

Abstract
Human immunodeficiency virus (HIV) protease inhibitors (PIs) act as reversible noncompetitive inhibitors of GLUT4 with binding affinities in the low micromolar range and are known to contribute to alterations in glucose homeostasis during treatment of HIV infection. As aspartyl protease inhibitors, these compounds all possess a core peptidomimetic structure together with flanking hydrophobic moieties. To determine the molecular basis for GLUT4 inhibition, a family of related oligopeptides containing structural elements found in PIs was screened for their ability to inhibit 2-deoxyglucose transport in primary rat adipocytes. The peptide oxybenzylcarbonyl-His-Phe-Phe-O-ethyl ester (zHFFe) was identified as a potent inhibitor of zero-trans glucose flux with a Ki of 26 μM. Similar to PIs, transport inhibition by this peptide was acute, noncompetitive, and reversible. Within a Xenopus oocyte expression system, zHFFe acutely and reversibly inhibited GLUT4-mediated glucose uptake, whereas GLUT1 activity was unaffected at concentrations as high as 1 mM. The related photoactivatable peptide zHFF-p-benzoylphenylalanine-[125I]Tyr-O-ethyl ester selectively labeled GLUT4 in rat adipocytes and indinavir effectively protected against photolabeling. Furthermore, GLUT4 bound to a peptide affinity column containing the zHFF sequence and was eluted by indinavir. These data establish a structural basis for PI effects on GLUT4 activity and support the direct binding of PIs to the transport protein as the mechanism for acute inhibition of insulin-stimulated glucose uptake.
The clinical use of HIV1 protease inhibitors (PIs) has led to dramatic improvements in HIV-related morbidity and mortality (1). However, it is now recognized that these drugs directly contribute to several metabolic changes that significantly increase the risk of developing diabetes mellitus and cardiovascular disease (2, 3). Insulin resistance appears early in the development of this dysmetabolic syndrome and can occur in the absence of lipodystrophy or hyperlipidemia (4). Although the complexity of drug regimens used during highly active antiretroviral therapy has complicated efforts to characterize adverse effects that are the direct result of PI use, a definitive link to the development of insulin resistance has been established by both in vitro and in vivo studies (5-7).
Several mechanisms have been proposed to mediate PI-induced insulin resistance including changes in insulin signaling (8, 9), SREBP processing (10), and adipokine secretion (11). Our previous studies establishing that the insulin-responsive facilitative glucose transporter GLUT4 is acutely inhibited by PIs at pharmacologically relevant drug levels (12) have identified a direct molecular target for the cellular effects of these drugs. Several observations support the hypothesis that GLUT4 inhibition is produced by direct, noncovalent binding of PIs to a unique structural domain within the transport molecule. 1) Inhibition of glucose transport by low micromolar concentrations of PIs is observed following maximal insulin stimulation with GLUT4 already translocated to the cell membrane. 2) Inhibition is also observed in a heterologous Xenopus oocyte expression system that is not insulin-responsive. 3) In this same system, GLUT1-mediated transport is unaffected by millimolar concentrations of the PI indinavir. 4) These effects are observed on a time scale of seconds to minutes and would thus be incompatible with changes in gene or protein expression. 5) The inhibitory effects in vitro and in vivo are readily reversible following removal of the drug.
Despite these data, without direct evidence that PIs bind to GLUT4, it remains possible that the effects of PIs on GLUT4 activity are indirect. For example, the drugs could interact with a unique regulatory molecule that either binds to GLUT4 or reversibly alters its structure such as through phosphorylation. Elucidation of the specific structural features of PIs that confer their ability to inhibit GLUT4 would not only facilitate efforts to define the molecular mechanism for this effect but could also provide a rationale for a way to design newer generations of PIs that retain their efficacy in treating HIV infection without producing insulin resistance. We report here the identification of acute, potent, and isoform-selective peptide inhibitors of GLUT4 and provide evidence that this inhibition is because of direct binding of these compounds to the transporter protein.
EXPERIMENTAL PROCEDURES
Materials—Xenopus laevis frogs were purchased from Xenopus Express (Plant City, FL). Iodobeads, BCA reagent and aminolink beads were obtained from Pierce. Indinavir was acquired from Merck (White-house City, NJ). Na125I and [3H]2-deoxyglucose were purchased from Amersham Biosciences and American Radiolabeled (St. Louis, MO), respectively. Sep-Pak cartridges were obtained from Waters (Milford, MA). Dinonylphthalate was purchased from VWR Scientific (Westchester, PA). z-His-Phe-Phe-Bpa-Tyr-O-Et was synthesized by Anaspec (San Jose, CA) and z-His-Phe-Phe-Phe-Lys was made by Genosys (The Woodlands, TX). All other peptides were obtained from Bachem (King of Prussia, PA). Affinity-purified anit-GLUT4 antibody produced from partially purified GLUT4 was purchased from Chemicon (Temecula, CA). Polyclonal antibody directed toward the carboxyl terminus of GLUT4 was a kind gift from Mike Mueckler (Washington University, St. Louis, MO). Unless otherwise specified, all other reagents were acquired from Sigma (St. Louis, MO).
