

Several metalloenzymes that catalyze essential reactions in Nature have dinuclear active sites where the two metal ions are coordinated by carboxylate oxygen and histidine nitrogen atoms.1 First row transition metals, including manganese, iron, and zinc, typically occupy such positions. Although alkali and alkaline earth metal ions are present in high levels during the biosynthesis of these enzymes,2 little is known about their ability to become incorporated within these active centers. Metal-for-metal substitution in leucine aminopeptidase has been documented.3 However, substitution of one metal for another within synthetic polymetallic complexes is rare; a handful of examples are known in cluster chemistry.4 In this communication, we report the synthesis and structure of a carboxylate-bridged, heterodinuclear complex composed of iron and sodium,5 the metal-for-metal substitution of sodium by iron in this complex, and kinetic experiments that suggest a dissociative mechanism for the reaction. The results provide synthetic precedent of possible relevance to the assembly of carboxylate-bridged diiron active sites in proteins.6
The heterodinuclear sodium-iron complex 2 (Scheme 1) was initially identified by X-ray crystallography as a minor byproduct from the reaction of NaO2CTrp, Fe(BF4)2·6H2O, and the dipicolinic ester diethynyltriptycene ligand PIC2DET (1).7 With knowledge of its composition, a higher-yielding, rational synthetic route to the complex was devised. Reaction of 1 with 1 equiv of Fe(OTf)2·2MeCN and 3 equiv of NaO2CTrp, where Trp is 9-triptycenyl, in 1,2-dichloroethane resulted in the immediate formation of a blue-green solution with concomitant precipitation of 2 equiv of NaOTf.8 Crystallization of the crude product from chlorobenzene/Et2O furnished dark blue blocks of the complex [NaFe(PIC2DET)(μ-O2-CTrp)3](2) in 71% yield, the elemental analysis of which confirmed the presence of both iron and sodium (Supporting Information).
Scheme 1.
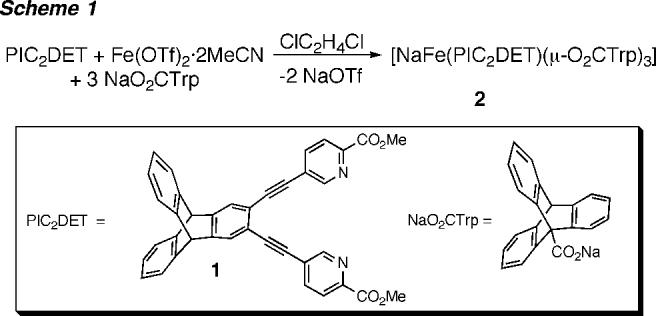
Dichroic blue-yellow blocks of 2 suitable for X-ray analysis were obtained by diffusion of Et2O into a CH2Cl2 solution of the compound. The crystal structure revealed that the iron and sodium atoms of 2 each have distorted trigonal-bipyramidal coordination geometries. The metal ions are bridged by three triptycene carboxylates with an Fe⋯Na distance of 3.181(2) Å (Figure 1), and each is coordinated to a pyridine nitrogen (N1 or N2) and carbonyl oxygen (O2 or O3) atom of 1, forming a five-membered chelate ring. Two of the bridging carboxylates in 2 are bidentate and the third, monodentate.9
Figure 1.
ORTEP diagram of [NaFe(PIC2DET)(μ-O2CTrp)3] (2) showing 30% probability thermal ellipsoids for all non-hydrogen atoms. Ligand 1 and the triptycene units of the bridging carboxylates are abbreviated for clarity. Selected interatomic distances (Å): Fe1⋯Na1, 3.181(2); Fe1-N1, 2.196(3); Fe1-O2, 2.184(3); Fe1⋯O5, 2.604(4); Fe1-O6, 2.131(3); Fe1-O7, 2.039(3); Fe1⋯O9, 2.320(4); Fe1-O10, 2.181(4); Na1-N2, 2.508-(4); Na1-O3, 2.348(4); Na1-O6, 2.308(4); Na1-O8, 2.282(4); Na1-O9, 2.285(4).
Because the triply bridged structure shown in Figure 1 is typical of cationic diiron(II) complexes,10 the identity of the Na atom in 2 was initially assigned using several indicators. First, the metal-ligand bond distances are considerably longer for Na1 than for Fe1. The Na1-N2 and Na1-O3 distances are 2.508(4) and 2.348(4) Å, respectively, whereas the respective Fe1-N1 and Fe1-O2 values are 2.196(3) and 2.184(3) Å. Second, during the X-ray analysis, attempts to assign Na1 as an iron atom gave an unusually large atomic displacement factor (U), which typically occurs when a lighter atom is incorrectly identified as a heavier one. Third, no counterion was found by X-ray analysis. A counterion would be required if 2 were a cationic diiron(II) complex.
