

Abstract
The crystal structures, dimer stabilities, and kinetics have been analyzed for wild-type human immunodeficiency virus type 1 (HIV-1) protease (PR) and resistant mutants PRL24I, PRI50V, and PRG73S to gain insight into the molecular basis of drug resistance. The mutations lie in different structural regions. Mutation I50Valters a residue in the flexible flap that interacts with the inhibitor, L24I alters a residue adjacent to the catalytic Asp25, and G73S lies at the protein surface far from the inhibitor-binding site. PRL24I and PRI50V, showed a 4% and 18% lower kcat/Km, respectively, relative to PR. The relative kcat/Km of PRG73S varied from 14% to 400% when assayed using different substrates. Inhibition constants (Ki) of the antiviral drug indinavir for the reaction catalyzed by the mutant enzymes were about threefold and 50-fold higher for PRL24I and PRI50V, respectively, relative to PR and PRG73S. The dimer dissociation constant (Kd) was estimated to be approximately 20 nM for both PRL24I and PRI50V, and below 5 nM for PRG73S and PR. Crystal structures of the mutants PRL24I, PRI50V and PRG73S were determined in complexes with indinavir, or the p2/NC substrate analog at resolutions of 1.10–1.50 Å. Each mutant revealed distinct structural changes relative to PR. The mutated residues in PRL24I and PRI50V had reduced intersubunit contacts, consistent with the increased Kd for dimer dissociation. Relative to PR, PRI50V had fewer interactions of Val50 with inhibitors, in agreement with the dramatically increased Ki. The distal mutation G73S introduced new hydrogen bond interactions that can transmit changes to the substrate-binding site and alter catalytic activity. Therefore, the structural alterations observed for drug-resistant mutations were in agreement with kinetic and stability changes.
Keywords: aspartic protease, catalysis, non-active site mutants, indinavir, substrate analog
Abbreviations used: HIV-1, human immunodeficiency virus type 1; PR, wild-type HIV-1 protease; PRL24I, PR with L24I mutation; PRI50V, PR with I50V mutation; PRG73S, PR with G73S mutation; Nle, norleucine; DMSO, dimethylsulfoxide; RMS, root-mean-square
Introduction
Inhibitors of human immunodeficiency virus type 1 (HIV-1) protease (PR) are important components in clinical therapy for AIDS. However, the long-term therapeutic efficacy of PR inhibitors is limited, due to the rapid development of drug resistance. Extensive mutations in the 99 residue PR have been found in clinical isolates that provide resistance to inhibitors.1,2 PR acts during viral maturation to hydrolyze the peptide bond at specific cleavage sites in the viral Gag and Gag-Pol polyprotein precursors and produce the viral structural proteins and enzymes.3 Structurally, the PR dimer forms an extended binding site for about seven residues (P3–P4′) of a peptide substrate within subsites S3–S4′. Most clinical inhibitors bind primarily in PR subsites S2–S2′. Hence, the observed drug-resistant mutations have been classified either as substitutions in the active site (inhibitor-binding site) that directly influence inhibitor binding, or as substitutions of non-active site residues with indirect influences. Many commonly occurring mutations and specific combinations have been characterized.4 PR mutants have shown a range of structural and kinetic effects that depend on the specific combination of mutation with substrate or inhibitor. Variations in catalytic activity, inhibition constants, and stability relative to the wild-type enzyme were observed, independent of the location of the mutation.3,5–9 The mutations of non-active site residues have shown significant contributions to drug resistance.10,11 Previously, we analyzed structural differences among crystal structures of the mature HIV-1 PR bearing either single or double substitutions in complexes with substrate analogs.8,12
Indinavir was one of the first PR inhibitors in clinical use. High levels of resistance to indinavir were associated with substitutions of up to 11 PR residues in different combinations.13 The crystal structures of drug-resistant HIV PRs with multiple mutations have been reported in complexes with indinavir.14–16 Our analysis of the high-resolution crystal structures of HIV PR, and the common indinavir-resistant mutants PRV82A and PRL90M in complexes with indinavir, showed structural changes consistent with differences in their enzymatic activity.17 However, biochemical and biophysical analyses have not been performed for other mutations that are consistently observed at lower frequency. Mutations L24I and G73S are observed in about 10% and 5%, respectively, of patients exposed to indinavir. These rare mutations are generally observed in combination with other resistant mutations. Mutations of Gly73 appear in patients exposed to multiple PR inhibitors, and are often found in combination with L90M.4 The effects of these mutations have been compared to that of I50V, which is rarely observed on exposure to indinavir (0.2%); however, it is found in 30% of patients exposed to amprenavir as their first PR inhibitor. PRI50V showed higher Ki values for saquinavir, indinavir, and nelfinavir in biochemical studies.19 Mutations L24I, I50V, and G73S alter residues in different regions of the PR dimer structure, as shown in Figure 1. I50V alters a residue at the tip of the flexible flap that forms part of the inhibitor-binding site. L24I is next to the catalytic Asp25 but has no direct contact with inhibitor, while G73S is located far from the inhibitor-binding site. These mutants provide good models to help dissect the varied molecular mechanisms of drug resistance.
Figure 1.
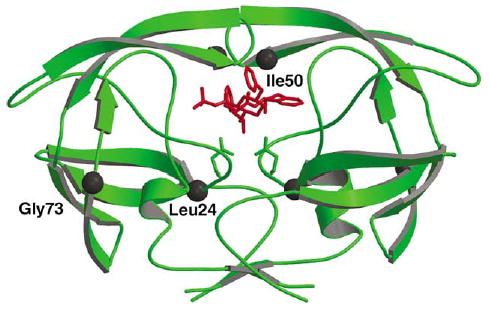
PR dimer structure (green ribbons) with indinavir (red bonds). The sites of mutation are indicated by black spheres for Leu24, Ile50 and Gly73. Only one subunit is labeled.
Here, we report the kinetics, dimer stability, and crystal structures of the HIV drug-resistant mutants PRL24I, PRI50V, and PRG73S. Crystal structures were determined for PRL24I, PRI50V, and PRG73S in complexes with indinavir, while PRL24I and PRI50V structures were determined also with a peptide analog of the p2/NC cleavage site in order to analyze the interactions with both substrate and inhibitor. Atomic details from these new crystal structures will be important for the design of second-generation inhibitors to circumvent the development of drug resistance.
Results and Discussion
Kinetics and stability
Kinetic parameters were determined for the resistant mutants using the spectrophotometric substrate (K-A-R-V-Nle-p-nitroPhe-E-A-Nle-amide) (Nle is norleucine), which is an analog of the HIV-1 CA/p2 cleavage site (Table 1). The two mutants PRL24I and PRI50V showed lower kcat/Km values of 3.7% and 18%, respectively, relative to PR. The decrease in kcat/Km for PRI50V was primarily due to an increase in Km, whereas for PRL24I, it was due to both an increase in Km and a decrease in kcat. PRG73S and PR showed similar kcat/Km values for this substrate. Therefore, PRG73S was tested for the hydrolysis of three other peptide substrates, representing different cleavage sites in Gag and Gag-Pol polyproteins: K-A-R-V-L*A-E-A-M-S (CA/p2) and V-S-F-N-F*P-Q-I-T-K-K (p6Pol/PR) and E-R-Q-A-N*F-L-G-K-I (NC/p1) (where * indicates the hydrolyzed peptide bond) (Table 2). PRG73S showed more variation in the Km values than in the kcat values. The relative kcat/Km values were 14%, 27.5% and 390% for the CA/p2, NC/p1 and p6Pol/PR peptides, respectively, suggesting significant differences in substrate specificity compared to PR.