2-Deoxyglucose Uptake Measurements in Primary Rat Adipocytes— Adipocytes were prepared from rat epididymal fat pads as described previously (8). Adipocytes were stimulated with the addition of 1 μM insulin for 30 min at 37 °C immediately prior to use. 2-Deoxyglucose uptake measurements were performed by adding 200 μl of the cell suspension to 200 μl of Krebs-Ringer bicarbonate Hepes buffer (KRB) (120 mM NaCl, 4 mM KH2 PO4, 1 mM MgSO4, 1 mM CaCl2, 10 mM NaHCO3, 200 nM adenosine, 30 mM Hepes, pH 7.4) containing 1% bovine serum albumin together with the indicated concentration of peptide or indinavir in 5-ml plastic scintillation vials. After incubating the mixture for 1 min at 37 °C, the assay was initiated with the addition of 20 μl of [3H]2-deoxyglucose (final 2-deoxyglucose concentration of 50-200 μM; 0.5 μCi/assay). After 1 min at 37 °C, the assay was quenched by adding cytochalasin B to a final concentration of 40 μM. The cells were then immediately separated from labeling mix by spinning the reaction through dinonylphthalate as described previously (8), and the intracellular radioactivity was quantified by liquid scintillation counting.
Glucose Transport Activity in X. laevis Oocytes—X. laevis oocytes were prepared and microinjected as described previously with 50 ng of Glut isoform mRNA synthesized in vitro (9). Measurement of [3H]2-deoxyglucose uptake was performed on groups of 15-20 oocytes in Barth's saline at 22 °C for 30 min. All assays were performed using 50 μM 2-deoxyglucose, 0.5 μCi/assay unless otherwise indicated. Peptides were added to the assay mixture 6 min prior to the initiation of uptake assays. Reactions were terminated by washing the oocytes with ice-cold Barth's saline containing 20 mM phloretin. Each oocyte was then transferred to an individual scintillation vial, solubilized in 1% SDS, and incorporated radioactivity was determined by liquid scintillation counting.
Radioiodination of z-His-Phe-Phe-Bpa-Tyr-O-Et—A 100 μM solution of z-His-Phe-Phe-Bpa-Tyr-O-Et in 0.1 M phosphate buffer (2.5 ml) was incubated with two Iodobeads and 100 μCi of Na125I at room temperature with gentle agitation for 30 min. The beads were then removed and the solution was applied to a C18 Sep-Pak cartridge preequilibrated with distilled water. The cartridge was washed 3 times with 5 ml of water, and the peptide was eluted with 1 ml of Me2SO. Yield and purity were assessed by high pressure liquid chromatography analysis in comparison to a known amount of z-His-Phe-Phe-O-Et labeled with cold NaI. The amount of 125I-labeled peptide was assessed by liquid scintillation counting, and the efficiency of labeling was estimated from the yield of radiolabeled peptide divided by the total peptide concentration.
Affinity Labeling of Primary Rat Adipocytes—Primary rat adipocytes were incubated with 1-20 μM radiolabeled z-His-Phe-Phe-Bpa[125I]Tyr-O-Et in KRB at room temperature in a 60-mm Petri dish. The photolabel was activated by exposure to 254 nm UV light with three successive cycles of 30 s of light followed by 30 s of darkness (total UV exposure 90 s). The adipocytes were then collected on ice into a 5-ml centrifuge tube containing PE200 tubing and spun at 200 rpm for 4 min. The layer below the floating adipocytes was removed via the PE200 tubing, and the adipocytes were washed three times with 4 ml of ice-cold KRB to remove any unreacted peptide. Samples were then stored at -20 °C.
Peptide Affinity Chromatography—The peptide z-His-Phe-Phe-Phe-Lys was attached via amide linkage to aminolink-agarose beads according to the manufacturer's specifications. Prior to each experiment, the column was equilibrated in phosphate-buffered saline at room temperature. Purified primary rat adipocytes were lysed by rapid freeze thawing in phosphate-buffered saline containing 0.1% decylmaltoside and Sigma protease inhibitor mixture, centrifuged at 4000 rpm for 30 min, and the supernatant fatty layer was carefully removed. The remaining cell lysate was applied to the peptide column, which was then washed with 3-column volumes of phosphate-buffered saline. The bound transporter was eluted with phosphate-buffered saline containing 500 μM indinavir. The column effluent was collected in 0.5-ml fractions. The protein in each fraction was quantified by the BCA assay. SDS PAGE was performed by loading 5 μg of protein onto a 10% polyacrylamide gel. Protein staining was assessed with Ruby Red dye. GLUT4 content was assessed by Western blot analysis using a polyclonal antibody directed toward the carboxyl terminus of rat GLUT4.
