary structure model of HIV-2 leader region RNA derived from biochemical data, comparative sequence analysis, and computational energy minimization (Berkhout, 1992; Berkhout and Schoneveld, 1993; Rhim and Rice, 1994). The viral RNA nucleotide numbers corresponding to the 5′ end of the oligonucleotides used in this study are indicated.
The existence and approximate location of the oligonucleotide-accessible sites was first determined by examining the DNA products resulting from the PCR amplification of cDNA strands primed from random oligonucleotides (Figure 2). The exact location of priming by these random tagged oligonucleotides was determined by extension of a radiolabeled detection oligonucleotide using the PCR products as templates. Since all of the cDNAs should have the same internal sequences, but vary with respect to their start site, the length of the product of the primer extension of the PCR product as determined alongside standard sequencing lanes on a sequencing gel gives an exact location of the original random oligonucleotide binding site/reverse transcription start site (Figure 3).
Figure 2.

Agarose gel analysis of PCR products derived from the random oligonucleotide priming and amplification using the RT-ROL protocol on HIV-2 leader region RNA. Lane L: DNA ladder (New England Biolabs, 100bp); Lane 1: negative control reaction containing no random oligonucleotide during the reverse transcription step; Lane 2: positive control reaction in which oligonucleotides complementary to the PBS and 145 regions were added to the reverse transcription reaction instead of the random oligonucleotides; Lane 3: complete RT-ROL reaction showing several amplification products arising from binding of random oligonucleotides and subsequent reverse transcription and PCR amplification.
Figure 3.

Determination of the exact hybridization sites of random oligonucleotides in the nucleotide 170-350 region by radiolabeled primer PCR analysis (see Materials and Methods). The RNA template (561 nucleotides long) was subjected to RT-ROL, then the exact sites of hybridization were determined by amplifying the RT-ROL products with radiolabeled primer sROD145 and unlabeled TAG oligonucleotide. Lanes C,U,G are sequencing lanes; Lane 1: RT-ROL reaction without any added oligonucleotides; Lane 2: positive control reaction in which an oligonucleotide complementary to the PBS region was added to the reverse transcription reaction instead of the random oligonucleotides; Lane 3: complete RT-ROL reaction showing several amplification products arising from binding of random oligonucleotides and subsequent reverse transcription and PCR amplification.
Interestingly, RT-ROL identified both putative single stranded and double stranded regions of viral RNA as depicted in the secondary structure model (the model was adapted from Berkhout and Schoneveld (1993) and is based upon comparative sequence analysis, computer assisted least energy folding, and chemical and enzymatic probing) as being efficient oligonucleotide binding/reverse transcription sites. The accessible sites were identified in potentially functionally important regions including the transactivation region (TAR), the 5′ polyadenylation signal, and the primer binding site domain (see Figure 1). Furthermore, several of these accessible sites are universally or nearly completely conserved (corresponding to the binding sites for oligonucleotides asROD32, asROD99, asROD258, asROD309, and asRODPBS) among published HIV-2/SIV viral sequences (Kuiken et al., 2001). The accessibility information obtained from RT-ROL was then used to design antisense oligonucleotides complementary to various regions of the viral RNA leader region (Table 1).
Oligonucleotide binding specificity to viral RNA by RNase H.
To determine whether the oligodeoxyribonucleotides we constructed based upon the RT-ROL results had unique binding sites on the viral RNA, we digested the RNA/oligodeoxyribonucleotide complexes with E. coli RNase H. The resulting digestion products were electrophoresed on a denaturing polyacrylamide gel. If the oligonucleotide binds to a single site and that site is subsequently cleaved by RNase H, the sizes of the resulting fragments gives a good approximation of the oligonucleotide binding site(s). Each of the antisense oligonucleotides we tested gave the expected digestion pattern and indicated a unique binding site while the control reactions using non-complementary oligonucleotide did not yield digestion products (Figure 4). The small band expected from digestion with asROD32 is too faint to be detected, but a band just slightly shorter than the full length 1-561 RNA is discernible near the top of the lane indicating the expected oligonucleotide placement.
Figure 4.

Determination of specificity of binding of oligonucleotides designed based upon RT-ROL results to viral RNA by cleavage with RNase H. HIV-2 1-561 RNA was incubated in monomer buffer with the oligonucleotides indicated and RNase H. Digestion products were loaded onto a denaturing polyacrylamide gel and fragments were visualized by ethidium bromide staining. Lanes L: RNA size ladder (Ambion); Lane 1: complete reaction except with no oligonucleotide added; Lanes 2-8: complete reactions with oligonucleotide asROD32, asROD99, asROD172, asROD258, asROD309, asRODPBS, and asROD344 added, respectively; Lane 9: control reaction with the non-complementary sense oligonucleotide sROD228.
