

Abstract
Microtubule (MT) destabilization promotes the formation of actin stress fibers and enhances the contractility of cells, however, mechanism involved in the coordinated regulation of MTs and the actin cytoskeleton is poorly understood. LIM kinase 1 (LIMK1) regulates actin polymerization by phosphorylating the actin depolymerization factor (ADF), cofilin. Here we report that LIMK1 is also involved in the MTs destabilization. In endothelial cells, endogenous LIMK1 co-localizes with MTs and forms a complex with tubulin via PDZ domain. MTs destabilization induced by thrombin or nocodazole resulted in decrease of LIMK1 colocalization with MTs. Overexpression of wild type LIMK1 resulted in MTs destabilization whereas kinase-dead mutant of LIMK1 (KD) did not affect MTs stability. Importantly, downregulation of endogenous LIMK1 by siRNA resulted in abrogation of the thrombin-induced MTs destabilization and the inhibition of thrombin-induced actin polymerization. Expression of ROCK2, which phosphorylates and activates LIMK1, decreases dramatically the interaction of LIMK1 with tubulin but increases its interaction with actin. Interestingly, expression of KD- or siRNA-LIMK1 prevents thrombin induced microtubule destabilization and Factin formation suggesting that LIMK1 activity is required for thrombin-induced modulation of microtubule destabilization and actin polymerization. Our findings indicate that LIMK1 may coordinate microtubules and actin cytoskeleton.
Abbreviation list: LIMK1, LIMK-domain-containing kinase 1; ROCK2, Rho kinase 2; ADF, actin depolymerization factor ; PAK, p21 activated kinases; HUVEC, human umbilical vein endothelial cells; MAPs, microtubule-associated proteins; GEF, guanine nucleotide exchange factor; HBSS, Hank’s balanced salt solution; PBS, phosphate buffered saline; BSA, bovine serum albumin; GST, glutathione S-transferase
Introduction
Microtubules, polymers of alpha- and beta-tubulins, are key component of the cytoskeleton and are involved in multiple cellular processes such as migration, mitosis, protein and organelle transport (1,2). Microtubule dynamics and their spatial arrangements are affected by a number of signaling molecules. Conversely, changes in microtubule dynamics modulate intracellular signal transduction (for review see (1)).
The actin cytoskeleton undergoes rearrangement under the control of various actin binding, capping, nucleating, and severing proteins, which are intimately involved in regulating the contractile status of the cells (3). Actin dynamics is regulated via transduction of extracellular signals to intracellular events primarily through the members of the Rho family of small GTPases. Rho is known to induce stress fiber formation, whereas Cdc42 and Rac are involved in formation of lamellipodia and filopodia, respectively (4).
Microtubule disassembly promotes the formation of actin stress fibers and enhances the contractility of cells (5). Agents such as nocodazole or vinblastine that disrupt microtubules induce rapid assembly of actin filaments and focal adhesions (6), whereas microtubule stabilization with taxol attenuated these effects. Regulation of the actin cytoskeleton by microtubules requires the Rho GTPases (for review see (7)). However, the mechanisms involved in the coordinated regulation of microtubules and the actin cytoskeleton remain poorly understood.
LIMK1 is a serine/threonine kinase that regulates actin polymerization by phosphorylating and inactivating its substrate, the actin depolymerization factor (ADF), cofilin (8,9). Cofilin regulates actin dynamics by severing actin filaments and sequestering actin monomer from the pointed end of actin filaments (10). However, once phosphorylated at serine 3 by LIMK1 (11,12), cofilin can no longer bind to actin (Morgan et al., 1993), resulting in accumulation of actin polymers. In turn, the activity of LIMK1 is regulated by the members of the Rho-GTPase family members, Rho, Rac and Cdc42 (13–15) through the activation of their effectors serine/threonine kinases; Rho kinase (ROCK) and the p21 activated kinases (PAK), PAK1 and PAK4, respectively. These kinases phosphorylate LIMK1 at Thr 508 located in the activation loop of the kinase domain resulting in its activation.
Here we demonstrate that LIMK1 coordinates both microtubules disassembly and actin polymerization. We provide experimental data to show that LIMK1 interacts with microtubules and the actin cytoskeleton in an agonistdependent manner. Stimulation of the cells with thrombin decreases the interaction of LIMK1 with tubulin and increases its interaction with actin. Moreover, we show that LIMK1 induces microtubule destabilization and actin stress fiber formation that requires the kinase activity of LIMK1.
Materials and methods
DNA and RNA constructs
Myc-LIMK1, kinase-dead Myc-LIMK, and GST-tagged cofilin, deletion mutants of LIMK1: Flag-LIM1,2, Flag-PDZ, Flag- KD were described previously (16), Rho kinase 2, ROCK2 was from Dr. R.Ye (University of Illinois at Chicago, Chicago, IL). Double-stranded small interference RNA-LIMK1 UGGCAAGCGUGGACUUUCAdTdT was from Dharmacorn (Chicago, IL). SiRNA-G13 was from Santa Cruz Biotechnology (Santa Cruz, CA).
Materials
Thrombin was purchased from Enzyme Research Laboratories (South Bend, IN). Taxol and nocodazole were purchased from Sigma (St. Louis, MO), glutaraldehyde and all the reagents used for immunostaining were purchased from Electron Microscopy Sciences (Ft.Washington, PA). Rat anti-LIMK1 antibody was described previously (16). Rabbit anti-LIMK1 and anti-phospho-LIMK1 antibodies were purchased from Cell Signaling Technology (Beverly, MA); mouse anti-α-tubulin , anti-Flag and anti-β-actin monoclonal antibodies (mAbs) were purchased from Sigma (St. Louis, MO); mouse anti-Myc mAb and rabbit anti-ROCK2 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); Phalloidin Alexa Fluor 594, donkey anti-rabbit Alexa Fluor 488, donkey anti-rabbit Alexa Fluor 594, donkey anti-mouse Alexa Fluor 594 and donkey anti-mouse Alexa Fluor 488 secondary antibodies used for immunofluorescent staining were purchased from Molecular Probes (Eugene, OR). [32P] γATP was obtained from DuPont NEN. All other reagents were obtained from Sigma.
Cell cultures and transfections
Human umbilical vein endothelial cells (HUVECs) were obtained at first passage from Cambrex (Walkersville, MD, culture line CC-2519) and were utilized at passages 6-10. Cells were cultured in EBM-2 medium (Cambrex) supplemented with 10% (v/v) fetal bovine serum (Cellgro) and EGM-2 SingleQuots (Cambrex) and maintained at 370C in humidified atmosphere of 5% CO2-95% air. Transient transfections of HUVECs were performed with SuperFect Transfection reagent (Qiagen) according to the manufacturer’s protocol with 10% transfection efficiency. Transient transfections of COS-7 cells were performed with LipofectAMINE 2000 (Life Technologies Inc.) according to manufacturer’s protocol. Transfection of siRNAs was performed using DharmaFect1 (Dharmacon), xperiments were performed 48 hours after transfection.
Immunocytochemistry
HUVECs grown to confluency on coverslips coated with gelatin were serum-starved for 3 hours before each experiment. Cells were washed with Hank’s balanced salt solution (HBSS) and fixed with 0.1% glutaraldehyde/0.1M sodium cacodilate, pH 7.3 for 10 min. Cells were permeabilized for 5 min with 0.1% Triton X-100/ PBS and washed extensively with HBSS. After blocking with 1%BSA/0.2% fish skin gelatin in HBSS for 1 h at room temperature (RT), cells were incubated with primary antibody in blocking solution for 1 h at RT followed by incubation with secondary antibodies. Slides were mounted using ProLong Antifade Kit (Molecular Probes). Microscopy was performed using Zeiss LSM 510 confocal microscope equipped with 63x water-immersion objective with appropriate filter sets.