Compound 2 contains a picolinic ester bound to iron or sodium.11 A linkage isomer involving the five-membered chelate ring of that unit was identified by X-ray crystallographic analysis of dichroic purple-colorless blocks that cocrystallized in small amounts with the blue-yellow blocks of 2. The full structure is similar to that of 2, with an Fe⋯Na distance of 3.160(2) Å. Details are provided in the Supporting Information (Structure B). In this isomer, the Fe1 is bound to O1, not by the carbonyl oxygen O2, with an Fe1-O1 distance of 2.363(2) Å.
The reaction of 2 with Fe(II) was examined (Scheme 2). Treatment of a solution of 2 in CH2Cl2 with 5 equiv of Fe(OTf)2·2MeCN under biphasic conditions12 resulted in a color change from blue to red within 10 min. UV-vis spectroscopy revealed a shift in λmax from 569 nm (ε ∼ 200 M-1 cm-1) in 2 to 472 nm (ε ∼ 310 M-1 cm-1)in 3.13 To establish the amount of iron required to drive the reaction to completion, a titration experiment was performed. Treatment of a MeCN solution of 2 (1 mM) with 4 equiv of Fe(OTf)2·2MeCN produced a change in the UV-vis spectrum up to the addition of 1.0 equiv. No further alteration occurred up to 4.0 equiv, consistent with only 1 equiv of iron reacting with 2.
Scheme 2.
The diiron complex 3 could be isolated as a microcrystalline purple solid by vapor diffusion of Et2O into a MeCN solution of 3, and its identity is supported by several measurements. Changes in the IR spectrum are consistent with iron substitution. In the carbonyl stretching frequency region, two absorptions appear at 1723 and 1694 cm-1 in 2, whereas one absorbance, at 1684 cm-1, occurs in 3, consistent with both picolinic esters binding to Fe(II).13 Moreover, strong absorptions at 1236, 1212, and 1030 cm-1, which did not appear in the spectrum of 2, indicate the presence of a triflate counterion. The structure of 3 was also assigned by ESMS, which revealed a molecular ion consistent with the formula [Fe2(PIC2DET)(O2CTrp)3]+. In addition, the molar conductivity of 3 measured in acetonitrile matches that of a 1:1 electrolyte.14
To gain insight into the mechanism of the metal-for-metal substitution shown in Scheme 2, the reaction was followed by stopped-flow spectroscopy in acetonitrile, a solvent in which both reactants are soluble. The kinetics of the reaction of 2 with Fe(OTf)2·2MeCN in MeCN at 24 °C (0.5-4 mM after mixing) were first order with respect to 2 and zero order with respect to Fe(OTf)2·2MeCN over the range of concentrations surveyed (kobs = 21 ± 2 s-1). This behavior is consistent with a dissociative mechanism (Scheme 3, kobs ≈ k1). A molar conductivity study of 2 confirms that it is partially dissociated in acetonitrile. Measurement of the temperature dependence of the first-order rate constant k1 over the range of -10 to 30 °C revealed a positive enthalpy of activation (ΔH‡ = 59 ± 6 kJ mol-1) and a negative entropy of activation (ΔSq = -20 ± 6 J mol-1 K-1). Negative entropies of activation are not typically observed for dissociative reactions, such as substitution of a ligand on a metal complex.15 However, the dissociation shown in Scheme 3 (2 f 4) is different because a metal cation is the leaving group. Negative entropies of activation have been reported for dissociation of alkali and alkaline earth metals from cryptands16 and may reflect the need for the metal cation to become partially solvated in the transition state.
Scheme 3.
In conclusion, the availability of the unusual carboxylate-bridged, heterodinuclear sodium-iron complex 2 has allowed the substitution of iron for sodium to be studied. To the best of our knowledge, this metal-for-metal exchange in a dinuclear coordination complex, which occurs by a dissociative mechanism, is the first of its kind. Finally, because the coordination environment of 2 resembles the cores of many dinuclear enzymes thought to bind only to transition metal ions, the possible occupancy of their active sites by abundant alkali or alkaline earth metal ions should be considered.
Acknowledgments
Acknowledgment. This work was supported by Grant GM32134 from the National Institute of General Medicine Sciences. J.J.K. is a National Institute of Health postdoctoral fellow (F32 GM069236-01). We thank Mr. Andy Tennyson for helpful discussions.