Oligonucleotide binding to viral RNA assayed by non-denaturing agarose gel electrophoresis.
To determine whether the antisense oligonucleotides bound to the viral RNA, they were 5′ end labeled with 32P, then a substoichiometric amount (approximately 1:20 oligonucleotide to RNA) was hybridized to viral RNA under buffer and temperature conditions that allowed the viral RNA to dimerize. The incubation mixture was then loaded onto a non-denaturing agarose gel and electrophoresed at 4°C to separate free oligonucleotide, monomeric RNA, and dimeric RNA. The products were visualized by ethidium bromide staining (Figure 5A) and by autoradiography (Figure 5B). Each of the oligonucleotides was supershifted into the RNA bands, but to varying extents. Oligonucleotides asROD172 and asROD309 were the most efficiently supershifted into the monomer and dimer RNA bands, while asROD344 was not efficiently recruited into a complex with the viral RNA in this assay.
Figure 5.

Antisense oligonucleotide binding to 1-381 RNA under native conditions. A) Ethidium bromide stained gel showing the effects of binding substoichiometric radiolabeled oligonucleotide (lanes 2, 4, 6, 8, 10, 12) or a 20:1 excess of unlabeled oligonucleotide (lanes 3, 5, 7, 9, 11, 13) to viral RNA under dimerization conditions and native electrophoresis (see Materials and Methods). Oligonucleotides used are indicated below lanes. Note that some oligonucleotides stained more or less efficiently with ethidium bromide (e.g. oligonucleotide 172 appears as a fainter band). However, oligonucleotide concentrations were accurately determined by spectrophotometric measurements. B) Autoradiogram of the same gel as in panel A showing the partitioning of the radiolabeled oligonucleotides with respect to the viral RNA. An asterisk (*) below the lane indicates presence of radiolabeled oligonucleotide, a plus sign (+) indicates the presence of a 20-fold excess of unlabeled oligonucleotide, and a minus sign (-) indicates that no oligonucleotide was added.
The partitioning of the radioactively labeled oligonucleotide with respect to the dimer or monomer bands of the viral RNA was of significant interest as well. Figure 5B shows that some of the oligonucleotides demonstrated preferential binding to either the monomer or dimer RNA bands. Whereas the asROD32, asROD99, and asROD344 partitioning approximately mirrors the relative intensities of the dimer and monomer RNA bands as visualized with ethidium bromide staining, asROD258 and asROD309 exhibited a strong preference for only monomeric RNA, suggesting that the sequences required for hybridization of these oligonucleotides were also required for dimerization. This observation is supported by the inhibition of dimerization by an excess of unlabeled oligonucleotides as ROD258 and asROD309 in lanes 9 and 11 of Figure 5A. Conversely, oligonucleotide asROD172 exhibited a preference for dimeric RNA. Interestingly, adding an excess of unlabeled asROD172 stimulated dimer formation as evidenced by the enhanced dimer band and reduced monomer band in Figure 5A, lane 7.
Oligonucleotide-mediated inhibition of HIV-2 RNA dimerization.
Certain of the oligonucleotides complementary to the PBS and flanking regions were observed to diminish dimerization of viral RNA, as seen in Figure 5A. Thus, we tested the ability of each oligonucleotide to inhibit dimerization by adding increasing amounts of oligonucleotide to a fixed amount of viral RNA during a dimerization reaction, then quantifying the resultant dimer and monomer bands on a native agarose gel stained with ethidium bromide. Because dimerization of HIV-2 leader region RNA is promoted by signal(s) upstream of SL-1 under these experimental conditions (Lanchy and Lodmell, 2002), we focused our dimerization inhibition assays on the 1-381 RNA. Two types of inhibition assays were used. In the first, oligonucleotide was added to the reaction mixture from the beginning of the experiment, before any RNA dimers were formed. Using this method, we determined that asROD258, asROD309, and asRODPBS could significantly inhibit dimerization (Figure 5A and data not shown). In the second type of assay, the RNA was allowed to dimerize first, then the dimers were challenged with increasing amounts of oligonucleotide. Using this assay, only asROD309 and asRODPBS were determined to be efficient inhibitors of dimerization (Figure 6). Notably, asROD258, which was a good inhibitor of dimerization when added at the beginning of the experiment, was almost completely devoid of inhibitory activity when used to challenge preformed dimers (data not shown). Oligonucleotides asROD32, asROD99, asROD172, and the non-complementary control oligonucleotide asROD418 did not inhibit dimerization (data not shown).