Extraction of cytosolic proteins from living cells
In order to quantitatively analyze the changes in F-actin and microtubules, we adapted a novel fixation approach (17) that allowed us to extract monomeric actin and tubulin from cells, preserving only polymeric cytoskeletal structures. Cytosolic proteins were extracted from living cells for 1 min at RT with extraction solution (0.5% Triton X-100, 4% polyethylene glycol 40,000) in PEM buffer (100 mM PIPES, pH 6.9, 1 mM EGTA, and 1 mM MgCl2), protease inhibitors cocktail (Sigma-Aldrich) supplemented where needed with 2 μM taxol and/or 2 μM phalloidin followed by fixation in 0.1% glutaraldehyde/0.1M sodium cacodilate, pH 7.3 for 10 min and quenching with 1 mg/ml NaBH4. Cells were rinsed with PEM buffer and stained as described above. For Western blot analysis, extracted cells were rinsed with PEM buffer and lysed in a buffer containing 20 mM Tris, pH7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail (Sigma), and 1 μM Na3VO4 supplemented with 20 μM taxol. Intact cells and nuclei were removed by centrifugation for 5 min at 800 g and the supernatants were loaded on 1 ml of buffered cushion (80 mM PIPES pH 7, 1 mM MgCl2, 1 mM EGTA, 50% glycerol) supplemented with 20 μM taxol and centrifuged for 40 min at 100,000 g. The pellet and supernatant were subjected to Western Blot analysis and the membrane was probed with anti-LIMK1 and anti-α-tubulin antibodies.
Immunoprecipitation and Western blotting
COS-7 cells grown in DMEM/10%CS were transfected with cDNA constructs using LipofectAMINE according to the manufacturer’s protocol. Forty-eight hours later, cells were lysed in lysis buffer (20 mM Tris, pH7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail (Sigma), and 1 μM Na3VO4 unless specified otherwise). Lysates were sonicated on ice, and cell debris were removed by centrifugation for 5 min at 800 g. Lysates were pre-cleared with protein A/G agarose beads (Santa Cruz) and the proteins were immunoprecipitated with the appropriate antibody overnight at 40 C followed by incubation with protein A/G agarose for 1 h at 40C. Immunoprecipitates were washed 3x with lysis buffer and proteins were separated on SDS-polyacrlyamide gel electrophoresis (PAGE). Immunoblotting analysis was performed as described previously (18).
Calculations of the co-localization coefficients and total intensities of protein staining
Images of Alexa Fluor 488- or 594 – stained HUVECs monolayers stimulated with thrombin were captured as described above and analyzed using Zeiss Enhanced Colocalization Tool software. Images were differentially segmented between cytosol (black) and microtubules or Factin (highest gray value) based on image grayscale levels. The microtubule disassembly and actin stress fiber formation were expressed as a ratio of the cytoskeletal polymer area to the area of the whole image and normalized against controls. Colocalization coefficients and the number of pixels in each channel were calculated using Zeiss Enhanced Colocalization Tool software. Data were collected from 20 cells for each experiment. Three independent experiments were performed. The values were statistically processed using Sigma Plot 7.1 (SPSS Science, Chicago, IL) software.
In vitro kinase assays
LIMK1 activity was determined as described previously (19). Briefly, Myc-tagged LIMK1 (Myc-LIMK1) was transfected with various cDNA constructs as described in Results. Forty-eight hours later the cells were lysed in lysis buffer containing 1% Triton X-100, 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol (DTT), 1 μM sodium orthovanadate with 0.1 mM phenylmethanesulfonyl fluoride, 1 μg/ml leupeptine, and 1 μM pepstatin for 30 min at 40 C. Debris were removed by centrifugation at 12,000g for 15 min at 40 C. Myc-LIMK1 was immunoprecipitated with anti-Myc antibody and protein A-agarose for 24 hours at 40 C. To measure the kinase activity of Myc-LIMK1, 0.2 μg of GST-cofilin was incubated with the immune complexes for 20 min at 300 C in buffer containing 40 mM HEPES (pH 8.0), 5 mM MgAc, 2 mM DTT, 1 mM EGTA, 50 μM ATP, and 1 μCi of [32P]γ ATP. The kinase reaction was terminated by addition of 6x Laemmli buffer, and the proteins were analyzed by SDS-PAGE. Phosphorylated cofilin was visualized by autoradiography, and radioactivity was measured by PhosphorImager, BioRad. Aliquots of whole cell lysates were subjected to immunoblotting analysis to confirm the appropriate expression of the transfected proteins as described in Results.
Statistical analysis
For statistical analysis, Student's t-test was used to compare data between two groups. Values are expressed as mean ± SD of three independent experiments. p < 0.05 was considered statistically significant.
Results
LIMK1 co-localizes with microtubules in endothelial cells
To examine the relative intracellular distribution of LIMK1 in endothelial cells, human umbilical vein endothelial cells (HUVECs), were serum starved, fixed with glutaraldehyde, and costained with rabbit anti-LIMK1 and mouse anti-α-tubulin antibodies. Optical sections (0.5 μm-thick confocal sections) of stained cells revealed a striking co-localization of LIMK1 and MTs (Figure 1A, upper panel), where most of the LIMK1 protein was found along microtubules.
Figure 1. LIMK1 co-localizes with microtubules in HUVECs.
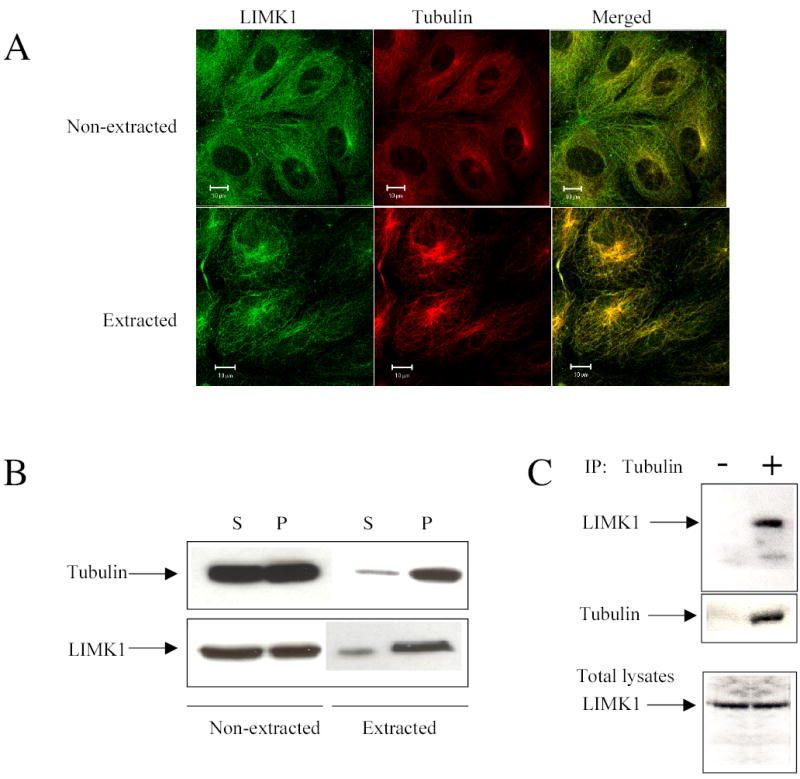
(A) Immunofluorescence analysis of LIMK1 and tubulin colocalization in HUVECs (top panels). To remove soluble cytosolic proteins, including monomeric tubulin, living cells were extracted with 0.5% Triton X-100 supplemented with 2 μM taxol before fixation with 0.1% glutaraldehyde/0.1 M sodium cacodilate and staining with anti-LIMK1 and anti-α-tubulin antibodies (bottom panels). Images were captured using dual-wavelength laser scanning confocal microscope Zeiss LSM 510. Bars 10 μm. Shown are representative images from three independent experiments.