Footnotes
Supporting Information Available: Details for preparing 2 and 3; analytical, UV-vis, stopped-flow, molar conductivity, and X-ray crystallographic data, including tables, ORTEP diagrams, and CIF files. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- Messerschmidt A, Huber R, Poulos T, Wieghardt K, editors. Handbook of Metalloproteins. Wiley; Chichester, U.K.: 2001. [Google Scholar]
- 2.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. [Google Scholar]
- 3.Allen MP, Yamada AH, Carpenter FH. Biochemistry. 1983;22:3778–3783. doi: 10.1021/bi00285a010. See for the substitution of one Zn(II) by Mg(II) and Co(II) in the dizinc(II) enzyme leucine aminopeptidase. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zhang Y-H, Liu P, Xia C-H, Hu B, Yin Y-Q. J. Organomet. Chem. 2003;676:55–61. [Google Scholar]; (b) Sterenberg BT, Spivak GJ, Yap GPA, Puddephatt RJ. Organometallics. 1998;17:2433–2439. [Google Scholar]; (c) Konchenko SN, Virovets AV, Podberezskaya NV. Polyhedron. 1997;16:1689–1691. [Google Scholar]; (d) Konchenko SN, Virovets AV, Tkachev SV, Podberezskaya NV. Polyhedron. 1997;16:1549–1554. [Google Scholar]; (e) Brown SSD, Salter ID. J. Organomet. Chem. 1988;377:C31–34. [Google Scholar]
- 5.(a) Steinhauser S, Heinz U, Sander J, Hegetschweiler K. Z. Anorg. Allg. Chem. 2004;630:1829–1838. Compound 2 is one of a few crystallographically characterized, dinuclear sodium-iron complexes described to date. [Google Scholar]; (b) Hernández-Molina R, Mederos A, Dominguez S, Gili P, Ruiz-Pérez C, Castiñeiras A, Solans X, Lloret F, Real J. A. Inorg. Chem. 1998;37:5102–5108. Compound 2 is one of a few crystallographically characterized, dinuclear sodium-iron complexes described to date. [Google Scholar]; (c) Kornev AN, Chesnokova TA, Semenov VV, Zhezlova EV, Zakharov LN, Klapshina LG, Domrachev GA, Rusakov VS. J. Organomet. Chem. 1997;547:113–119. Compound 2 is one of a few crystallographically characterized, dinuclear sodium-iron complexes described to date. [Google Scholar]
- 6.(a) Bollinger JM, Jr., Chen S, Parkin SE, Mangravite LM, Ley BA, Edmondson DE, Huynh BH. J. Am. Chem. Soc. 1997;119:5976–5977. [Google Scholar]; (b) Bollinger JM, Jr., Tong WH, Ravi N, Huynh BH, Edmondson DE, Stubbe J. Methods Enzymol. 1995;258:278–303. doi: 10.1016/0076-6879(95)58052-2. [DOI] [PubMed] [Google Scholar]
- 7.Kodanko JJ, Morys AJ, Lippard SJ. Org. Lett. 2005;7:4585–4588. doi: 10.1021/ol051700h. The synthesis of 1 is described elsewhere. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vardhan HB, Bach RD. J. Org. Chem. 1992;57:4948–4954. The identity of NaOTf was confirmed by IR. [Google Scholar]
- 9.Rardin RL, Tolman WB, Lippard SJ. New J. Chem. 1991;15:417–430. [Google Scholar]
- 10.(a) Kuzelka J, Farrell JR, Lippard SJ. Inorg. Chem. 2003;42:8652–8662. doi: 10.1021/ic034928e. See for examples of triply carboxylate-bridged diiron complexes. [DOI] [PubMed] [Google Scholar]; (b) Lee D, Lippard SJ. Inorg. Chem. 2002;41:2704–2719. doi: 10.1021/ic020186y. See for examples of triply carboxylate-bridged diiron complexes. [DOI] [PubMed] [Google Scholar]; (c) He C, Lippard SJ. Inorg. Chem. 2001;40:1414–1420. doi: 10.1021/ic000975k. See for examples of triply carboxylate-bridged diiron complexes. [DOI] [PubMed] [Google Scholar]
- 11.March R, Clegg W, Coxall RA, Cucurull-Sánchez L, Lezama L, Rojo T, González-Duarte P. Inorg. Chim. Acta. 2003;353:129–138. Only a few metal-bound picolinic esters have been characterized by X-ray crystallography. See for recent examples and references therein. [Google Scholar]
- 12.Fe(OTf)2·2MeCN and NaOTf are insoluble in CH2Cl2
- 13.Hay RW, Clark CR. Transition Met. Chem. 1979;4:28–31. (Compare) The observed wavelength and extinction coefficients are consistent with a d-d transition, as measured for an ethyl picolinate Fe(II) complex. [Google Scholar]
- 14.Geary WJ. Coord. Chem. ReV. 1971;7:81–122. [Google Scholar]
- 15.Crabtree RH. The Organometallic Chemistry of the Transition Metals. John Wiley & Sons; New York: 2001. pp. 96–106. [Google Scholar]
- 16.(a) Cox BG, Firman P, Schneider H. Inorg. Chim. Acta. 1983;69:161–166. [Google Scholar]; (b) Loyola VM, Pizer R, Wilkins RG. J. Am. Chem. Soc. 1977;99:7185–7188. [Google Scholar]; (c) Ceraso JM, Smith PB, Landers JS, Dye JL. J. Phys. Chem. 1977;81:760–766. [Google Scholar]; (d) Cahen YM, Dye JL, Popov AI. J. Phys. Chem. 1975;79:1292–1295. [Google Scholar]