Figure 6.
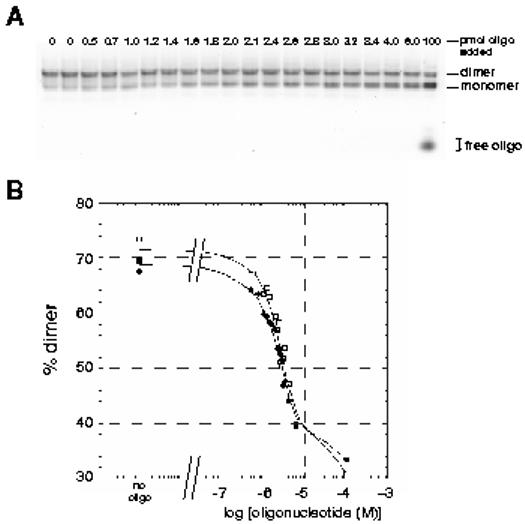
Effect of challenging pre-formed HIV-2 RNA dimers with increasing concentrations of oligonucleotide asRODPBS and asROD309. A) Increasing amounts of oligonucleotide asRODPBS were added to pre-formed dimers of RNA 1-381 and incubated for an additional 37°C for 15 minutes. One microliter aliquots of oligonucleotide stock solutions were added to five picomoles of RNA in a final reaction volume of 10μl (final oligonucleotide concentrations ranged between 0 and 10 μM). Reactions were loaded onto TBM agarose gels and electrophoresis was carried out at 4°C. Ethidium bromide staining was used to visualize the monomer and dimer RNA bands. B) Quantification of dimer yield from the gel shown in panel A and for the same experiment using oligonucleotide asROD309. A Fuji FLA-3000G Fluorescence Imager and Image Gauge software were used to determine the fraction of RNA in the dimer band of each lane. These values were plotted versus the log of the oligonucleotide concentration for the two oligonucleotides found to be effective inhibitors of dimerization, namely asRODPBS (open squares) and asROD309 (closed squares). Other oligonucleotides tested did not effectively inhibit dimerization in this assay, even at high concentrations.
Discussion:
Identification of potential therapeutic targets in the replication cycle of pathogens is of utmost importance given the widespread emergence of multiple drug resistant strains of bacteria and viruses. Among the retroviruses, including the causative agents of AIDS, HIV-1 and HIV-2, an essential step in the replication cycle is the dimerization of two copies of genomic RNA prior to or during encapsidation. In both HIV-1 and HIV-2, the principal dimerization signals lie in the 5′ untranslated region of the viral genome upstream of the gag open reading frame (Laughrea and Jetté, 1994; Skripkin et al., 1994; Dirac et al., 2001; Jossinet et al., 2001; Lanchy and Lodmell, 2002). Also found in the 5′ untranslated region are functionally important signals for transactivation, primer binding, encapsidation, and splicing. In this study we characterized the 5′ leader region of HIV-2 viral RNA with respect to its accessibility for hybridization with antisense oligonucleotides and tested the effects of these oligonucleotides for their ability to inhibit dimerization of viral RNA in vitro. The results reported here demonstrate several excellent binding sites for complementary oligonucleotides, which may represent good potential targets for antisense-based antiretroviral therapies. Furthermore, the locations of these binding sites and the effects of binding oligonucleotides to them lend insight into the structure of the 5′ untranslated region and the mechanism of dimerization of HIV-2 RNA.