(B) LIMK1 co-sediments with tubulin after extraction of cytosolic proteins from living cells. Cytosolic proteins were extracted from living cells with extraction solution (see Materials and Methods). Cells lysates were loaded on buffered cushion and centrifuged for 40 min at 100,000 g. The pellet (P) and supernatant (S) were assayed by Western Blotting and the membrane was probed with anti-LIMK1 and anti-α-tubulin antibodies. (C) Endogenous LIMK1 co-immunoprecipitates with tubulin. Cell lysates of confluent HUVECs (grown in 100 mm dishes) immunoprecipitated (IP) with anti-α-tubulin antibody or protein A/G agarose as indicated. Immunoprecipitates and total lysates were analyzed by Western blotting and the membrane was probed with anti-α-tubulin and anti-LIMK1 antibodies. This experiment was repeated three times with similar results.
As high levels of actin monomer, tubulin subunits and cytoskeletal binding proteins are present in the cytoplasm, the resolution of cytoskeletal polymers could be reduced, making the detailed analysis of the localization of polymer binding proteins rather difficult. Therefore, to determine whether a certain pool of LIMK1 protein is associated with microtubule cytoskeleton, we have adapted a novel fixation approach that allows the extraction of cytosolic proteins, including monomeric tubulin, from living cells, preserving only polymeric cytoskeletal structures (17). The extraction of cytosolic LIMK1 and tubulin from living HUVECs was confirmed by Western blotting (Figure 1B). Importantly, we showed that a certain fraction of endogenous LIMK1 could not be extracted from the living cells and was associated with MTs (Figure 1A, lower panel). To quantify the relative amount of the co-localized LIMK1 and tubulin, we used the Zeiss Enhanced Co-localization Tool software. Relative cell surface area was selected for each cell. Co-localization coefficient was calculated as (c1(%) =100%* pixelsCh1, coloc/ pixelsCh1, total) and found to be equal to 85±4.5% suggesting a high extent of colocalization.
To demonstrate that endogenous LIMK1 and tubulin can form a complex in vitro, tubulin was immunoprecipitated from HUVECs lysates and the immunoprecipitates were analyzed by SDS-PAGE followed by probing with anti-LIMK1 antibody (Figure 1C). The data showed that tubulin was specifically immunoprecipitated together with endogenous LIMK1, whereas Protein A/G agarose (Figure 1C) and nonimmune sera did not precipitate tubulin or LIMK1 (data not shown), suggesting that the interaction between tubulin and LIMK1 was specific.
The PDZ domain mediates the interaction between LIMK1 and tubulin
LIMK1 interacts with F-actin via its kinase domain, as it was determined using an in vitro cosedimentation assay (9). To identify the LIMK1 domain(s) that mediate the interaction with tubulin, we transfected COS-7 cells with Myc-tagged full length LIMK1 or Flag-tagged deletion mutants consisting of the two LIM domains, the PDZ domain, or the kinase domain of LIMK1 (Figure 2A). Tubulin was immunoprecipitated from cell lysates and the presence of the LIMK1 proteins was determined using either anti-Myc or anti-Flag antibodies. Tubulin was immunoprecipitated together with Myc-tagged LIMK1 but not with Myc-tagged zyxin used as a negative control (Figure 2B, left panel). In addition, Myc-tagged LIMK1 was immunoprecipitated together with endogenous tubulin, whereas non-immune serum did not precipitate Myc-tagged LIMK1 or tubulin (Figure 2B, right panel).
Figure 2. LIMK1 interacts with tubulin via its PDZ domain.
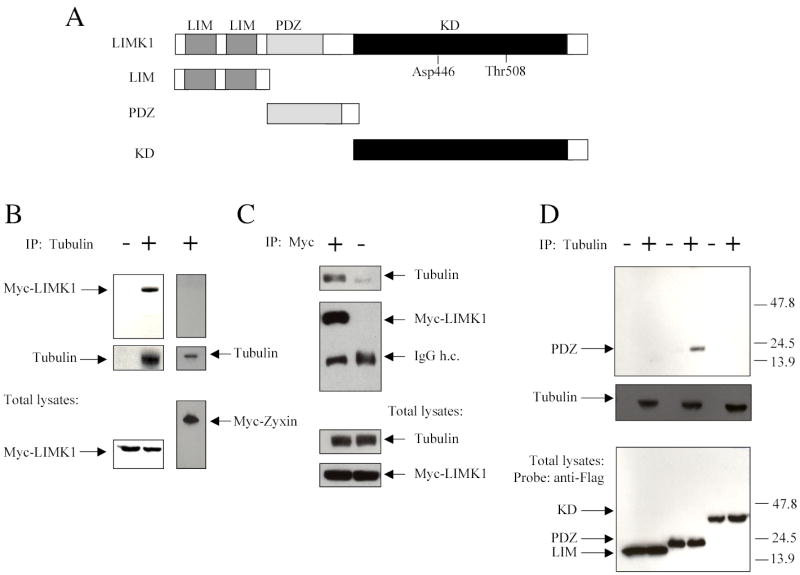
(A) Schematic representation of LIMK1 domain structure and its deletion mutants. Asp 446 is located in the kinase domain and is important for its kinase activity (13). Thr 508 is phosphorylated and activated by ROCK (40). KD, kinase domain.
(B) Overexpressed LIMK1 co-immunoprecipitates with endogenous tubulin. Lysates of COS-7 transfected with full-length Myc-tagged LIMK1 (Myc-LIMK1) or Myc-tagged Zyxin (Myc-Zyxin) were immunoprecipitated (IP) with anti-α-tubulin antibody (left panel) or anti-Myc antibody (right panel). Immunoprecipitates and total lysates were analyzed by Western blotting with anti-α-tubulin or anti-Myc antibodies.
(C) The PDZ domain is responsible for the interaction with tubulin. Lysates of COS-7 transfected with Flag-tagged deletion mutants of LIMK1 were immunoprecipitated with anti-α-tubulin antibody. Immunoprecipitates and total lysates were analyzed by Western blotting with anti-α-tubulin or anti-Flag antibodies. Shown are representative blots from three independent experiments that gave similar results.
Immunoprecipitation of cell lysates with anti-tubulin antibody followed by Western blotting with anti- Flag antibody revealed that the PDZ and not the LIM or the kinase domain interacted with tubulin (Figure 2C).
Modulation of the microtubule cytoskeleton induces changes in LIMK1 localization
We analyzed the intracellular distribution of LIMK1 in HUVECs stimulated with thrombin, a multifunctional enzyme that plays a central role in the regulation of biochemical, transcriptional, and functional responses of endothelial cells (reviewed in (20)). To test the effect of thrombin on microtubule organization in endothelial cells, we treated HUVECs with 25 nM thrombin for 10 min and found pronounced MTs destabilization resulting in disassembly of the peripheral microtubule network (Figure 3A). Changes in the relative amount of MTs were measured using Zeiss Enhanced Colocalization Tool software. The degree MTs disassembly was expressed as a ratio of the MTs area to the area of the whole cell. The data showed that approximately 44% of the MTs underwent disassembly upon thrombin stimulation (Figure 3A), consistently with previously published results (21). In addition, the level of acetylated tubulin, representing the stable microtubule pool, was decreased, further confirming the destabilization of MT upon thrombin stimulation (Figure 3A).
Figure 3. Thrombin induced LIMK1 translocation requires MTs destabilization.