RESULTS
Aromatic Peptide Effects on GLUT4 Activity—All HIV protease inhibitors share a similar core structure that resembles the aromatic peptide backbone that acts as the native substrate for the HIV type 1 protease (Fig. 1). We therefore hypothesized that synthetic peptides containing this basic structure would serve as effective inhibitors of glucose transport. To test this hypothesis we screened a family of aromatic di- and tripeptides for their affects on insulin-stimulated glucose uptake in primary rat adipocytes, which predominately express the insulin-responsive facilitative glucose transporter GLUT4 (13). With the addition of 200 μM each peptide to primary rat adipocytes, four compounds were identified with significant inhibitory effects on zero-trans 2-deoxyglucose flux (Fig. 2). Like PIs, all four of these peptides contain a highly aromatic core peptide structure flanked by hydrophobic moieties. None of the peptides with charged amino or carboxyl termini affected transport activity. The most potent inhibitor of glucose transport identified in this screen was z-His-Phe-Phe-O-Et (zHFFe), which is also a known substrate for the aspartyl protease pepsin (14).
FIG. 1.
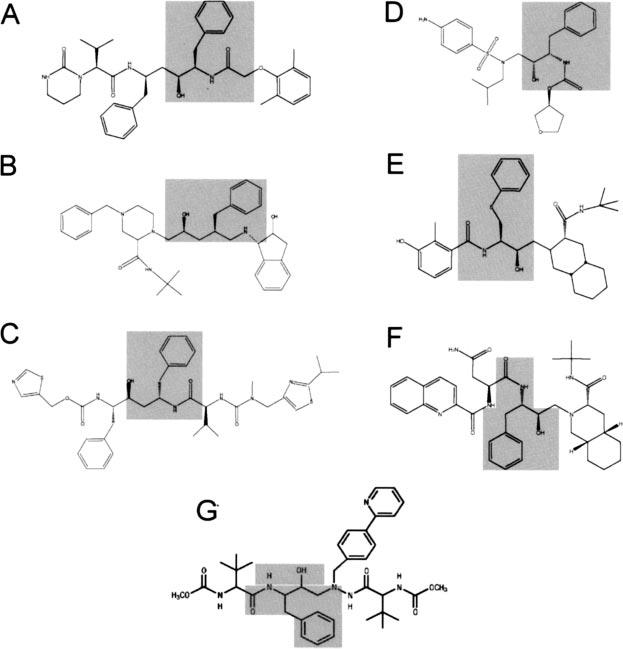
Structure features of HIV protease inhibitors. The core peptidomimetic structure found within all HIV protease inhibitors is shown in the shaded regions. A, amprenavir; B, indinavir; C, ritonavir; D, lopinavir; E, nelfinavir; F, saquinavir; G, atazanavir.
FIG. 2.
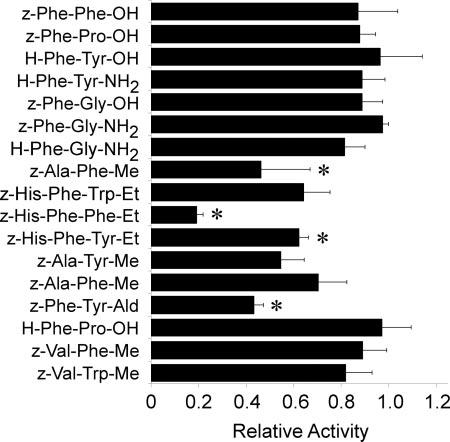
Inhibition of 2-deoxyglucose uptake by aromatic peptides in primary rat adipocytes. Aromatic peptides (200 μM) were added 1 min prior to measuring [3H]2-deoxyglucose transport activity (50 μM, 1 min at 37 °C). All assays were performed in triplicate and are expressed as the percent of a vehicle-treated control ±S.E. Asterisks (*) indicate p < 0.05 as determined by an analysis of variance.
Kinetic Characterization of the Inhibition of GLUT4 by z-His-Phe-Phe-O-Et—The ability of zHFFe to impair glucose transport was further investigated by studying the concentration dependence for inhibition in both primary rat adipocytes and Xenopus oocytes heterologously expressing the rat GLUT4 transporter isoform. As the Dixon plots shown in Fig. 3 demonstrate, this peptide acutely inhibited glucose transport in a concentration-dependent manner in both cell types. The apparent binding affinities for the peptide differed considerably between the adipocytes and oocytes, with Kis of 26 ± 2 and 205 ± 5 μM, respectively. This difference is comparable with that seen with indinavir-mediated inhibition of glucose uptake in these two cell types (5, 12). The intercept on the negative x-axis is indicative of noncompetitive inhibition under the kinetic conditions used, which is also identical to the inhibition pattern observed for indinavir in primary rat and 3T3-L1 adipocytes (12). Because of the rapidity of the inhibitory effects of zHFFe (occurring within minutes after adding the drug) in adipocytes following insulin stimulation, it is unlikely that zHFFe produces its effect through alterations in insulin signaling or GLUT4 translocation. This is further supported by the inhibitory effect of this peptide in the Xenopus oocyte system, in which cell surface expression of the glucose transporter is independent of insulin signaling (15).