Table 1.
Kinetic parameters for hydrolysis of spectroscopic substrate (KARV-Nle-p-nitroPhe-EA-Nle-amide) (CA/p2#)
| Protease | Substrate | Km (μM) | kcat (min−1) | kcat/Km (min−1μM−1) | Relative kcat/Km |
|---|---|---|---|---|---|
| PRa | CA/p2# | 55±7 | 290±10 | 5.2±0.2 | 100 |
| PRL24I | CA/p2# | 310±45 | 61±4 | 0.19±0.03 | 3.7 |
| PRI50V | CA/p2# | 500±36 | 480±19 | 0.93±0.08 | 18 |
| PRG73S | CA/p2# | 46±4 | 280±65 | 6.1±0.6 | 117 |
PR values were taken from Mahalingam et al.17
Table 2.
Kinetic parameters from the HPLC assay for hydrolysis of peptides KARVL*AEAMS (CA/p2) and VSFNF*PQITKK (p6Pol/PR) and ERQAN*FLGKI (NC/p1)
| Protease | Substrate | Km (μM) | kcat (min−1) | kcat/Km (min−1μM−1) | Relative kcat/Km |
|---|---|---|---|---|---|
| PRa | CA/p2 | 164±9 | 26±2 | 0.16 | 100 |
| PRG73S | CA/p2 | 680±170 | 15±1 | 0.022 | 14 |
| PRa | p6Pol/PR | 253±15 | 105±2 | 0.42 | 100 |
| PRG73S | p6Pol/PR | 61±11 | 100±4 | 1.64 | 390 |
| PR | NC/p1 | 234±68 | 187±35 | 0.80 | 100 |
| PRG73S | NC/p1 | 347±25 | 76.2±3.6 | 0.22 | 27.5 |
PR values were taken from Mahalingam et al.8
The stability of these three mutants, as assessed by urea-denaturation, was reduced to 50–60% for PRL24I and PRI50V, and about 80% for PRG73S relative to the PR value (Figure 2(a)). Consistent with the lower catalytic activity and susceptibility to urea-denaturation, the dissociation constant (Kd) was approximately 20 nM for both PRL24I and PRI50V (Figure 2(b)). In contrast, PRG73S exhibited no significant decrease in specific proteolytic activity at the lowest measured concentration of protein similar to PR, which showed no dissociation at a concentration of 5 nM.20,21
Figure 2.
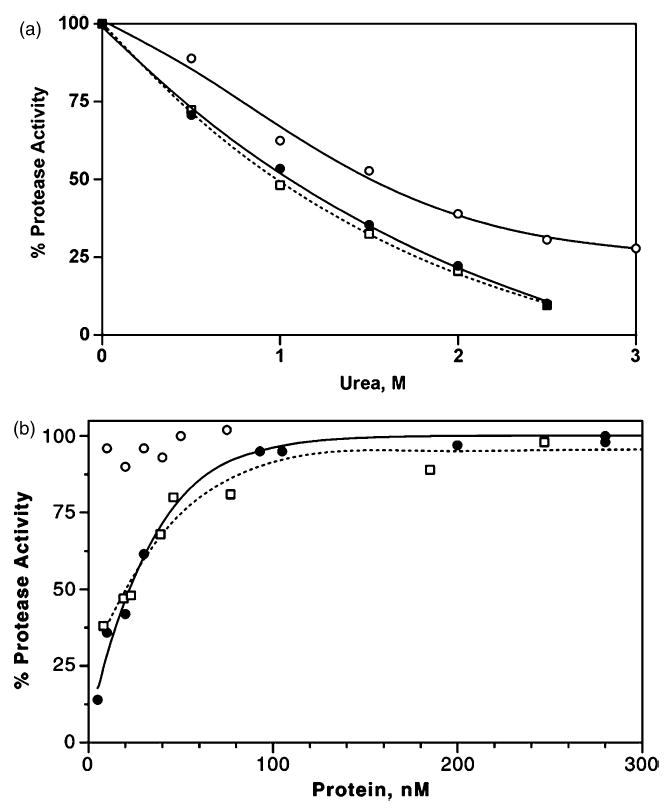
Protease stability. (a) Sensitivity to urea: L24I, filled circles and continuous line (UC50 = 1.05M); I50V, open squares and dotted line (UC50 = 0.97 M); G73S, open circles and continuous line (UC50 = 1.54M). (b) Dimer dissociation: L24I, filled circles and continuous line (Kd = 22 nM); I50V, open squares and dotted line (Kd = 19 nM); G73S, open circles (no dissociation observed).
Inhibition
The three mutants were assayed for inhibition by the clinical inhibitor indinavir, and two substrate analog inhibitors, R-V-L-r-F-E-A-Nle (CA/p2) and Ace-T-I-Nle-r-Nle-Q-R (p2/NC) (r is the reduced peptide bond), that represent two cleavage sites in Gag (Table 3). PRI50V was poorly inhibited by indinavir with about 50-fold higher inhibition constant (Ki), and about 19-fold higher Ki with the p2/NC analog, and about threefold with the CA/p2 analog as compared to PR. In contrast, PRL24I showed relatively strong inhibition by the CA/p2 analog with Ki of 0.05-fold of the PR value, while the inhibition by indinavir was 2.6-fold and by p2/NC was similar to the PR value. Inhibition of the hydrolytic reaction catalyzed by PRG73S was similar to PR for indinavir and p2/NC, and was 4.4-fold of the PR value for CA/p2 (Table 2). Therefore, the mutant PRI50V had the largest effect on inhibition, while PRG73S was most similar to wild-type PR.
Table 3.
Inhibition constants for indinavir (IDV), the reduced peptide analogs CA/p2 (RVL-r-FEA-Nle) and p2/NC (Ace-TI-Nle-r-Nle-QR), where r is the reduced peptide bond
| Inhibition constant (Ki)
|
|||
|---|---|---|---|
| Protease | IDV (nM) | CA/p2 (nM) | p2/NC (μM) |
| PR | 0.54 | 75 | 2.17 |
| PRL24I | 1.40 (2.6) | 3.5 (0.05) | 2.0 (0.9) |
| PRI50V | 27.0 (50) | 230 (3.0) | 41 (19) |
| PRG73S | 0.55 (1.0) | 330 (4.4) | 3.3 (1.6) |
Values relative to PR are given in parentheses.