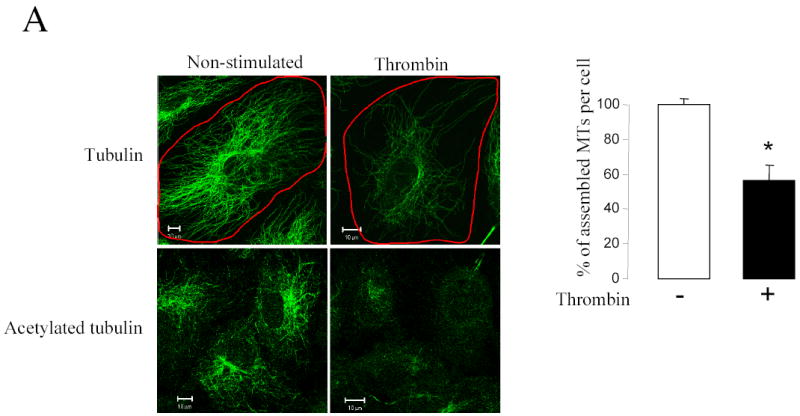
(A) Thrombin induces microtubule disassembly and decreases the amount of acetylated microtubules. HUVECs grown on gelatin-coated coverslips and serum starved for 3 h were stimulated with 25 nM thrombin for 10 min. Soluble cytosolic proteins, including monomeric tubulin, were extracted from the living cell. The cells were fixed and stained with either anti-α-tubulin or anti-acetylated tubulin antibodies. Shown are representative images from three independent experiments. Changes in the relative amount of microtubules were determined using Zeiss Enhanced Colocalization Tool software. Data were collected from 20 cells for each experiment. An average of the percentage of assembled microtubules (MTs) per cell was calculated from three independent experiments. Bars, 10 μm. * p<0.05
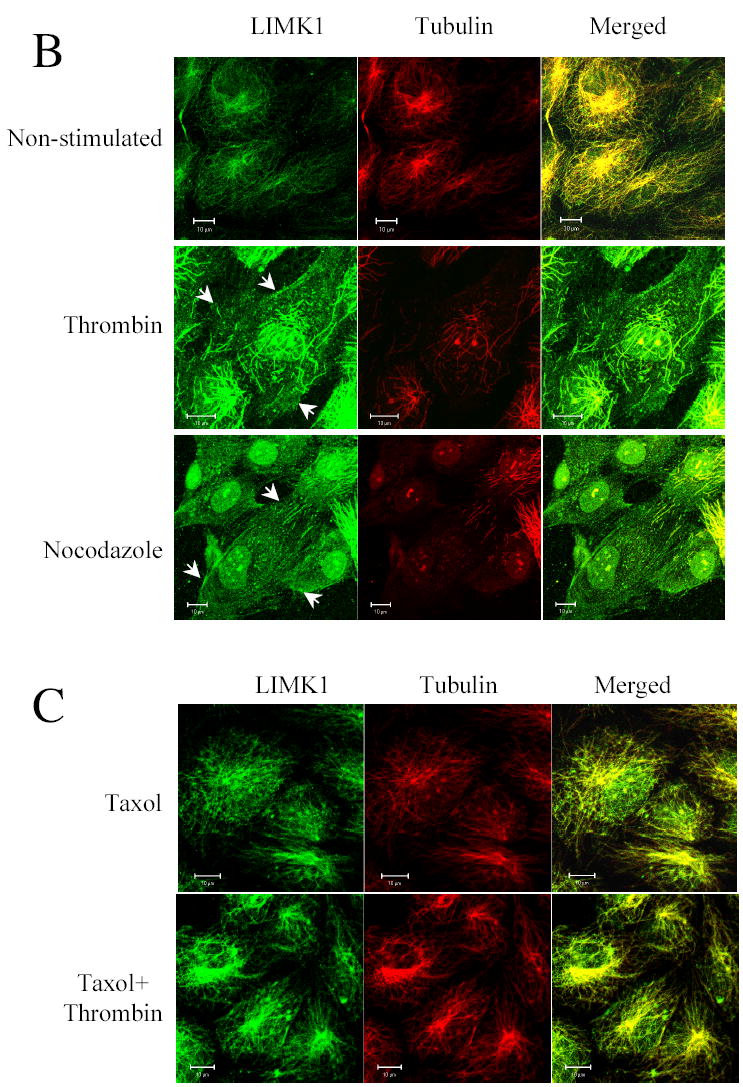
(B) Thrombin and nocodazole induce MTs destabilization and LIMK1 translocation. Serum starved HUVECs were stimulated with 25 nM thrombin for 10 min or with 1 μM nocodazole for 5 min. Soluble cytosolic proteins, were extracted from the living cells and the cells were fixed and stained with anti-α-tubulin and anti-LIMK1 antibodies. Bars, 10 μm. Shown are representative images from three independent experiments.
(C) Stabilization of microtubules with taxol prevents thrombin-dependent microtubules disassembly and LIMK1 translocation. HUVECs were pre-treated with 20 nM taxol for 20 min and stimulated with 25 nM thrombin for 10 min as indicated. Soluble cytosoli c proteins were extracted and the cells were fixed and stained with anti-α-tubulin and anti-LIMK1 antibodies. Images were captured by dual-wavelength laser scanning confocal microscope. Bars, 10 μm. Shown are representative images from three independent experiments.
Importantly, upon thrombin treatment the cell morphology changed together with the pattern of LIMK1 staining; the apparent filamentous staining of LIMK1 became more homogenous as it translocated to the periphery of the cell (Figure 3B). Colocalization coefficient calculated as (c1 (%)=100%* pixelsCh1, coloc/ pixelsCh1, total) of LIMK1 and tubulin significantly decreased from 85±4.5% to 47±6.2%. To determine if thrombin-induced changes to LIMK1 staining pattern and localization was due to MTs destabilization, we treated endothelial cells with 1 μM nocodazole for 5 min. Treatment with nocodazole resulted in MTs destabilization similar to that seen after incubation with thrombin (Figure 3B). Similar to cells treated with thrombin, addition of nocodazole also resulted in more homogenous LIMK1 staining and its re-distribution to the periphery of the cell (Figure 3B). Colocalization coefficient of LIMK1 and tubulin was significantly from 85±4.5% to 25±7.9%.
To determine if MTs destabilization was required for thrombin-dependent changes of LIMK1 intracellular distribution, microtubule cytoskeleton was stabilized by taxol, an agent that binds microtubules and counteracts the effects of GTP hydrolysis (22). In cells treated with taxol, LIMK1 was co-localized with MTs (Figure 3C). Stimulation of these cells with thrombin did not affect microtubule structure and did not change the pattern of LIMK1 localization (Figure 3C). Together, these results indicate that modulation of the microtubule cytoskeleton induces changes in LIMK1 localization.
Thrombin enhances the interaction of LIMK1 with F-actin
We have determined that MTs and F-actin are located at different intracellular compartments with greater proportion of MTs found in the apical part of the cell, whereas F-actin is found in the basal part of the cell (Figure 4A). Thrombin that induced MTs destabilization and actin polymerization did not promote the co-localization of MTs and F-actin (Figure 4A). Moreover, co-localization coefficient calculated as (c1 (%)=100%* pixelsCh1, coloc/ pixelsCh1, total) was equal to ~1.61% suggesting the absence of colocalization.
Figure 4. LIMK1 co-localizes with F-actin upon thrombin stimulation.