Using a newly developed combinatorial method we have empirically identified oligonucleotide binding sites on the HIV-2 viral RNA 5′ leader region without any a priori assumptions about which sites might provide the best binding (Allawi et al., 2001). Such combinatorial techniques have helped in recent years to eliminate much of the tedious trial and error oligonucleotide selection associated with antisense strategies. Another combinatorial method for elucidating good hybridization sites on RNA molecules makes use of RNase H directly (Ho et al., 1998; Ho et al., 2000). The selection and identification of sites using this method relies on the recognition and cleavage of DNA:RNA heteroduplexes by RNase H, then using primer extension from an interior site on the RNA to detect the exact sites of cleavage. On the other hand, the RT-ROL method used in this study relies on the recognition of the DNA:RNA heteroduplex by reverse transcriptase, which then extends the complex to the 5′ end of the RNA molecule. This newly formed cDNA can then be amplified by PCR, and hybridization sites are identified by size analysis of the resultant PCR products. An advantage of this technique that was important for the present application is that fewer RNA molecules are required in the initial pool since cDNAs are subsequently amplified by PCR. Because HIV-2 leader region RNA can dimerize, it was advantageous for us to keep the RNA concentration low so that dimerization would interfere minimally with oligonucleotide hybridization. The characterization of these hybridization-competent sites has the potential to be applied to various antisense strategies to inhibit viral replication. In addition, these results help us to answer some fundamental questions about the structure and function of the 5′ leader region of HIV-2 viral RNA.
The RT-ROL produced some surprising results when compared to a secondary structure model of the leader region of HIV-2 RNA derived from solution structure probing, comparative sequence analysis, and folding energy minimization (Berkhout and Schoneveld, 1993). For example, RT-ROL showed that apparently double-stranded segments of the TAR stem and the stem immediately 5′ to the PBS were good hybridization sites. On the other hand RT-ROL selected several other regions that seemed like obvious candidates for hybridization based upon the secondary structure model, including the PBS and the apical loop of the polyadenylation signal (see Figure 1). These results emphasize the caution with which one must proceed in designing complementary oligonucleotides based solely on a secondary structure model.
There are several possible reasons that oligonucleotides hybridized successfully to apparently double stranded regions of the viral RNA. First, it is important to note that RT-ROL depends not only on hybridization, but also on the subsequent reverse transcription event. It is possible that some sequence or structural features are favored in this assay that are only secondarily related to efficiency of oligonucleotide binding. Second, it is possible that the secondary structure model is not entirely correct, or that structural dynamics of the HIV-2 transcripts allows these apparently double stranded regions to unpair. The leader region of HIV-2 RNA is indeed prone to conformational changes, as has been documented recently, although the exact nature of these changes at the nucleotide level is only beginning to emerge (Dirac et al., 2002; Lanchy and Lodmell, 2002; Lanchy et al., 2003). The TAR region of HIV-2 RNA is depicted in the secondary structure model, based upon biochemical data, as a bifurcated stem-loop structure (Berkhout and Schoneveld, 1993; Rhim and Rice, 1994). However, the thermodynamically most stable folding for this region of RNA based upon the software Mfold (Mathews et al., 1999; Zuker et al., 1999) depicts the region as a single long stem-loop structure. Therefore, it is perhaps not surprising that multiple conformations could exist in this region and that one or more of the conformations (or even the transition states between the conformations) would be competent for oligonucleotide binding.
It is noteworthy that RT-ROL identified the PBS as a hybridization-competent region inasmuch as the PBS has been implicated not only in tRNAlys,3 annealing for use as a primer for reverse transcription, but also for RNA dimerization (Jossinet et al., 2001; Lanchy and Lodmell, 2002). For both of these functions, it is expected that the PBS would need to be exposed to the solvent and accessible for hybridization. The RT-ROL results corroborate these expectations. Furthermore, the finding that oligonucleotides complementary to the PBS region identified by RT-ROL are very efficient inhibitors of this essential replication step demonstrates the utility of the approach for discovering functionally active antisense oligonucleotides.
There has been some debate in the literature with respect to the role of the PBS in dimerization of HIV-2 RNA (Dirac et al., 2001; Jossinet et al., 2001; Dirac et al., 2002; Lanchy and Lodmell, 2002). The results of the radioactively labeled oligonucleotide binding studies as well as the dimerization inhibition assays reported here show that in the 1-381 HIV-2 RNA construct, the PBS is directly involved in dimerization. The radiolabeled oligonucleotide partitioning experiments demonstrated that oligomer binding to the PBS region and dimerization were mutually exclusive events, while oligonucleotide binding to other parts of the RNA molecule was not affected by (or did not affect) the monomeric or dimeric state of the RNA molecule. This observation was further supported by the observation that PBS-region antisense oligonucleotides inhibited RNA dimerization, while other oligonucleotides could not. The ability of asROD258 to inhibit dimerization, but only if it was added prior to the high-temperature step of the dimerization procedure, suggests that the PBS domain normally attains a stable overall structure that is conducive to dimerization in the absence of oligonucleotide. However, an oligonucleotide that is able to disrupt this structure, even if it does not directly compete with another strand of viral RNA for hybridization at the PBS, is still capable of disrupting dimerization.