Crystal structures
We determined the crystal structures of mutants PRL24I, PRI50V, and PRG73S in complex with indinavir, and mutants PRL24I and PRI50V with the substrate analog p2/NC in order to identify any structural changes compared to the wild-type PR. We did not obtain good diffraction data for crystals of PRG73S with p2/NC. The crystallographic data collection and refinement statistics are shown in Table 4. The crystal structures of PRL24I-p2/NC, PRL24I-IDV and PRI50V-IDV were refined to R-factors of 10.6–10.8% at the highest resolution of 1.10 Å. The structures of PRI50V-p2/NC, and PRG73S-IDV were refined to an R-factor of 11.1–14.4% at resolutions of 1.30–1.50 Å. The crystal structures had one dimer in the asymmetric unit of space groups P212121 or P21212, except for PRG73S-IDV, which had two dimers in the asymmetric unit of space group P21, as observed previously.7 Overall, the main-chain structure of the dimers was very similar and superimposed with root-mean-square (RMS) differences of <0.3 Å for pairs in the same space group, and 0.5–0.6 Å for comparison of dimers in two different space groups. The indinavir was bound in two alternate conformations in the dimers of PRI50V and PRG73S with relative occupancies of 0.8/0.2 and 0.6/0.4, respectively. Both dimers of PRG73S had similar relative occupancies for the inhibitors. The inhibitor showed one orientation in all the other structures. The electron density map of indinavir in PRL24I-IDV is shown in Figure 3.
Table 4.
Crystallographic data statistics
| Protease mutant | L24I | I50V | L24I | I50V | G73S | |
| Inhibitors | p2/NC | p2/NC | IDV | IDV | IDV | |
| Space group | P21212 | P21212 | P212121 | P212121 | P21 | |
| Unit cell dimensions | a (Å ) | 58.2 | 57.9 | 51.5 | 51.3 | 51.3 |
| b (Å ) | 85.9 | 86.0 | 58.6 | 58.4 | 62.7 | |
| c (Å ) | 46.5 | 46.5 | 61.6 | 61.0 | 59.2 | |
| β (deg.) | 90.0 | 90.0 | 90.0 | 90.0 | 98.15 | |
| Unique reflections | 91,188 | 57,183 | 75,564 | 72,654 | 56,309 | |
| Rmerge overall (%) (final shell) | 6.8 (13.2) | 5.8 (28.1) | 5.7 (12.2) | 5.6 (17.7) | 6.5 (23.9) | |
| I/σ(I) overall (final shell) | 25.24 (9.9) | 26.79 (4.14) | 28.23 (7.38) | 33.81 (5.07) | 17.42 (4.51) | |
| Resolution range for refinement (Å) | 10–1.10 | 10–1.30 | 10–1.10 | 10–1.10 | 10–1.50 | |
| Rwork (%) | 10.62 | 11.14 | 10.84 | 10.72 | 14.43 | |
| Rfree (%) | 13.22 | 14.41 | 13.81 | 14.12 | 21.62 | |
| No. water molecules | 252.0 | 313.0 | 202 | 240.5 | 257.5 | |
| Completeness overall (%) (final shell) | 95.9 (84.0) | 92.2 (93.9) | 98.9 (90.0) | 98.0 (85.9) | 94.1 (70.5) | |
| RMS deviation from ideality | ||||||
| Bond lengths (Å) | 0.016 | 0.013 | 0.015 | 0.015 | 0.008 | |
| Angle distance (Å) | 0.035 | 0.029 | 0.034 | 0.036 | 0.027 | |
| Average B-factors (Å2) | ||||||
| Main-chain | 7.8 | 10.1 | 11.0 | 9.3 | 18.3 | |
| Side-chain | 10.9 | 12.7 | 13.2 | 11.3 | 23.4 | |
| Inhibitor | 9.8 | 14.8 | 10.5 | 8.8 | 15.2/18.5a | |
| Solvent | 24.1 | 27.3 | 24.2 | 24.5 | 28.4 |
The two numbers represent the average B-factors for inhibitors in the two dimers in an asymmetric unit.
Figure 3.
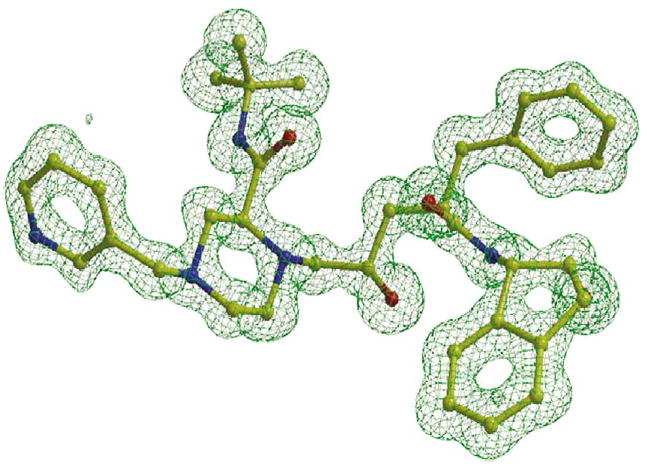
Omit map for indinavir in the crystal structure of PRL24I–IDV contoured at a level of 3.5σ.
The atomic B-factors were especially low for the protein and inhibitor atoms in the structures at 1.10 Å resolution; the average B values were 8–11 Å2 for main-chain and inhibitor atoms, and 11–13 Å2 for side-chain atoms. The average B-factors increased as the resolution decreased. The 1.5 Å resolution structure of PRG73S-IDV had average B-factors of 18.3 Å2 and 23.4 Å2 for main-chain and side-chain atoms, respectively, and 15.2 Å2 and 18.5 Å2 for the atoms of the two inhibitors, consistent with the lower resolution and greater difference (7%) between Rwork and Rfree. From 202 to 313 solvent molecules were modeled for the different structures, with average B-factors of 24.1–28.4 Å2.
Alternate conformations were observed for many amino acid side-chains and some main-chain atoms, especially for the highest-resolution structures (Figure 4(a)). There were 42 side-chains modeled with alternate conformations for PRL24I-IDV, 39 for PRL24I-p2/NC, 33 for PRI50V-IDV, 28 for PRI50V-p2/NC, and 44 for the two dimers in the PRG73S-IDV structure. Main-chain atoms with alternate conformations were modeled for residues in the surface turns of 39–41 in PRI50V-IDV and 66–69 in PRL24I-p2/NC. There were alternate conformations for the side-chains of Nle P1′ and ArgP3′ of the peptide analog p2/NC in the two mutant structures. The crystal structures that were refined at 1.1 Å resolution had the most alternate conformations. However, none of the residues consistently showed alternate conformations in both subunits of all structures. Moreover, the structures can show different relative occupancies for the alternate conformations of the same residue. The side-chains of Glu21, Met46, Val82 and Leu97 had alternate conformations in both subunits of most structures. Met46 and Val82 are located near the inhibitor and frequently are mutated in drug-resistant variants. All the mutated residues showed alternate conformations of the side-chains (Figure 4(b)). The side-chain of Ile24 had alternate conformations in both subunits of both the PRL24I structures, Val50 had alternate conformations in one subunit of each of the PRI50V structures, and Ser73 showed alternate conformations in three subunits of the two dimers in the PRG73S-IDV structure.