FIG. 3.
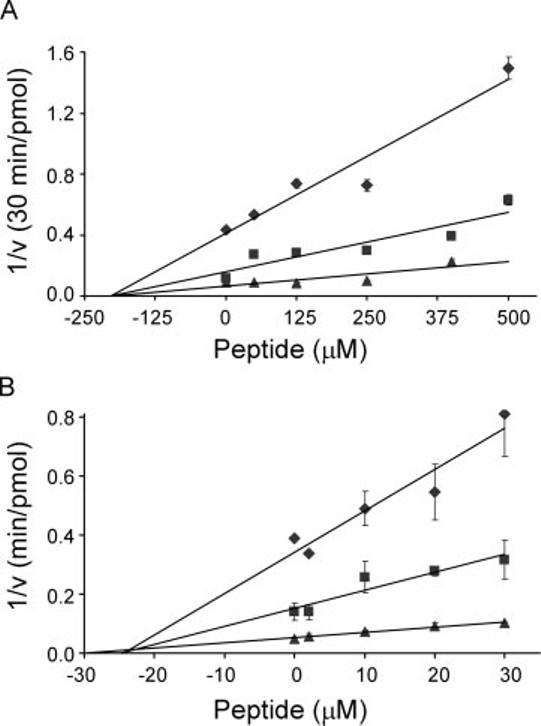
Kinetic analysis of GLUT4 inhibition by z-His-Phe-Phe-O-Et. A, the uptake of [3H]2-deoxyglucose (◆, 50μM; ■, 100μM; ▲, 200 μM) was measured in Xenopus oocytes 3 days after injecting GLUT4 mRNA. The peptide zHFFe was added 6 min prior to the start of each 30-min assay performed at 22 °C. B, [3H]2-deoxyglucose uptake was measured in insulin-stimulated primary rat adipocytes 1 min following the addition of zHFFe. Assays were carried out for 1 min at 37 °C. The data are expressed as Dixon plots where the vertical axis represents the reciprocal of the transport velocity. All points represent the mean ± S.E. of three determinations.
Isoform Selectivity and Reversibility of zHFFe Effects on Glucose Transport—One of the distinguishing features of PI-mediated effects on glucose transport is their selectivity for inhibition of GLUT4. In the Xenopus oocyte system, GLUT1 is resistant to inhibition by indinavir at millimolar concentrations, whereas GLUT4 is highly sensitive with a Ki of ∼50 μM (12). To determine the isoform selectivity of zHFFe, oocytes injected with mRNA for GLUTs 1-4 were tested for their sensitivity to acute and reversible inhibition of zero-trans 2-deoxyglucose transport. As shown in Fig. 4A, only GLUT4 was inhibited by 250 μM peptide, whereas concentrations of zHFFe up to 1 mM had no effect on GLUT1 or GLUT3. A small (32%) but statistically insignificant (p = 0.087) reduction in GLUT2 activity was observed with 1 mM zHFFe. This compares favorably with the behavior of PIs, which have been shown to inhibit GLUT2 at high concentrations in Xenopus oocytes (12). As shown in Fig. 4B, acute inhibition of GLUT4 was fully reversible following removal of the peptide. Full restoration of transport activity was observed irrespective of the time in which the oocytes were exposed to drug (1-30 min, data not shown).
FIG. 4.
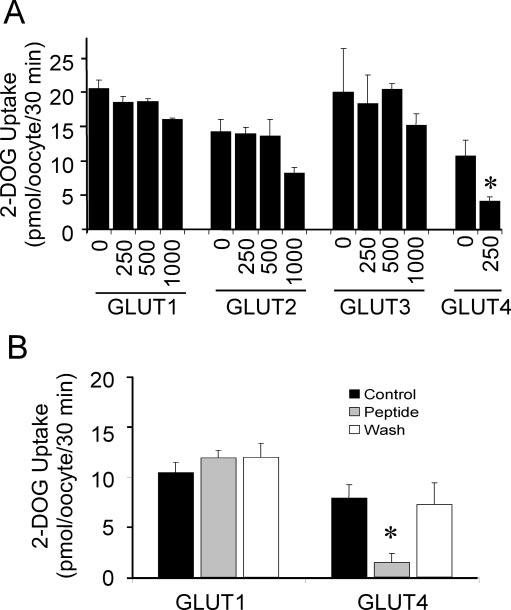
Reversible isoform-selective inhibition of GLUT4 activity by z-His-Phe-Phe-O-Et.A, [3H]2-deoxyglucose uptake (30 min at 22 °C) was measured 3 days following the injection of mRNA for each GLUT isoform into stage 5 Xenopus oocytes. The zHFFe peptide (dis-solved in dimethylformamide) was added 6 min prior to initiating the uptake assay. An equivalent volume of vehicle was added to the zero peptide control. Results represent the mean ± S.E. of three independent experiments. Asterisk (*) indicates p < 0.05. B, oocytes (15-20/group) were incubated for 60 s with 200 μM of z-His-Phe-Phe-O-Et (Peptide + Wash) or Bath's saline (Control). Oocytes were then either washed with peptide-free Barth's saline (Wash) or incubated in the continued presence of peptide (Peptide) during the 2-deoxyglucose up-take assay. Results represent the mean ± S.E.; asterisk (*) indicates p < 0.01 versus control.