That an oligonucleotide is capable of inhibiting dimerization in one set of experimental conditions and not in another (e.g. asROD258) raises interesting questions about how an oligonucleotide hybridizes to its target sequence. In recent studies, the energetic and kinetic relationships between the competing forces of oligonucleotide folding/unfolding, RNA structural rearrangements, and oligonucleotide hybridization with and dissociation from the RNA were examined theoretically and experimentally (Patzel and Sczakiel, 2000; Walton et al., 2002). These studies emphasized the idea that using thermodynamic parameters alone to predict antisense efficacy is not realistic, and that the stronger determinant is the kinetics of hybridization. In the present case, we may surmise that when the oligonucleotide asROD258 is added to the RNA prior to dimerization, the kinetics of hybridization are faster than those of dimerization and it is effective in inhibiting dimerization. By contrast, when the RNA has already dimerized, the resulting target site is less accessible for hybridization thermodynamically (with respect to requisite RNA restructuring upon hybridization) and/or kinetically.
In summary, we have identified several potentially useful oligonucleotide hybridization sites on HIV-2 viral RNA. These sites could eventually be exploited as antiretroviral targets using antisense strategies, and they may also indicate an overall vulnerability of these regions of viral RNA. In addition, the combinatorial approach used here has yielded important structural and mechanistic information concerning the dimerization of HIV-2 viral RNA that emphasizes the dual-functionality of the PBS in its roles as a primer hybridization site as well as a dimerization promoting site.
Acknowledgements
We thank Marcela Majda for her efforts during the early phases of this work and John Ivanovitch and Hector Valtierra for critical reading of the manuscript and fruitful discussions. The proviral DNA clone pROD10 was provided by Drs. J.-M. Bechet and A. M. L. Lever through the Centralised Facility for AIDS Reagents supported by EU Programme EVA (contract QLK2-CT-1999-00609) and the UK Medical Research Council. E. L. D. and B. D. were supported by NSF-EPSCoR Undergraduate Research Fellowships (Grant number EPS-0091995). This research was supported by a National Institutes of Health Grant number AI45388 to J.S.L.
References
- Allawi HT, Dong F, Ip HS, Neri BP, Lyamichev VI. Mapping of RNA accessible sites by extension of random oligonucleotide libraries with reverse transcriptase. RNA. 2001;7:314–327. doi: 10.1017/s1355838201001698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B. Structural features in TAR RNA of human and simian immunodeficiency viruses: a phylogenetic analysis. Nucleic Acids Res. 1992;20:27–31. doi: 10.1093/nar/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B, Schoneveld I. Secondary structure of the HIV-2 leader RNA comprising the tRNA-primer binding site. Nucleic Acids Res. 1993;21:1171–1178. doi: 10.1093/nar/21.5.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B, van Wamel JL. Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J Virol. 1996;70:6723–6732. doi: 10.1128/jvi.70.10.6723-6732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clever JL, Parslow TG. Mutant human immunodeficiency virus type 1 genomes with defects in RNA dimerization or encapsidation. J Virol. 1997;71:3407–3414. doi: 10.1128/jvi.71.5.3407-3414.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirac AM, Huthoff H, Kjems J, Berkhout B. The dimer initiation site hairpin mediates dimerization of the human immunodeficiency virus, type 2 RNA genome. J Biol Chem. 2001;276:32345–32352. doi: 10.1074/jbc.M103462200. [DOI] [PubMed] [Google Scholar]
- Dirac AM, Huthoff H, Kjems J, Berkhout B. Regulated HIV-2 dimerization by means of alternative RNA conformations. Nucleic Acids Res. 2002;30:2647–2655. doi: 10.1093/nar/gkf381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrick M, Lear AL, Cann AJ, Heaphy S. Evidence that a kissing loop structure facilitates genomic RNA dimerisation in HIV-1. J Mol Biol. 1996;259:58–68. doi: 10.1006/jmbi.1996.0301. [DOI] [PubMed] [Google Scholar]