Figure 4.
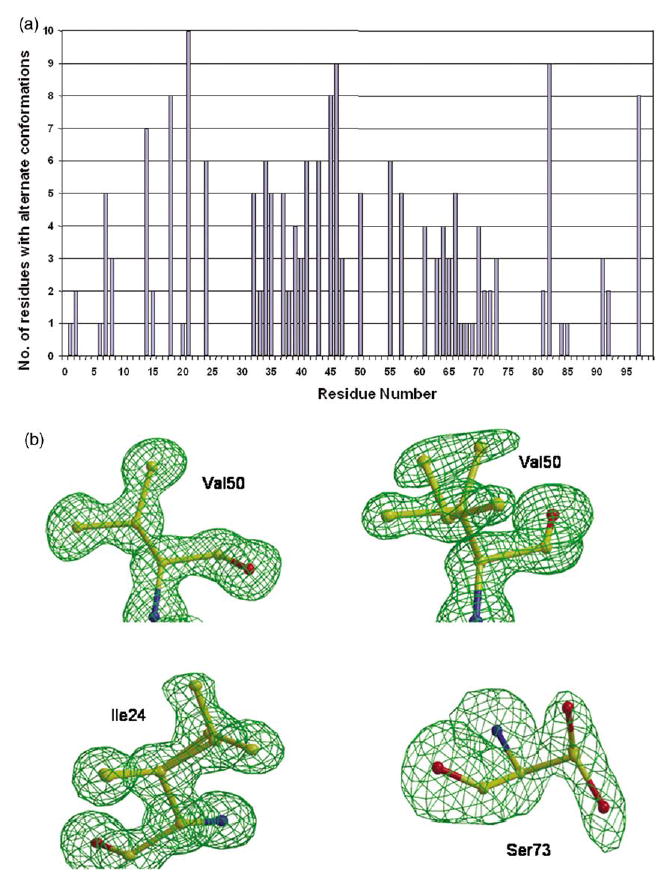
(a) Residues with alternate conformations. Alternate conformations of residues in six dimers of five new crystal structures. Alternate conformations for both side-chain and main-chain atoms were included. (b) Omit maps for mutated residues contoured at a level of 3.5σ. Val50′ in PRI50V–IDV had a single conformation for the side-chain. Two alternate conformations are shown for the side-chains of Val 50 (relative occupancy of 0.7/0.3) in PRI50V–IDV, Ile24 (relative occupancy of 0.6/0.4) in PRL24I–p2/NC, and Ser73 (relative occupancy of 0.5/0.5) in PRG73S–IDV structures.
Structural differences at sites of mutation
Each mutation introduced distinct structural changes compared to PR that can propagate to the inhibitor-binding site and the dimer interface. The new mutant structures were compared to structures of PR-IDV and PR-p2/NC.17,18 Leu/Ile24 lies in a hydrophobic internal pocket formed by residues from both subunits: Ile3, Val11, Ile66, Ile85, Leu90, Leu97′, and Phe99′ in all the structures. The PRL24I showed two alternate conformations for the Ile side-chain in both subunits of the two crystal structures of PRL24I–IDV and PRL24I–p2/NC. The relative occupancies were about 0.8/0.2 for Ile24 and about 0.6/0.4 for Ile24′. Similar interactions were observed in both structures. The two alternate conformations of the side-chains of Ile24 and Ile24′ in the mutant enabled the formation of van der Waals contacts similar to those of the side-chains of Leu24/24′ in PR. One exception was the new interaction of the CG side-chain atom of Ile24 with the side-chain of Leu90, which was not observed for the wild-type PR (Figure 5(a)). The mutant Ile24/24′ showed reduced interactions with Ile85/85′ and Phe99′/99; for example, the shortest interatomic distance increased from 3.8–4.4 Å between the side-chain atoms of residue 24 and Phe99′ (Figure 5(a)). Leu/Ile24 interacted with Leu97′ and Phe99′ at the C terminus of the other subunit. Therefore, structural changes can propagate from the mutated residue 24/24′ to the dimer interface between the two C-terminal β strands formed by residues 95–99. PRL24I had one less intersubunit contact of residues 24/24′ compared to PR in the complexes with p2/NC, and two less in the indinavir complexes. The altered contacts at the dimer interface appeared to be unfavorable, consistent with the lowered stability in urea and increased dissociation of the dimer.
Figure 5.
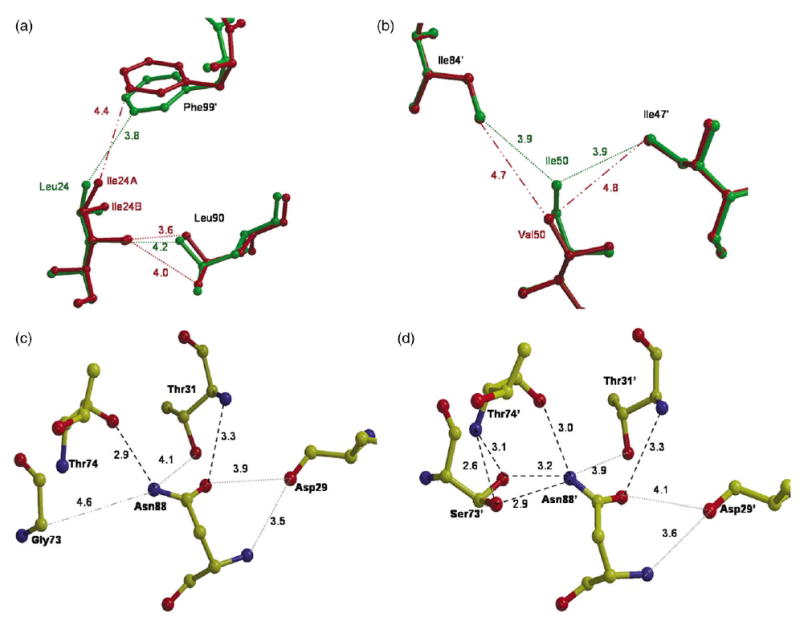
Structural differences at sites of mutation. Broken lines indicate hydrogen bond interactions (2.6–3.3 Å). Dotted lines indicate van der Waals interactions (3.5–4.2 Å). Dash-dot lines indicate distances over 4.2 Å for comparison. (a) Interactions of Leu/Ile24 with Leu90 and Phe99′ in the indinavir complexes. PR is green and PRL24I is red. (b) Interactions of Ile/Leu 50 with Ile 47′ and Ile 84′ in the complexes with p2/NC. PR is in green and PRI50V is in red. (c) Interactions of Gly73 with residues 74, 31, 29 and 88 in PR-IDV structure. (d) Interactions of Ser73 with residues 74, 31, 29 and 88 in the second dimer of PRG73S-IDV crystal structure. The side-chain of Ser73′ has two conformations.