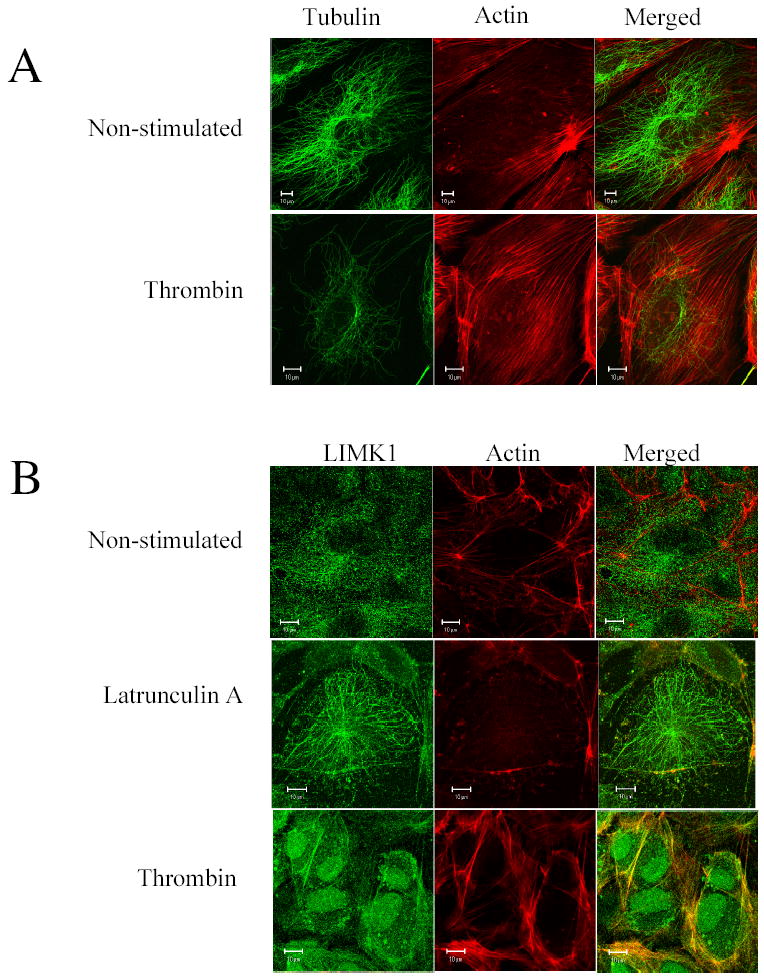
(A) Microtubule and F-actin are located at different cellular compartments in resting and thrombin-stimulated HUVECs. Serum starved HUVECs grown on gelatin-coated coverslips were stimulated with 25 nM thrombin for 10 min. Soluble cytosolic proteins were extracted from the living cells in the presence of 20 nM taxol and 2 μM phalloidin and then fixed and stained with anti-α-tubulin antibody and Alexa Fluor 594 phalloidin. Images were captured by dual-wavelength laser scanning confocal microscope. Bars, 10 μm. Shown are representative images from three independent experiments.
(B) LIMK1 colocalizes with F-actin upon thrombin stimulation. Serum starved HUVECs grown on gelatin-coated coverslips were treated either with 25 nM Latrunculin A or with 25 nM thrombin for 10 min, as indicated. Soluble cytosolic proteins were extracted from the living cells in the presence of 2 μM phalloidin. Cells were fixed and stained with anti-LIMK1 antibody and Alexa Fluor 594 phalloidin. Bars, 10 mm.
As LIMK1 was reported to interact with F-actin in in-vitro (23) and in-vivo (23), we analyzed the relative cellular distribution of endogenous Factin and LIMK1 in HUVECs using confocal microscopy. Surprisingly, in unstimulated cells very little, if any, colocalization of LIMK1 with F-actin was observed (Figure 4B). Co-localization coefficient of LIMK1 and actin was equal 11±6.4. Latrunculin A, an inhibitor of actin polymerization, dramatically decreased F-actin staining but had no effect on the pattern of LIMK1 staining, supporting the notion that LIMK1 did not interact with Factin in these cells. Interestingly, upon thrombin stimulation, we detected pronounced co-localization between LIMK1 and F-actin, especially at the cell periphery (Figure 4B). Colocalization coefficient was significantly increased from 11±6.4 to 43±6.4.
The kinase activity of LIMK1 is required for the interaction with tubulin and actin
In endothelial cells, thrombin was shown to activate Rho and its target Rho kinase (25). The Rho-associated kinase ROCK activates LIMK1 by phosphorylation at threonine 508 within the kinase activation loop (26). Inhibition of Rho kinase activity by the pharmacological inhibitor, Y27632, prevented thrombin-induced actin stress fiber formation in endothelial cells (21). Similarly, inhibition of Rho kinase activity prevented thrombin-induced depolymerization of microtubules (21). Initially, we determined whether thrombin could induce activation of LIMK1 by ROCK. Endogenous LIMK1 was immunoprecipitated from HUVECs treated with or without 25 nM thrombin and was used in in vitro kinase assay using cofilin as a substrate. Data showed that stimulation of HUVECs with thrombin for 10 min significantly increased LIMK1- dependent cofilin phosphorylation, as it was detected using specific antiphospho-cofilin antibodies (Figure 5A). Pre-treatment of HUVECs with ROCK inhibitor Y27632 for 10 min completely abolished thrombin-induced cofilin phosphorylation (Figure 5A), which suggest that LIMK1 is activated upon thrombin stimulation by ROCK. Therefore, we have tested the possibility that inhibition of ROCK would also prevent the thrombindependent change of LIMK1 localization in endothelial cells. Pretreatment of HUVECs with Y27632 for 10 min inhibited thrombin-induced actin polymerization and microtubule depolymerization (Figure 5B). Interestingly, Y27632 also abolished LIMK1 translocation in cells challenged with thrombin (Figure 5B), suggesting that ROCK was involved in the thrombin-dependent LIMK1 translocation.
Figure 5. LIMK1 activity is required for its interaction with tubulin and actin.
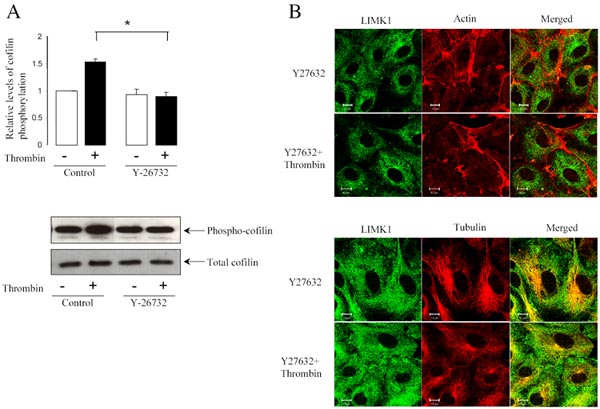
(A) Inhibition of ROCK activity prevents thrombin induced cofilin phosphorylation. HUVECs were pretreated with or without 5 μM Y27632 for 30 min and incubated with 25 nM thrombin for 10 min. Total lysates were analyzed by Western blotting with anticofilin and ant-phospho-cofilin antibodies. Data represent mean ± S.E. of triplicate determinations obtained from three experiments.
(B) Inhibition of ROCK activity prevents thrombin-induced actin stress fiber formation and microtubule depolymerization. HUVECs pretreated with 5 μM Y27632 for 30 min, were incubated with 25 nM thrombin for 10 min, and then fixed, and stained with anti-LIMK1 and anti-α-tubulin antibodies or with Alexa Fluor 594 phalloidin, as indicated.(B) ROCK2 stimulates LIMK1 activity. In vitro kinase assay of immunoprecipitated Myc-tagged wild type LIMK1 or LIMK1 (D446A) proteins in the absence or presence of ROCK2, using 0.2 μg of recombinant GST-cofilin as a substrate. After 20 min incubation with 50 μM ATP and 1 μCi [32P]-γ ATP, the proteins were separated on SDS-PAGE. Phosphorylated cofilin was visualized by autoradiography, and its phosphorylation level was determined by a PhosphorImager. LIMK1 activity is expressed as percentage increase in cofilin phosphorylation over its basal level. Basal level of phosphorylation was defined as the amount of [32P] incorporated into cofilin in the absence of overexpressed Myc-LIMK1. Data represent mean ± S.E. of triplicate determinations obtained from five experiments.