Photolabeling with a Reactive Peptide Derivative—Having identified zHFFe as a peptide-based inhibitor of GLUT4 that shares inhibitory characteristics identical to those observed with the PI indinavir, a radiolabeled and photoreactive derivative of this peptide was synthesized and used to test the hypothesis that transport inhibition is due to a direct binding of the drug to the transport protein. The peptide z-His-Phe-Phe-Bpa-Tyr-O-Et was found to effectively inhibit glucose transport in primary rat adipocytes with a binding affinity comparable with the parent zHFFe compound (Fig. 5A). When the adipocytes were exposed to UV light to activate the Bpa residue, a concentration-dependent and irreversible inhibition of transport activity by the photactivatable peptide was observed (Fig. 5B). As shown in Fig. 6A, UV light exposure resulted in the photolabeling of a 50-kDa protein that corresponded precisely with the molecular mass of GLUT4. The degree of labeling directly correlated with the time of exposure to the UV light and was specific for this protein with up to 3 min of light exposure (data not shown). Because a large radioactive signal was present at the bottom of the gel, reflecting unbound peptide, it was not possible to definitively exclude the additional modification of a small molecular mass protein <10 kDa. Although initial attempts to immunoprecipitate the radiolabeled protein with polyclonal antibody directed toward the carboxyl terminus of GLUT4 were unsuccessful, the radiolabeled protein was effectively immunoprecipitated by affinity-purified polyclonal GLUT4 antibody recognizing epitopes throughout the transporter Fig. 6B. Moreover, indinavir effectively protected against photolabeling. Together, these observations support the conclusion that the photolabel is bound to the same GLUT4 site as PIs.
FIG. 5.
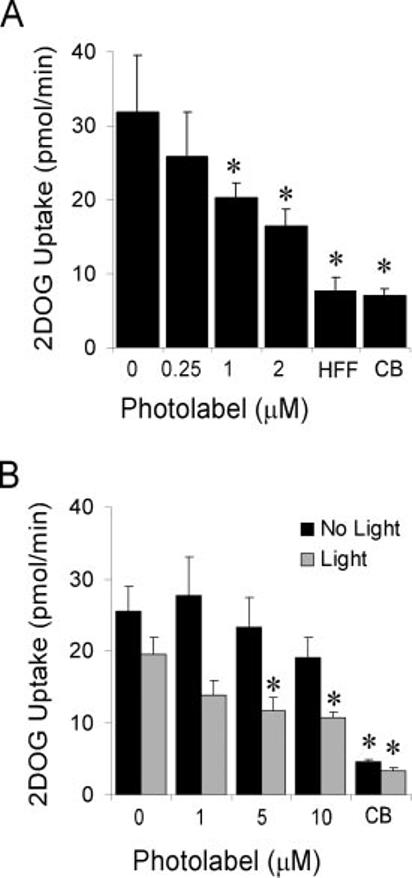
Inhibition of glucose uptake in rat adipocytes with z-His-Phe-Phe-Bpa-[125I]Tyr-O-Et. A, the indicated concentrations of peptide were added to insulin-stimulated primary rat adipocytes 1 min prior to measuring [3H]2-deoxyglucose uptake (1 min at 37 °C). HFF and CB represent positive controls in which 200 μM of the peptide zHFFe or 20 μM cytochalasin b, respectively, were added to the assay mixture. B, insulin-stimulated primary rat adipocytes were treated with the indicated concentrations of peptide and exposed to 254 nm light (black bars) or kept in the dark (gray bars) for 3 min at 37 °C. The adipocytes were then immediately washed three times with KRB to remove unreacted peptide before measuring residual 2-deoxyglucose transport activity. All assays were performed in triplicate and the values shown represent the mean ± S.E. Asterisks (*) indicate p < 0.05 compared with untreated control as determined by the Student's t test.
FIG. 6.