Residue 50 lies at the tip of the flap and interacts closely with the other flap of the dimer and the inhibitor (Figure 1). The interactions with inhibitor are described in the following section. The carbonyl oxygen atom of Ile/Val50 from one subunit formed a conserved hydrogen bond interaction with the amide group of Gly51 from the other subunit. The mutated residue in one of the subunits, Val50 in PRI50V-IDV and Val50′ in PRI50V-p2/NC, showed two alternate conformations for the side-chain, with relative occupancies of about 0.7/0.3 (Figure 4(b)). Ile/Val50 and 50′ showed slightly asymmetric van der Waals interactions (<4.2 Å) in all the structures (Table 5). In general, residue 50 interacted with residues from both flaps in the dimer (Gly51, Gly52, Ile47′, Gly48′, Gly49′, Ile/Val50′, Ile54′) and residues Thr80′, Pro81′ and Ile84′ in the 80 s loop from the other subunit. Residue 50′ in the other subunit interacted with similar residues except for Ile47. PR-IDV differed slightly from the mutant PRI50V-IDV in the loss of contact of Ile50 with Thr80′, fewer interactions with Pro81′, more interactions of Ile50 with Gly47′ and Gly48′, and additional interactions with Gly49 and Gly49′. The interactions in the PR-p2/NC structure also showed small differences from the PRI50V-p2/NC structure, such as the improved interactions of Ile50 with Ile84′ and Ile47′ (Figure 5(b)). Val50/50′ in both PRI50V-IDV and PRI50V-p2/NC had approximately ten fewer intersubunit contacts than Ile50/50′ in the PR complexes (considering only major conformations). The loss of intersubunit interactions of Val50/50′ compared to those of Ile50/50′ was consistent with the lower stability and higher dimer dissociation constant of PRI50V.
Table 5.
The van der Waals contacts (<4.2 Å) of residue 50 with other residues in PR and PRI50V
| PR-inhibitor | Residue | Intrasubunit | Intersubunit | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT-IDV | I50 | 49 | 51 | 52 | 47′ | 48′ | 49′ | 50′ | 54′ | 81′ | 84′ | |||
| I50V-IDV | V50 | 51 | 52 | 47′ | (48′) | (49′) | 50′ | 54′ | (80′) | 81′ | 84′ | |||
| WT-p2/NC | I50 | 51 | 52 | 47′ | 48′ | 49′ | 50′ | 51′ | 52′ | 54′ | 80′ | 81′ | 84′ | |
| I50V-p2/NC | V50 | 49 | 51 | 52 | 47′ | 48′ | 49′ | 50′ | 51′ | 52′ | 54′ | 80′ | 81′ | |
| WT-IDV | I500 | 49′ | 51′ | 52′ | 49 | 50 | 51 | 52 | 54 | 80 | 81 | 84 | ||
| I50V-IDV | V500 | 51′ | 52′ | 49 | 50 | 51 | 52 | 54 | 80 | 81 | 84 | |||
| WT-p2/NC | I500 | 51′ | 52′ | 48 | 49 | 50 | 52 | 54 | 80 | 81 | 84 | |||
| I50V-p2/NC | V500 | (49) | 51′ | 52′ | 48 | 49 | 50 | 52 | 54 | 80 | 81 | (84) | ||
Contacts involving minor conformations of Val 50/50′ are in parentheses.
The mutant PRG73S showed two alternate conformations for the side-chain of Ser73 in three of the four subunits in the crystal structure with indinavir (Figure 4(b)). In all four subunits, at least one conformation of the Ser side-chain hydroxyl group formed a new hydrogen bond interaction with the side-chain of Asn88, a hydrogen bond interaction with the amide group of Thr74, and new van der Waals contacts with Leu89 (Figure 5(c) and (d)). The side-chain of Asn88 formed conserved hydrogen bond interactions with the carbonyl oxygen atom and the amide group of Thr74, and van der Waals contact with the carbonyl oxygen atom of Asp29 in both mutant and wild-type PR structures.22 The new interactions of Ser73 can propagate to the active site via the Asn88 interaction with Asp29 and Thr31, since Asp29, Asp30 and Val32 interacted directly with the substrate or inhibitor. This network of hydrogen bond and van der Waals interactions provides a mechanism for non-active site mutations to transmit energetic effects to the binding site for substrates and inhibitors. Little difference was observed in the inhibitory effect of indinavir on PRG73S compared with PR, probably because indinavir did not form hydrogen bond interactions with Asp30, unlike the peptide analogs. Therefore, the new interactions of Ser73 in PRG73S are likely to be responsible for the observed differences in inhibition by the CA/p2 analog and the relative kcat/Km values for different substrates.
Protease–inhibitor interactions
Generally, the mutants and wild-type PR showed similar interactions with inhibitors. Indinavir was bound by a set of seven direct hydrogen bond interactions to protease residues (Figure 6(a)), as described for PR.17 There were also conserved interactions mediated by four distinct water molecules. The mutants showed changes in the hydrogen bond interactions with the pyridyl group of indinavir. In PRL24I-IDV, the major indinavir conformation in PRI50V–IDV, and in the first dimer of PRG73S–IDV, the pyridyl group of indinavir can form a hydrogen bond with the side-chain of Arg8′ (Figure 6(b)). A similar interaction has been observed in the indinavir complex with PRL90M but not for PR and PRV82A.17 The pyridyl group of indinavir appeared to have two possible positions, and the side-chain of Arg8′ was observed frequently in alternate conformations. Consequently, indinavir can form a hydrogen bond interaction with Arg8′ when the two groups are close enough. The minor conformations of indinavir in the complexes with PRI50V and PRG73S had lost interactions with Asp29 and Gly27. The first dimer of PRG73S had lost interactions via water molecule C, although it is possible that this water molecule was not visible due to the lower resolution of the crystal structure.
Figure 6.
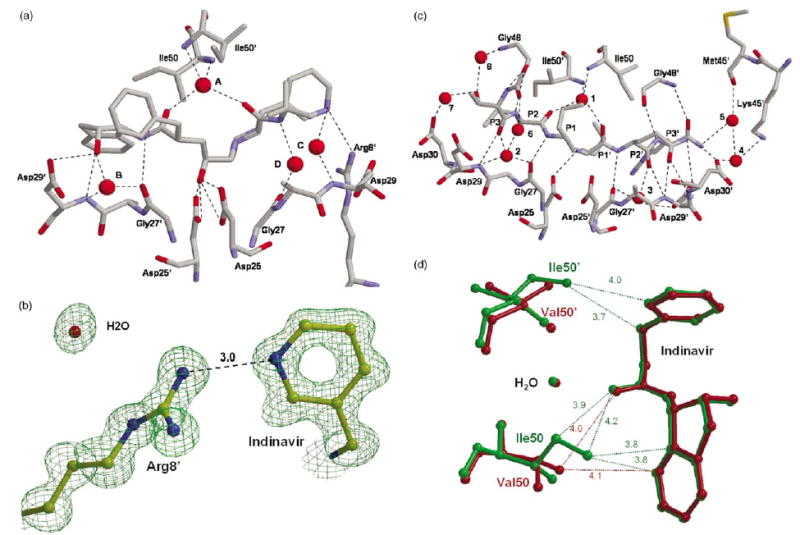
Protease-inhibitor interactions. Only the residues involved in hydrogen bond interactions are shown. Water molecules are represented as spheres. Hydrogen bonds are indicated by broken lines. (a) PRL24I hydrogen bond interactions with indinavir. Water molecules are labeled A–D. (b) Interactions of Arg8′ with the pyridyl group of indinavir in PRI50V–IDV. The omit map is contoured at 3.5σ. (c) PRL24I interactions with p2/NC. Water molecules are labeled 1–8. Arg8 and Arg8′ are omitted for clarity. (d) Selected interactions of the side-chains of Ile/Leu50 and 50′ with indinavir in the PRI50V and PR indinavir complexes. PR-IDV is in green and PRI50V–IDV is red. Only the central portion of indinavir is shown with van der Waals contacts indicated by dotted lines with distances in Å.