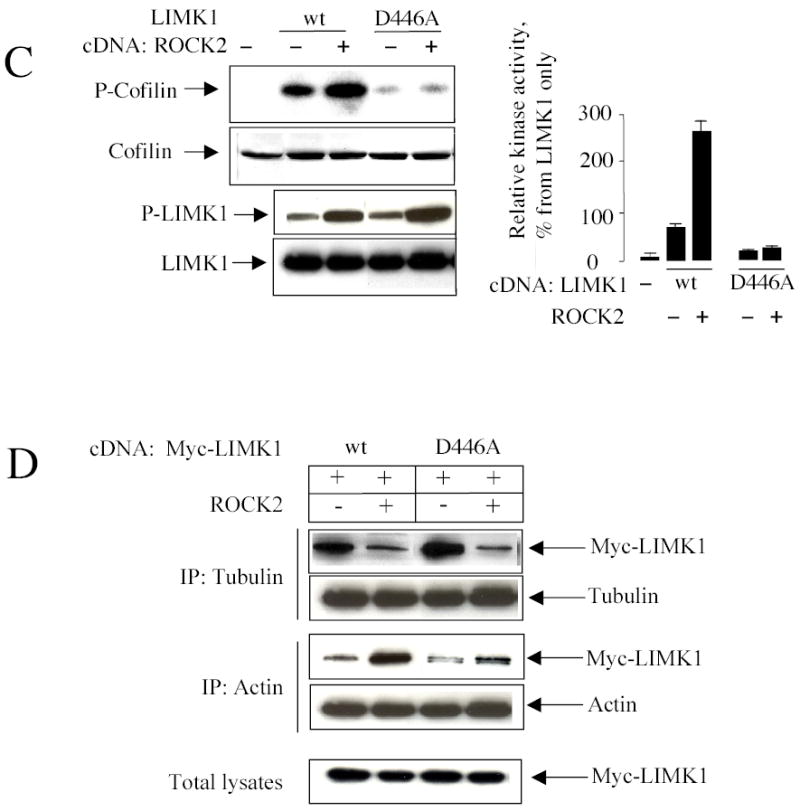
(C) LIMK1 phosphorylation by ROCK2 decreases its interaction with tubulin and increases complex formation of LIMK1 with actin. Lysates of COS-7 cells (60-mm dishes) expressing wild type LIMK1 or LIMK1 (D446A) in the absence or presence ROCK2 protein were immunoprecipitated with anti-α-tubulin or anti-β-actin antibodies. Immunoprecipitates and total cell lysates were analyzed by Western blotting with anti-ROCK2, -tubulin, -actin, -Myc, and -phospho-LIMK antibodies, as indicated. Shown are representative blots from three independent experiments.
Initially, we determined the ability of overexpressed wild type LIMK1 or kinase-dead LIMK1 (D446A) to phosphorylate cofilin and to be phosphorylated by the Rho kinase, ROCK2. In vitro kinase assay showed that wild-type LIMK1 phosphorylated cofilin (Figure 5C) and that this phosphorylation was significantly increased by ROCK2. As previously demonstrated, LIMK1 (D446A) could not phosphorylate cofilin (Figure 5C). Similarly, antibody specific to phosho-T508 of LIMK1 detected basal phosphorylation of wild-type LIMK1 and kinase-dead LIMK1 (D446A) that was further enhanced by ROCK2 (Figure 5C).
To determine whether ROCK2 could affect the interaction between wild-type LIMK1 and kinase-dead mutant with tubulin and actin, COS-7 were transfected with wild-type LIMK1 or LIMK1 (D446A) in the absence or presence of ROCK2. Forty-eight hours later, the cells were lysed and immunoprecipitated with either anti-α-tubulin or anti-β-actin antibodies. Western blot analysis showed that the amount of wild-type LIMK1 and LIMK1 (D446A) associated with tubulin was greatly decreased in the presence of ROCK2 (Figure 5D). These data suggest that kinase activity is not required for complex formation with tubulin and for the modulation of LIMK1 association with tubulin upon activation.
In contrast, the amount of wildtype LIMK1 associated with actin was greatly increased in the presence of ROCK2 (Figure 5D), supporting the notion that activation of LIMK1 led to increased association with actin. Importantly, while the interaction between LIMK1 (D446A) and actin is similar to that of wild type LIMK1, no changes were observed in the presence of ROCK2 (Figure 5D). These data suggest that LIMK1 activation increases its association with actin.
LIMK1 is required for thrombininduced MTs destabilization and actin polymerization
To determine the ability of LIMK1 to modulate the microtubule and actin cytoskeleton in HUVECs, we studied the effects of overexpressed wild type LIMK1, LIMK1 (D446A), and small interference RNA (siRNA). Overexpression of wild type LIMK1 induced MTs destabilization; with the relative amount of microtubules decreased by ~52 percent (Figure 6A). Similarly, acetylated tubulin was dramatically decreased in cells expressing wild type LIMK1 (Figure 6A). Thrombin that induced MTs destabilization in non-transfected cells did not cause any further changes in microtubule organization in the cells transfected with wild-type LIMK1 (Figure 6A). In contrast, expression of kinase-dead LIMK1 did not induce MTs destabilization in endothelial cells (Figure 6B). Importantly, it attenuated thrombin-induced MTs destabilization and preserved ~80% of acetylated microtubules (Figure 6B).
Figure 6. LIMK1 activity is required for MTs destabilization.
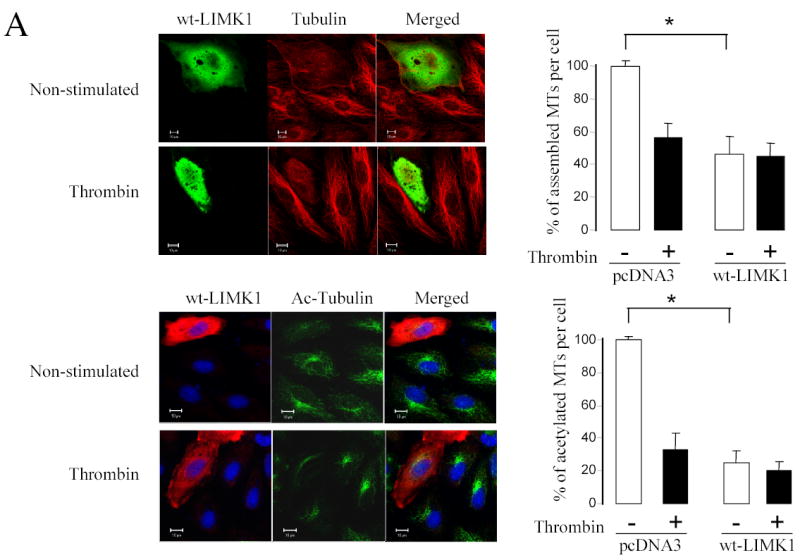
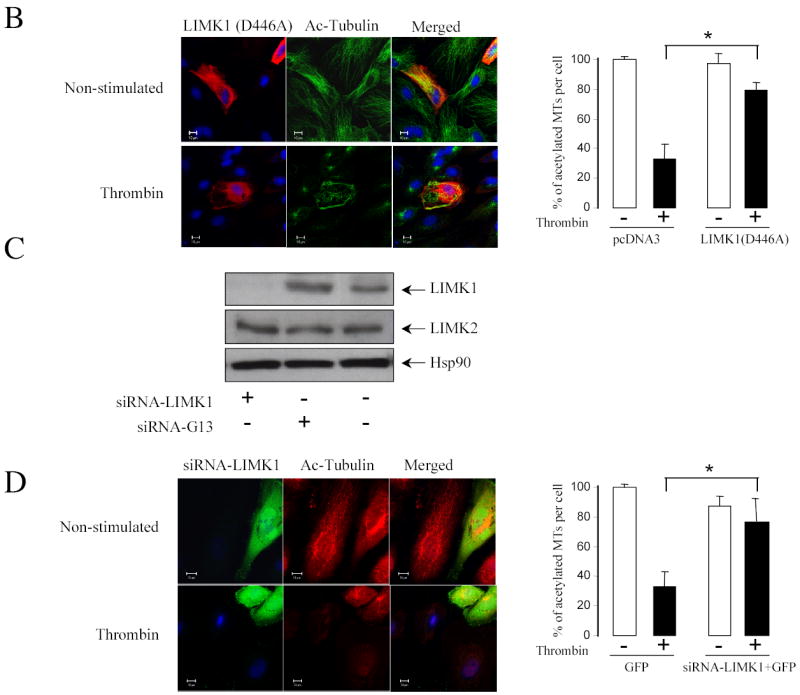
HUVECs grown on gelatin-coated coverslips were transfected with (A) Myc-tagged wild-type LIMK1 or (B) Myc-tagged LIMK1 (D446A) cDNAs. Twenty-four hours later, cells were serum starved for 3 h, stimulated with 25 nM thrombin for 10 min and then fixed and stained with anti-Myc, anti-α-tubulin, or anti-acetylated tubulin antibodies. Images were captured by dual-wavelength laser scanning confocal microscope. Changes in the relative amount of microtubules were quantified using Zeiss Enhanced Colocalization Tool software. The MTs stability was expressed as the ratio between the area containing the microtubules and that of the whole cell. Data was collected from 20 cells for each experiment. *p<0.05 Shown are representative images from three independent experiments. (B) HUVECs were transfected with siRNALIMK1 or siRNA-G13 for 24 hours. Total cell lysates were analyzed by Western blotting using anti-LIMK1, anti-Hsp90, and anti-LIMK2 antibodies. (D) HUVECs were transfected with plasmid encoding for green fluorescent protein (GFP) only or together with siRNA-LIMK1. Twenty-four hours after transfection, HUVECs were serum starved for 3 h, stimulated with 25 nM thrombin for 10 min and then fixed and stained with anti-acetylated tubulin antibody. Images were captured by dual-wavelength laser scanning confocal microscope. Changes in the relative amount of acetylated tubulin were quantified using Zeiss Enhanced Colocalization Tool software. The MTs stability was expressed as the ratio between the area containing the acetylated tubulin and that of the whole cell. Data was collected from 100 cells for each experiment. *p<0.05 Shown are representative images from three independent experiments.