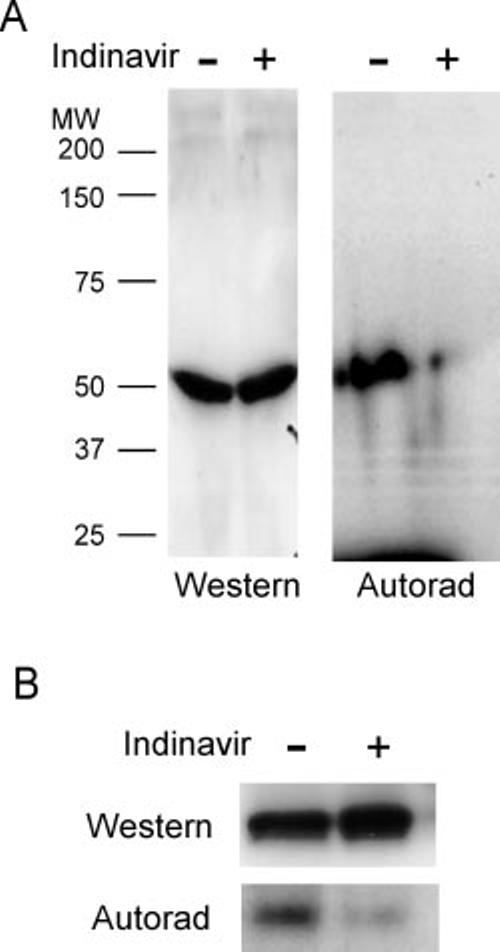
Photoaffinity labeling of GLUT4 with z-His-Phe-Phe-Bpa-[125I]Tyr-O-Et. A, SDS-PAGE analysis of primary rat adipocytes treated with 4 μM of photoactivated z-His-Phe-Phe-Bpa-[125I]Tyr-O-Et in the presence or absence of 500 μM indinavir. Total cell homogenates (10 μg/lane) were analyzed by Western blot analysis with polyclonal antibody directed toward the carboxyl terminus of GLUT4 (Western) or autoradiography (Autorad) to determine incorporated 125I label. B, the total cellular homogenates from photolabeled rat adipocytes were immunoprecipitated using affinity-purified polyclonal antibody directed toward partially purified GLUT4 and subjected to either Western blot analysis using the GLUT4 carboxyl-terminal antibody or autoradiography. Each lane contains 20 μg of protein.
Peptide Affinity Chromatography—The relative ease in modifying the core zHFF epitope that confers selective binding to GLUT4 provided a means to further investigate the relationship between aromatic peptide and PI-mediated inhibition of glucose transport. The peptide z-His-Phe-Phe-Phe-Lys, which contains the zHFF epitope, was linked to an agarose resin to generate an affinity column which could bind GLUT4. As shown in Fig. 7, when homogenates of primary rat adipocytes were applied to this resin, immunoreactive GLUT4 was found to bind to this column and could be selectively eluted with 500 μM indinavir. The native structure of GLUT4 appeared to be necessary for this interaction, because boiling of the adipocytes homogenate prior to loading the peptide column resulted in no immunodetectable GLUT4 binding to the column (data not shown).
FIG. 7.

Peptide affinity chromatography of GLUT4. Primary rat adipocytes homogenized and passed over a 1-ml peptide affinity column containing the peptide z-His-Phe-Phe-Phe-Lys linked via the lysine residue to an agarose resin. Shown is a Western blot of column fractions analyzed using polyclonal antibody directed toward the carboxyl terminus of rat GLUT4. Lane 1 represents the adipocyte homogenate (5 μg) prior to loading onto the peptide column. Column wash fractions are shown in lanes 2-4, and protein eluted with buffer containing 500 μM indinavir is shown in lanes 5-8 (50 μl/lane). Rat GLUT4 heterologously expressed in Xenopus oocytes is shown in lane C as a positive control.
DISCUSSION
Given the crystal-structure based rationale design of PIs, which target an aspartyl protease that selectively cleaves PhePro and Tyr-Pro bonds in a large polyprotein precursor of the developing HIV virus, it is not surprising that all of the currently available PIs contain some elementary aromatic peptidomimetic structure. The ability of these compounds to alter the intrinsic activity of facilitative glucose transporters, however, was an entirely unforeseen effect of these drugs. Because of the indisputable clinical utility of PIs in reducing HIV-associated morbidity and mortality (17), PIs are likely to remain a cornerstone of highly active antiretroviral therapy. Continuing efforts are clearly needed to design newer generations of PIs that retain their clinical utility without producing their adverse effects on glucose disposal.
Although millimolar concentrations of dipeptide metalloproteinase substrates have previously been reported to inhibit glucose uptake into rat adipocytes (18), the relatively high concentrations of peptides required to inhibit transport in these studies made it difficult to determine whether this effect was because of nonspecific cellular changes. The identification of high affinity, isoform-selective, reversible peptide-based inhibitors of facilitative glucose transport proteins together with the successful photolabeling of GLUT4 with a derivative of this peptide identifies a direct molecular mechanism for the inhibition of GLUT4 by these peptides and PIs.