PR interacted with p2/NC substrate analog by 12 direct hydrogen bonds from PR residues Asp25, Gly27, Asp29, Asp30, and Gly48 from both subunits that extended from the amide group of P3 to the NH2 of P4′ (Figure 6(c)). Seven of these interactions involved main-chain amide groups and carbonyl oxygen atoms in both PR and inhibitor, as described.23 Additional PR–p2/NC interactions were mediated by eight water molecules. The highly conserved water 1 that interacted with the flaps and inhibitor was equivalent to water A in the indinavir complexes. Water 2 was structurally equivalent to water B in the indinavir complexes, while the others were not in equivalent positions. Water 2 mediated interactions of P3 C═O with Gly27 and Asp29, while water 3 had pseudo-symmetric interactions with P2′ C═O, Gly27′ and Asp29′. Water 4–8 interacted with the P3 Thr side-chain and with the termini of the inhibitor. The hydrogen bond interactions of PR with p2/NC substrate analog were highly similar in the mutant structures (differences in length of 0.1 Å or less) (Figure 6(c)), with only minor changes in the interactions with water. The hydroxyl group of P3 Thr had one interaction with water in PR and two interactions in the mutants, while the P4′ NH2 only had one hydrogen bond with water in PRI50V.
Residue 50/50′ was the only mutated residue that had direct contacts with inhibitor. Val50 cannot form the van der Waals interactions of the Ile CD atom with indinavir. The loss of interactions was partially compensated by movement of the PRI50V flaps toward indinavir by 0.4 Å at the Cα atoms of residues 50 and 50′. Both structures of PRI50V showed two alternate conformations for inhibitor and for the side-chain of Val50 in one subunit of the dimer. The presence of alternate conformations clearly increased the number of protease–inhibitor contacts. However, the occupancy ratio was 0.8/0.2 for alternate conformations of indinavir, and about 0.7/0.3 for p2/NC and the Val50/50′ side-chains. Therefore, the contacts involving major conformations were expected to be more significant. The side-chain atoms of Ile50 and Ile50′ showed nine van der Waals contacts with indinavir in PR-IDV, while Val50 and Val50′ had five van der Waals contacts with indinavir for the major conformers. Some differences are illustrated in Figure 6(d). Similarly, PR-p2/NC showed six van der Waals contacts between the side-chains of Ile50 and Ile50′ and the p2/NC, while the mutant had three contacts of Val50 and the major conformer of Val50′ side-chains with p2/NC. In both cases the mutant had lost three or four contacts with inhibitor, consistent with the increased relative Ki values for PRI50V of 50-fold for indinavir and 20-fold for p2/NC.
Catalytic sites
The 1.1 Å resolution crystal structures of PRL24I-IDV and PRI50V-IDV showed more asymmetrical interactions between the carboxylate oxygen atoms of the catalytic Asp25 and Asp25′ and the hydroxyl group of indinavir than observed for PR. The PR-IDV structure showed interatomic distances of 2.7–2.9 Å, while the mutant structures had two shorter interactions of 2.6–2.7 Å and two longer interactions of 3.0–3.2 Å. The peptide analog did not have a carbonyl group at the catalytic site and there was only one hydrogen bond formed between the amide group of P1′ and the OD2 of Asp25, unlike the four possible with the hydroxyl group of indinavir.
Crystal structures of HIV PR–inhibitor complexes at a resolution of at least 1.1 Å have shown potential difference density for the hydrogen atom associated with the catalytic aspartate residues.24,25 Positive difference density was observed near the catalytic aspartate residues in the 1.1 Å structure of PRL24I–p2/NC. This difference density was between the two closest inner carboxylate oxygen atoms of Asp25 and Asp25′ (Figure 7). A smaller peak was observed between the Asp25 and the CH2 of the reduced peptide group after adding hydrogen. The peak representing the proton between the Asp25 and Asp25′ oxygen atoms in the PRL24I–p2/NC complex was not in exactly the same position as that in the PRV82A–UIC94017 complex.25 Therefore, the location of the proton may depend on the chemistry of the inhibitor.
Figure 7.
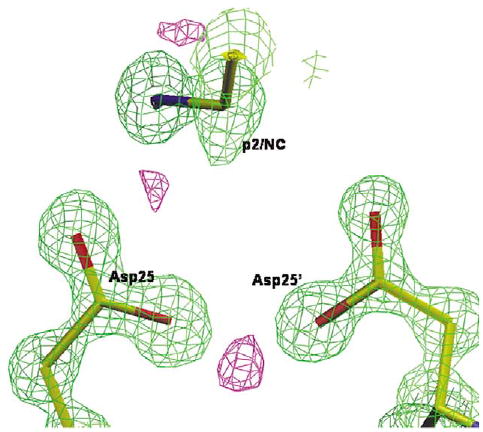
The catalytic site of PRL24I–p2/NC at 1.1 Å resolution. The 2Fo – Fc map is in green and contoured at 2.6σ, and the positive Fo – Fc map is in purple, contoured at 3.5σ.
Conclusion
Structural changes due to mutations may result in reduced affinity for inhibitor, altered protease activity or stability, and consequently provide resistance to drugs. All three protease variants showed distinct structural changes near the site of mutation and changes in catalytic activity or inhibition relative to wild-type protease. The substantially reduced catalytic activity of PRL24I agreed with the sensitive location of the mutation next to the catalytic Asp25. Although this mutation has been observed in about 10% of patients exposed to indinavir, there was only a small (2.6-fold) increase in Ki relative to PR. Hence, the drug resistance is expected to arise from the effect of L24I on reducing catalytic activity and dimer stability, which is consistent with the observed presence of this mutation only in combination with other indinavir-resistant mutations. In contrast, PRI50V exhibited a dramatic 50-fold increase in Ki for indinavir, although this mutation is observed very rarely in isolates resistant to indinavir. The weaker inhibition appeared to arise from reduced van der Waals interactions of inhibitors with Val50 in PRI50V compared to Ile50 in PR. PRG73S was similar to PR in dimer dissociation and inhibition, consistent with the location of residue 73 at the protein surface and far from the active site. Interestingly, Ser73 in PRG73S formed new hydrogen bond networks that can transmit changes to the substrate-binding site, consistent with the variation in activity for different substrates. The rarity of this mutation, and its selection in combination with other resistant mutations, are consistent with the relatively minor effects on protease structure and catalysis.