We used small interfering RNA targeted against LIMK1 to determine LIMK1 role in the thrombin-induced modulation of microtubule cytoskeleton. HUVECs were transfected with or without siRNA against LIMK1 or Gα13 (negative control). Twenty-four hours later, cells were lysed and probed with antibodies against LIMK1, LIMK2, and Hsp90 (Figure 6C). Data showed that siRNALIMK1 but not siRNA-Gα13 induced significant down-regulation of the LIMK1 protein. Importantly, LIMK2 expression was not affected under any experimental condition suggesting that LIMK1 siRNA was specific (Figure 6C).
To demonstrate that LIMK1 is required for MTs destabilization induced by thrombin, HUVECs were transfected with GFP in the absence or presence of siRNA-LIMK1 and MTs stability was analyzed in the cells expressing GFP. In control experiment using oligonucleotide conjugated to a fluorescent probe, we determined that the cotransfection efficiency with GFP was ~95%. Data showed that siRNALIMK1 did not affected MTs stability in non-stimulated HUVECs. Importantly, downregulation of LIMK1 inhibited MTs destabilization induced by thrombin (Figure 6D).
Similar to its effect on fibroblasts and epithelial cells, wildtype LIMK1 induced formation of actin stress fibers in endothelial cell (Figure 7A), whereas stimulation of the HUVECs with thrombin did not further increase F-actin staining. LIMK1 (D446A) did not promote stress fiber formation, but it attenuated actin stress fiber formation upon thrombin stimulation (Figure 7B). Importantly, down-regulation of endogenous LIMK1 using siRNA also attenuated stress fiber formation upon thrombin stimulation (Figure 7C). Together, these data indicate that LIMK1 is required for thrombin-induced MTs destabilization and actin polymerization.
Figure 7. LIMK1 activity is required for actin polymerization.
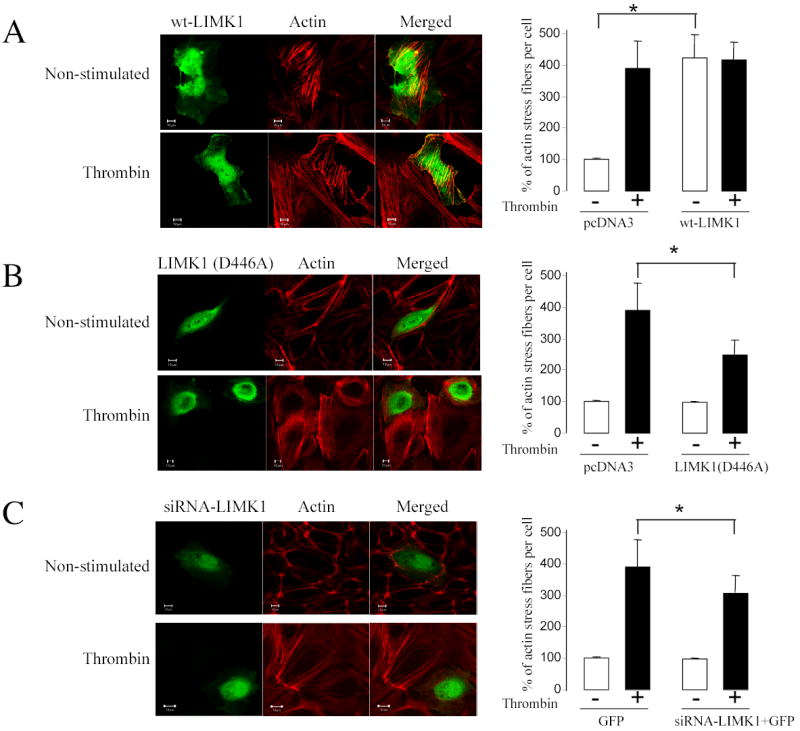
HUVECs were grown on gelatin-coated coverslips and transfected with (A) Myc-tagged wild-type LIMK1, (B) LIMK1 (D446A) DNA or (C) GFP with or without siRNALIMK1. Twenty-four hours later cells were serum starved for 3 h, stimulated with 25 nM thrombin for 10 min, fixed and stained with anti-Myc antibody and Alexa Fluor 594 phalloidin. The amount of F-actin was measured using Zeiss Enhanced Colocalization Tool software. Shown are representative images from three independent experiments. The changes in the relative amount of F-actin were expressed as the ratio of the F-actin area to the area of the whole cell. Data was collected from 100 cells for each experiment. *p<0.05
Discussion
LIMK1 interacts with tubulin
We have demonstrated here that in endothelial cells LIMK1 is co-localized with MTs and can form a complex with tubulin via its PDZ domain. Depolymerization of MTs induced by thrombin or nocodazole resulted in decreased LIMK1 interaction with tubulin and changes LIMK1 localization to sites with increased actin dynamics. This thrombin-dependent translocation of LIMK1 was prevented by stabilization of MTs with taxol.
In contrast to previous findings, we did not detect any significant colocalization of LIMK1 with F-actin in non-stimulated endothelial cells; however, co-localization was dramatically increased upon stimulation with thrombin. Finally, LIMK1 was required for the thrombin-induced modulation of MTs destabilization and actin polymerization. These results indicate that LIMK1 may coordinate microtubules and actin cytoskeleton in endothelial cells.
Microtubules and LIMK1
In endothelial cells, MTs destabilization is associated with stress fiber formation, contraction, and endothelial cell barrier dysfunction (27). Recent studies suggest direct involvement of MTs in the regulation of endothelial integrity and wound repair, as depolymerization by the microtubule inhibitors nocodazole and vinblastine results in rearrangement of the actin cytoskeleton, increased stress fiber formation, cell contraction, and permeability (6,28). Here, we showed that LIMK1 could induce MTs destabilization (Figure 6A). As kinase-dead mutant of LIMK1 and LIMK1 downregulation using siRNA, prevented thrombin-induced MTs destabilization, this suggests that LIMK1 is required for the thrombininduced MTs destabilization.