Because the peptidomimetic phenylalanine moiety is the only feature that is consistently shared between PIs and the newly identified peptide inhibitors, this structure is likely to be a primary determinant in conferring the ability to inhibit GLUT4 activity. The shared participation of this structural feature in the binding of PIs to both the HIV protease and GLUT4 may present a challenge in new drug design. Attempts to change this core structure to prevent GLUT4 inactivation may render the candidate drug ineffective in inactivating the HIV protease. The recent development of atazanavir, a PI that does not appear to alter glucose uptake in 3T3-L1 adipocytes (19), provides optimism that the problem of GLUT4 inactivation by PIs can be circumvented. Reflecting the close correlation between the effects of PIs on GLUT4 activity in vitro and insulin sensitivity in vivo, atazanavir does not appear to contribute to changes in glucose homeostasis in HIV-infected patients who have been treated with this drug (20). Atazanavir differs from the other available PIs in several respects. Although atazanavir contains a similar peptide-like core, with two phenylalanine-like structures arranged in a unique C-2 axis of symmetry, an additional pyridine ring is attached to one of the phenylalanine residues (Fig. 1). Although the location of the GLUT4 domain involved in PI binding remains to be identified, it is possible that this additional ring structure sterically prevents access of the drug to this region of the protein.
Atazanavir also differs from other PIs by lacking the same degree of hydrophobicity in the flanking structural domains. Although the mechanism by which these hydrophobic moieties contribute to GLUT4 inhibition remains to be determined, all of the peptide inhibitors identified in this study contain an oxobenzylcarbonyl group at the amino terminus of the peptide. It is possible that this hydrophobicity facilitates entry of the drugs into the cell, thereby allowing access to a GLUT4 binding domain on the cytosolic surface of the transporter. The involvement of a cytosolic binding domain is suggested by our observation that the affinity-labeled transporter could not be immunoprecipitated using an antibody directed toward the carboxyl terminus. This could either be because of direct steric interference of the affinity label or an inhibitor-induced change in the protein conformation that leads to masking of this epitope. Because the epitope was recognized in the denatured protein on Western blots, the latter appears more likely. Unmasking of the carboxyl terminus of GLUT4 in response to insulin treatment has previously been reported in both primary rat adipocytes (21) and skeletal muscle (22). It has been postulated that this unmasking may contribute, together with GLUT4 trans-location, to the activation of this transporter following insulin stimulation. The identification of peptide inhibitors thus raises the interesting possibility that an endogenous protein or peptide interacts at this same location to regulate GLUT4 intrinsic activity.
Although the current study supports the hypothesis that PIs directly bind to GLUT4 in vitro, it remains possible that these drugs also interact with other molecules in vivo to alter glucose homeostasis and/or contribute to the other adverse metabolic effects observed during PI containing highly active antiretroviral therapy. Several recent studies have implied direct effects of PIs on glucose stimulated insulin release (16, 23). Although the differences observed in the ability of PIs and the peptide inhibitors to inhibit GLUT4 when measured in primary adipocytes versus oocytes may be merely reflective of differences in the assay procedures used, it is also possible that GLUT4 sensitivity is dependent upon the membrane environment of the transporter. If this is true, significant effects of zHFFe on GLUT2 activity in β-cells and/or hepatocytes may still occur at pharmacologically relevant drug levels. Future studies will be needed to determine whether these newly identified peptide inhibitors or related proteins are capable of photoaffinity labeling glucose transporters and/or other proteins in other tissues involved in glucoregulation, including skeletal muscle.
In addition to their utility in assisting the development of newer and safer PIs, the relative ease in designing and modifying peptide-based GLUT inhibitors provides a new approach to the development of reagents to study facilitative glucose transport. The unique selectivity of PIs and the newly identified peptide inhibitors of GLUT4 provide a facile way to acutely and reversibly induce insulin resistance in cell culture and whole animal model systems. With a more detailed understanding of the structural basis for PI binding to GLUT4, it may be possible to design additional reagents that selectively interact with other GLUT isoforms.
Footnotes
The abbreviations used are: HIV, human immunodeficiency virus; Bpa, benzoylphenylalanine; Me2SO, dimethyl sulfoxide; Et, ethyl ester; KRB, Krebs-Ringer bicarbonate Hepes buffer; PI, HIV protease inhibitor; SREBP, sterol response element-binding protein; z, oxybenzylcarbonyl; zHFFe, oxybenzylcarbonyl-His-Phe-Phe-O-ethyl ester.