- Hirota M, Koyanagi Y, An DS, Iwanaga Y, Yamamoto N, Shimotohno K. Mutational analysis of the 5′ noncoding region of human immunodeficiency virus type 1 genome. Leukemia. 1997;3:102–105. [PubMed] [Google Scholar]
- Ho SP, Bao Y, Lesher T, Malhotra R, Ma LY, Fluharty SJ, Sakai RR. Mapping of RNA accessible sites for antisense experiments with oligonucleotide libraries. Nat Biotechnol. 1998;16:59–63. doi: 10.1038/nbt0198-59. [DOI] [PubMed] [Google Scholar]
- Ho SP, Britton DH, Bao Y, Scully MS. RNA mapping: selection of potent oligonucleotide sequences for antisense experiments. Methods Enzymol. 2000;314:168–183. doi: 10.1016/s0076-6879(99)14102-8. [DOI] [PubMed] [Google Scholar]
- Jossinet F, Lodmell JS, Ehresmann C, Ehresmann B, Marquet R. Identification of the in Vitro HIV-2/SIV RNA Dimerization Site Reveals Striking Differences with HIV-1. J Biol Chem. 2001;276:5598–5604. doi: 10.1074/jbc.M008642200. [DOI] [PubMed] [Google Scholar]
- Kuiken C, Foley B, Hahn B, Marx P, McCutchan F, Mellors J, Wolinsky S, Korber B. HIV Sequence Compendium 2001. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; Los Alamos: 2001. [Google Scholar]
- Lanchy JM, Lodmell JS. Alternate usage of two dimerization sites in HIV-2 viral RNA in vitro. J Mol Biol. 2002;319:637–648. doi: 10.1016/S0022-2836(02)00369-8. [DOI] [PubMed] [Google Scholar]
- Lanchy JM, Rentz CA, Ivanovitch JD, Lodmell JS.Elements located upstream and downstream of the major splice donor site influence the ability of HIV-2 leader RNA to dimerize in vitro Biochemistry 2003. in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughrea M, Jetté L. A 19-nucleotide sequence upstream of the 5′ major splice donor is part of the dimerization domain of human immunodeficiency virus 1 genomic RNA. Biochemistry. 1994;33:13464–13474. doi: 10.1021/bi00249a035. [DOI] [PubMed] [Google Scholar]
- Laughrea M, Jetté L, Mak J, Kleiman L, Liang C, Wainberg MA. Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J Virol. 1997;71:3397–3406. doi: 10.1128/jvi.71.5.3397-3406.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughrea M, Shen N, Jetté L, Wainberg MA. Variant effects of non-native kissing-loop hairpin palindromes on HIV replication and HIV RNA dimerization: role of stem-loop B in HIV replication and HIV RNA dimerization. Biochemistry. 1999;38:226–234. doi: 10.1021/bi981728j. [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Paillart JC, Berthoux L, Ottmann M, Darlix JL, Marquet R, Ehresmann B, Ehresmann C. A dual role of the putative RNA dimerization initiation site of human immunodeficiency virus type 1 in genomic RNA packaging and proviral DNA synthesis. J Virol. 1996;70:8348–8354. doi: 10.1128/jvi.70.12.8348-8354.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzel V, Sczakiel G. In vitro selection supports the view of a kinetic control of antisense RNA-mediated inhibition of gene expression in mammalian cells. Nucleic Acids Res. 2000;28:2462–2466. doi: 10.1093/nar/28.13.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves JD, Doms RW. Human immunodeficiency virus type 2. J Gen Virol. 2002;83:1253–1265. doi: 10.1099/0022-1317-83-6-1253. [DOI] [PubMed] [Google Scholar]
- Rhim H, Rice AP. Functional significance of the dinucleotide bulge in stem-loop1 and stem-loop 2 of HIV-2 TAR RNA. Virology. 1994;202:202–211. doi: 10.1006/viro.1994.1336. [DOI] [PubMed] [Google Scholar]
- Skripkin E, Paillart JC, Marquet R, Ehresmann B, Ehresmann C. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc Natl Acad Sci U S A. 1994;91:4945–4949. doi: 10.1073/pnas.91.11.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis D, Gotte D, Sanders-Buell E, Ritchey D, Salminen M, Carr J, McCutchan F. Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J Mol Biol. 1998;72:3991–3998. doi: 10.1128/jvi.72.5.3991-3998.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton SP, Stephanopoulos GN, Yarmush ML, Roth CM. Thermodynamic and kinetic characterization of antisense oligodeoxynucleotide binding to a structured mRNA. Biophys J. 2002;82:366–377. doi: 10.1016/S0006-3495(02)75401-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M, Mathews DH, Turner DH. Algorithms and Thermodynamics for RNA Secondary Structure Prediction: A Practical Guide. In: Barciszewski J, Clark BFC, editors. RNA Biochemistry and Biotechnology. Kluwer Academic Publishers; 1999. pp. 11–43. [Google Scholar]