Two of the three mutants PRL24I and PRI50V appeared to have the major effect of reducing intersubunit interactions and increasing dimer dissociation. The subunit–subunit interface in the PR dimer is formed mainly by residues from the N and C termini (below the active site), the catalytic residues, and the flaps (above the active site) (Figure 1). Increased dimer dissociation was observed for both the PRI50V mutant that reduced the intersubunit interactions of the flaps, and PRL24I, which altered intersubunit interactions with the C-terminal residues located at the opposite side of the molecule from the flaps.
This analysis has confirmed that drug resistance can arise when mutations alter the PR dimer interface at the flaps or the terminal β sheet, as well as when mutations alter the inhibitor-binding site directly. Furthermore, distal mutations with relatively minor effects can transmit changes to the substrate-binding site and contribute to viral resistance.
Materials and Methods
Preparation of HIV-1 protease mutants
The HIV-1 PR (Genbank HIVHXB2CG) clone was constructed with the substitutions Q7K, L33I, and L63I to minimize the autoproteolysis of the PR, and C67A and C95A to prevent cysteine-thiol oxidation.21 The kinetic parameters and stability of this stabilized PR were indistinguishable from those of the mature enzyme.7,21 Plasmid DNA (pET11a; Novagen, Madison, WI) encoding PR was used with the appropriate oligonucleotide primers to generate the constructs PRL24I, PRI50V, and PRG73S using the Quick-Change mutagenesis kit (Stratagene, La Jolla, CA). The PR mutants were expressed using pET11a vector and Escherichia coli BL21 (DE3) and purified from inclusion bodies, as described.8 The mutations were confirmed by nucleic acid sequencing and protein mass spectrometry.
Enzyme kinetic assays
The chromogenic substrate K-A-R-V-Nle-p-nitroPhe-E-A-Nle-amide (Sigma, St. Louis, MO), a CA/p2 analog, was used to determine the kinetic parameters. PR at a final concentration of 70–120 nM was added to various concentrations of substrate (25–400 μM) maintained in 50 mM sodium acetate (pH 5.0), 0.1 M NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, and assayed by monitoring the decrease in absorbance at 310 nm using a Perkin Elmer Lambda 35 UV/Vis spectrophotometer. The absorbance was converted to substrate concentration via a calibration curve. PR hydrolysis of the peptides K-A-R-V-L-A-E-AM-S (CA/p2) and V-S-F-N-F-P-Q-I-T-K-K (p6Pol/PR) was assayed using HPLC as described.8 The enzyme concentrations were based on active site titration data. The Michaelis–Menten curves were fit using SigmaPlot 8.0.2 (SPSS Inc.).
The reduced peptide analogs R-V-L-r-F-E-A-Nle (CA/p2) and Ace-T-I-Nle-r-Nle-Q-R (p2/NC) (r is the reduced peptide bond and Nle replaces M) were purchased from BACHEM. Indinavir was a gift from Merck & Co. The Ki values were obtained from the IC50 values estimated from an inhibitor dose-response curve with the spectroscopic assay and the chromogenic substrate using the equation:
where [E] and [S] are the concentrations of PR and substrate, respectively.26
Urea-denaturation assay
The effect of urea-denaturation was measured using the spectroscopic assay. PR activity was measured with increasing concentration of urea (0–4.0 M) at a final concentration of enzyme of 300–500 nM and concentration of substrate of 400 μM.7 The UC50 values for half-maximal velocity were obtained by plotting the initial velocities against concentration of urea and fitting to a curve for solvent-denaturation of protein using SigmaPlot 8.02 software.
Kd determination
Specific activity was measured as a function of dimeric enzyme concentration at a final substrate concentration of 375 μM in 50 mM sodium acetate (pH 5.0), 0.1 M NaCl at °5 C.20,21
Crystallographic analysis
Crystals were grown at room temperature by the vapor-diffusion, hanging-drop method. PRL24I, PRI50V, and PRG73S were co-crystallized with indinavir. PRL24I and PRI50V were co-crystallized with the reduced peptide analog of the p2/NC Gag cleavage site. The protein (1.8–3.5 mg/ml) was preincubated with the inhibitor at a molar ratio of 1:5–20. For PRL24I -IDV, the reservoir contained 0.1 M sodium citrate/0.2 M sodium phosphate buffer (pH 5.0–6.0), 10% (v/v) dimethylsulfoxide (DMSO), and 20–40% saturated ammonium sulfate as precipitant. For PRI50V and PRG73S complexes with indinavir, the reservoir contained 0.1 M sodium citrate/0.2 M sodium phosphate buffer (pH 4.6–5.8), 10% DMSO, 6–10% methyl-2,4-pentanediol, and 20–40% saturated ammonium sulfate. The complexes with p2/NC were crystallized using a reservoir containing 0.1 M sodium citrate/0.2 M sodium phosphate buffer (pH 5.4–5.8), 8–13% DMSO, and 6–20% saturated ammonium sulfate as precipitant. The crystallization drops had a 1:1 (v/v) ratio of reservoir solution and protein solution. The crystals grew in two to seven days and were frozen in liquid nitrogen with a cryoprotectant of 20–35% (v/v) glycerol.
X-ray diffraction data for all the complexes were collected on the SER-CAT beamline of the Advanced Photon Source, Argonne National Laboratory. Data were processed using HKL2000.27 The structures were solved by molecular replacement using AMoRe,28 refined using SHELX,29 and refitted using O.30 Alternate conformations were modeled for residues when obvious in the electron density maps. The solvent was modeled with over 300 water molecules, and ions present in the crystallization solutions, as described.25 Anisotropic B-factors were refined for all the structures. Hydrogen atom positions were included in the last stage of refinement, using all data once all other parameters, including disorder, had been modeled.
The mutant crystal structures were compared with the wild-type by superimposing their Cα atoms on each other as described.25 Structural Figures were made using MOLSCRIPT,31 Bobscript,32 and RasMol.33
Protein Data Bank accession codes
The structures have been submitted to the Protein Data Bank with accession code 2AVO for PRl24I-IDV, 2AVS for PRI50V-IDV, 2AVV for PRG73S-IDV, 2AVW for PRl24I-p2/NC, and 2AVQ for PRI50V-p2/NC.
Acknowledgments
I.W. and R.H. are Distinguished Cancer Scholars. We thank Xianfeng Chen for assistance with analysis of protease–indinavir interactions. We thank Merck & Co. for providing the indinavir used for the crystallographic analysis. We thank the staff at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract no. W-31-109-Eng-38. The research was supported, in part, by the Georgia Research Alliance, the Georgia Cancer Coalition, the National Institutes of Health grants GM62920, AIDS-FIRCA TW01001, Hungarian OTKA F35191 and T43482.