This research was supported by Grants AI 49747 and DK064572 from the National Institutes of Health, the American Diabetes Association, and by the Diabetes Research Training Center at Washington University School of Medicine. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Palella FJ, Jr., Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. N. Engl. J. Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 2.Carr A, Samaras K, Burton S, Law M, Freund I, Chisholm DJ, Cooper DA. AIDS. 1998;12:F51–F58. doi: 10.1097/00002030-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Mulligan K, Grunfeld C, Tai VW, Algren H, Pang M, Chernoff DN, Lo JC, Schambelan M. J. Acquired Immune Defic. Syndr. 2000;23:35–43. doi: 10.1097/00126334-200001010-00005. [DOI] [PubMed] [Google Scholar]
- 4.Noor M, Lo JC, Mulligan K, Halvorsen R, Schwarz JM, Shambelan M, Grunfeld C. Antiviral Therapy. 2000;5:8. [Google Scholar]
- 5.Murata H, Hruz PW, Mueckler M. J. Biol. Chem. 2000;275:20251–20254. doi: 10.1074/jbc.C000228200. [DOI] [PubMed] [Google Scholar]
- 6.Hruz P, Murata H, Qiu H, Mueckler M. Diabetes. 2002;51:937–942. doi: 10.2337/diabetes.51.4.937. [DOI] [PubMed] [Google Scholar]
- 7.Noor MA, Seneviratne T, Aweeka FT, Lo JC, Schwarz JM, Mulligan K, Schambelan M, Grunfeld C. AIDS. 2002;16:F1–F8. doi: 10.1097/00002030-200203290-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cammalleri C, Germinario RJ. J. Lipid Res. 2003;44:103–108. doi: 10.1194/jlr.m200245-jlr200. [DOI] [PubMed] [Google Scholar]
- 9.Schutt M, Meier M, Meyer M, Klein J, Aries SP, Klein HH. Diabetologia. 2000;43:1145–1148. doi: 10.1007/s001250051505. [DOI] [PubMed] [Google Scholar]
- 10.Caron M, Auclair M, Vigouroux C, Glorian M, Forest C, Capeau J. Diabetes. 2001;50:1378–1388. doi: 10.2337/diabetes.50.6.1378. [DOI] [PubMed] [Google Scholar]
- 11.Xu A, Yin S, Wong L, Chan KW, Lam KS. Endocrinology. 2004;145:487–494. doi: 10.1210/en.2003-1140. [DOI] [PubMed] [Google Scholar]
- 12.Murata H, Hruz P, Mueckler M. AIDS. 2002;16:859–863. doi: 10.1097/00002030-200204120-00005. [DOI] [PubMed] [Google Scholar]
- 13.Holman GD, Kozka IJ, Clark AE, Flower CJ, Saltis J, Habberfield AD, Simpson IA, Cushman SW. J. Biol. Chem. 1990;265:18172–18179. [PubMed] [Google Scholar]
- 14.Inouye K, Voynick IM, Delpierre GR, Fruton JS. Biochemistry. 1966;5:2473–2483. doi: 10.1021/bi00871a044. [DOI] [PubMed] [Google Scholar]
- 15.Keller K, Strube M, Mueckler M. J. Biol. Chem. 1989;264:18884–18889. [PubMed] [Google Scholar]
- 16.Woerle HJ, Mariuz PR, Meyer C, Reichman RC, Popa EM, Dostou JM, Welle SL, Gerich JE. Diabetes. 2003;52:918–925. doi: 10.2337/diabetes.52.4.918. [DOI] [PubMed] [Google Scholar]
- 17.Schwarcz SK, Hsu LC, Vittinghoff E, Katz MH. Am. J. Epidemiol. 2000;152:178–185. doi: 10.1093/aje/152.2.178. [DOI] [PubMed] [Google Scholar]
- 18.Aiello LP, Wessling-Resnick M, Pilch PF. Biochemistry. 1986;25:3944–3950. doi: 10.1021/bi00361a031. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Mulvey R, Laing N, Leet J, Flint J, Parker RA. Antiviral Therapy. 2002;7:L6. [Google Scholar]
- 20.Murphy R, Pokrovsky V, Rozenbaum W. Proceedings and Abstracts of the 10th Conference on Retroviruses and Opportunistic Infections (Boston); Abstract 555, Foundation for Retrovirology and Human Health; Alexandria, VA. February 10-14, 2003.2003. [Google Scholar]
- 21.Smith RM, Charron MJ, Shah N, Lodish HF, Jarett L. Proc. Natl. Acad. Sci. U. S. A. 1991;88:6893–6897. doi: 10.1073/pnas.88.15.6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W, Hansen PA, Marshall BA, Holloszy JO, Mueckler M. J. Cell Biol. 1996;135:415–430. doi: 10.1083/jcb.135.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koster JC, Remedi MS, Qiu H, Nichols CG, Hruz PW. Diabetes. 2003;52:1695–1700. doi: 10.2337/diabetes.52.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]