References
- 1.Hertogs K, Bloor S, Kemp SD, Van den Eynde C, Alcorn TM, Pauwels R, et al. Phenotypic and genotypic analysis of clinical HIV-1 isolates reveals extensive protease inhibitor cross-resistance: a survey of over 6000 samples. AIDS. 2000;14:1203–1210. doi: 10.1097/00002030-200006160-00018. [DOI] [PubMed] [Google Scholar]
- 2.Wu TD, Schiffer CA, Gonzales MJ, Taylor J, Kantor R, Chou S, et al. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–4847. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis JM, Weber IT, Tozser J, Clore GM, Gronenborn AM. HIV-1 protease: maturation, enzyme specificity, and drug resistance. Advan Pharmacol. 2000;49:111–146. doi: 10.1016/s1054-3589(00)49025-3. [DOI] [PubMed] [Google Scholar]
- 4.Shafer R. Genotypic testing for human immunodeficiency virus type 1 drug resistance. Clin Microbiol Rev. 2002;15:247–277. doi: 10.1128/CMR.15.2.247-277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ridky TW, Kikonyogo A, Leis J, Gulnik S, Copeland T, Erickson J, et al. Drug-resistant HIV-1 proteases identify enzyme residues important for substrate selection and catalytic rate. Biochemistry. 1998;37:13835–13845. doi: 10.1021/bi980612k. [DOI] [PubMed] [Google Scholar]
- 6.Xie D, Gulnik S, Gustchina E, Yu B, Shao W, Qoronfleh W, et al. Drug resistance mutations can effect dimer stability of HIV-1 protease at neutral pH. Protein Sci. 1999;8:1702–1707. doi: 10.1110/ps.8.8.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahalingam B, Louis JM, Reed CC, Adomat JM, Krouse J, Wang YF, et al. Structural and kinetic analysis of drug resistant mutants of HIV-1 protease. Eur J Biochem. 1999;263:238–245. doi: 10.1046/j.1432-1327.1999.00514.x. [DOI] [PubMed] [Google Scholar]
- 8.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: high resolution crystal structures of the mutant protease/substrate analog complexes. Proteins: Struct Funct Genet. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 9.Gulnik SV, Suvorov LI, Liu B, Yu B, Anderson B, Mitsuya H, Erickson JW. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry. 1995;34:9282–9287. doi: 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- 10.Muzammil S, Ross P, Freire E. A major role for a set of non-active site mutations in the development of HIV-1 protease drug resistance. Biochemistry. 2003;42:631–638. doi: 10.1021/bi027019u. [DOI] [PubMed] [Google Scholar]
- 11.Clemente JC, Moose RE, Hemrajani R, Whitford LR, Govindasamy L, Reutzel R, et al. Comparing the accumulation of active- and non-active-site mutations in the HIV-1 protease. Biochemistry. 2004;43:12141–12151. doi: 10.1021/bi049459m. [DOI] [PubMed] [Google Scholar]
- 12.Mahalingam B, Boross P, Wang YF, Louis JM, Fischer CC, Tozser J, et al. Combining mutations in HIV-1 protease to understand mechanisms of resistance. Proteins: Struct Funct Genet. 2002;48:107–116. doi: 10.1002/prot.10140. [DOI] [PubMed] [Google Scholar]
- 13.Brown AJ, Korber BT, Condra JH. Associations between amino acids in the evolution of HIV type 1 protease sequences under indinavir therapy. AIDS Res Hum Retroviruses. 1999;15:247–253. doi: 10.1089/088922299311420. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z, Li Y, Schock HB, Hall D, Chen E, Kuo LC. Three-dimensional structure of a mutant HIV-1 protease displaying cross-resistance to all protease inhibitors in clinical trials. J Biol Chem. 1995;270:433–436. doi: 10.1074/jbc.270.37.21433. [DOI] [PubMed] [Google Scholar]
- 15.Munshi S, Chen Z, Yan Y, Li Y, Olsen DB, Schock HB, Galvin BB, et al. An alternate binding site for the P1-P3 group of a class of potent HIV-1 protease inhibitors as a result of concerted structural change in the 80s loop of the protease. Acta Crystallog sect D. 2000;56:381–388. doi: 10.1107/s0907444900000469. [DOI] [PubMed] [Google Scholar]
- 16.King NM, Melnick L, Prabu-Jeyabalan M, Nalivaika EA, Yang SS, Gao Y, et al. Lack of synergy for inhibitors targeting a multi-drug-resistant HIV-1 protease. Protein Sci. 2002;11:418–429. doi: 10.1110/ps.25502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahalingam B, Wang YF, Boross PI, Tozser J, Louis JM, Harrison RW, Weber IT. Crystal structures of HIV protease V82A and L90M mutants reveal changes in the indinavir-binding site. Eur J Biochem. 2004;271:1516–1524. doi: 10.1111/j.1432-1033.2004.04060.x. [DOI] [PubMed] [Google Scholar]
- 18.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, et al. Molecular basis for substrate recognition and drug resistance from 1.1–1.6 Å resolution crystal structures of HIV-1 protease mutants with substrate analogs. FEBS J. 2005;272:5265–5277. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Partaleis JA, Yamaguchi K, Tisdale M, Vlair EE, Falcione C, Maschera B, et al. In vitro selection and characteization of human immunodeficiency virus type 1 (HIV-1) isolates with reduced sensitivity to hydroxyethylamino sulfonamide inhibitors. J Virol. 1995;69:5228–5235. doi: 10.1128/jvi.69.9.5228-5235.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wondrak EM, Louis JM. Influence of flanking sequences on the dimer stability of human immunodeficiency virus type 1 protease. Biochemistry. 1996;35:12957–12962. doi: 10.1021/bi960984y. [DOI] [PubMed] [Google Scholar]
- 21.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nature Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 22.Weber IT. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J Biol Chem. 1990;265:10492–10496. [PubMed] [Google Scholar]
- 23.Gustchina A, Sansom C, Prevost M, Richelle J, Wodak SY, Wlodawer A, Weber IT. Energy calculations and analysis of HIV-1 protease-inhibitor crystal structures. Protein Eng. 1994;7:309–317. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- 24.Brynda J, Rezacova P, Fabry M, Horejsi M, Stouracova R, Sedlacek J, et al. A phenyl-norstatine inhibitor binding to HIV-1 protease: geometry, protonation, and subsite-pocket interactions analyzed at atomic resolution. J Med Chem. 2004;47:2030–2036. doi: 10.1021/jm031105q. [DOI] [PubMed] [Google Scholar]
- 25.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, et al. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 26.Maibaum J, Rich DH. Inhibition of porcine pepsin by two substrate analogues containing statine: the effect of histidine at the P2 subsite on the inhibition of aspartic proteinases. J Med Chem. 1988;31:625–629. doi: 10.1021/jm00398a022. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallog sect D. 1994;50:157–163. [Google Scholar]
- 29.Sheldrick GM, Schneider TR. High resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 30.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 31.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallog. 1991;24:946–950. [Google Scholar]
- 32.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crysallog sect D. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 33.Sayle RA, Milner-White EJ. Rasmol: Biomolecular graphics for all. Trends Biochem Sci. 1995;20:374–376. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]