The LIM and PDZ domains are known to mediate protein–protein interactions (reviewed in (29,30)). Deletion mutants lacking these domains show increased LIMK1 activity suggesting that they regulate LIMK1 activity (13). We have shown that LIMK1 interacts with tubulin via its PDZ domain (Figure 2C). Previous yeast two-hybrid and mammalian cell interaction analyses have revealed that LIMK1 interacts via its LIM domains with a number of proteins including protein kinase C (PKC) (31), the cytoplasmic domain of the transmembrane ligand neuregulin (32), the cytoplasmic tail of bone morphogenic protein receptor type II (BMPRII) (16) and LATS1 (33). While no function was assigned to the interactions between LIMK1 and PKC or neuregulin, the interaction with BMPRII and LATS1 resulted in down regulation of LIMK1 activity. However, the PDZ domain was not shown previously to mediate LIMK1 interactions.
Microtubule dynamics is regulated by two groups of proteins; one stabilizes microtubules and the other destabilizes them. The proteins belonging to the first group are known as structural microtubule-associated proteins (MAPs), such as MAP1B and tau (34). Proteins that are potent destabilizers of microtubules have been identified more recently (35,36) and include stathmin and SCG10, which belong to the same gene family. The mechanism by which LIMK1 induces MTs destabilization is not yet known. One possibility is that LIMK1 may phosphorylate proteins that either stabilize or destabilize microtubules thereby affecting their function. As cofilin is the only well studied LIMK1 substrate, it is of great importance to identify novel proteins that can be phosphorylated by LIMK1. Another possibility could be that LIMK1 directly affects microtubule dynamics. Such a scenario was described for the Gα subunits of heterotrimeric G proteins where the Gα subunits activate tubulin GTPase and modulate microtubule polymerization dynamics (37).
LIMK1 and crosstalk between microtubules and actin cytoskeleton –
The role of LIMK1 in regulation of actin dynamics is well established. LIMK1 regulates actin dynamics via phosphorylation and inactivation of cofilin (8,9,15). The functional significance of LIMK1 in actin organization was revealed in studies using LIMK1 (−/−) mice demonstrating that LIMK1 is essential for normal spine morphology, synaptic regulation and memory (38). In agreement with previous findings, our data also demonstrate that LIMK1 induces actin stress fiber formation. In addition, we have shown that kinase-dead mutant of LIMK1 and downregulation of LIMK1 with siRNA attenuated actin stress fiber formation induced by thrombin (Figure 7B) and that stabilization of microtubules with taxol, prevented both translocation of LIMK1 to F-actin (Figure 3C).
Crosstalk between microtubules and the actin cytoskeleton is essential for the regulation of many cellular functions such as migration, locomotion, cytokinesis, and cell polarity (reviewed in (39)). The Rho family of small GTPases was shown to participate in the regulation of both microtubules and actin (7). Importantly, microtubule disassembly was shown to induce Rho activation (40). Microtubule disassembly releases the microtubule-bound Rho guanine nucleotide exchange factor (GEF), GEF-H1 to activate RhoA (41). However, the mechanism of agonistdependent microtubule disassembly is not yet understood. We have shown here that LIMK1 induces MTs destabilization and actin polymerization and propose that LIMK1 may serve as a molecular switch that regulates both MTs destabilization and formation of actin stress fibers, thereby providing the molecular mechanism that explains how microtubule disassembly promotes the formation of actin stress fibers.
In conclusion, the results presented here suggest that the regulation of microtubules and actin polymerization by LIMK1 provides a mechanism for the coordination of microtubule and the actin cytoskeletons. We propose that in resting endothelial cells LIMK1 is associated with microtubules (Figure 8). Ligandinduced activation of Rho-ROCK pathway activates LIMK1, which in turn causes MTs destabilization and release of LIMK1. Consequently, activated LIMK1 associates with actin, thereby inducing its polymerization via cofilin phosphorylation.
Figure 8. A proposed model of LIMK1 regulation of the actin cytoskeleton and microtubules.
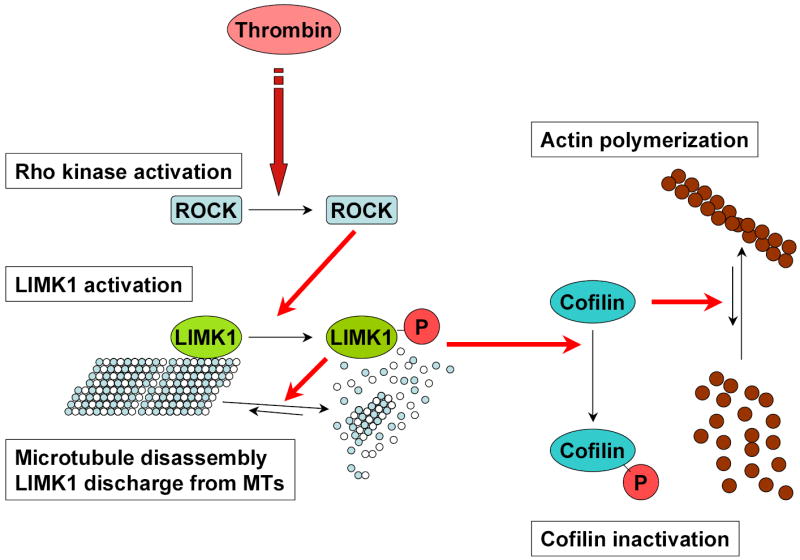
We propose that in resting endothelial cells LIMK1 is associated with microtubules. Ligand-induced activation of Rho-ROCK pathway activates LIMK1, which in turn causes MTs destabilization and release of LIMK1. Consequently, activated LIMK1 associates with actin, thereby inducing its polymerization via cofilin phosphorylation.
Acknowledgments
These studies were supported by NIH grants GM56159, GM65160 and HL06078 and by grant from American Heart Association to T.V.Y. T.V.Y. is an Established Investigator of the American Heart Association and AHA predoctoral fellowship to M.G. and J.P. Matvey Gorovoy thanks his parents for their constant help and support.
References
- 1.Gundersen GG, Cook TA. Curr Opin Cell Biol. 1999;11:81–94. doi: 10.1016/s0955-0674(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 2.Waterman-Storer CM, Salmon E. Curr Opin Cell Biol. 1999;11:61–67. doi: 10.1016/s0955-0674(99)80008-8. [DOI] [PubMed] [Google Scholar]
- 3.Dudek SM, Garcia JG. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 4.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 5.Danowski BA. J Cell Sci. 1989;93:255–266. doi: 10.1242/jcs.93.2.255. [DOI] [PubMed] [Google Scholar]
- 6.Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Curr Biol. 1996;6:1279–1289. doi: 10.1016/s0960-9822(02)70714-8. [DOI] [PubMed] [Google Scholar]
- 7.Wittmann T, Waterman-Storer CM. J Cell Sci. 2001;114:3795–3803. doi: 10.1242/jcs.114.21.3795. [DOI] [PubMed] [Google Scholar]
- 8.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 9.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 10.Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moriyama K, ida K, Yahara I. Genes Cells. 1996;1:73–78. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- 12.Agnew BJ, Minamide LS, Bamburg JR. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- 13.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 14.Dan C, Kelly A, Bernard O, Minden A. J Biol Chem. 2001;276:32115–32121. doi: 10.1074/jbc.M100871200. [DOI] [PubMed] [Google Scholar]
- 15.Maekawa M, shizaki T, Boku S, Watanabe N, Fujita A, wamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 16.Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O, Soosairaiah J. J Cell Biol. 2003;162:1089–1098. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Svitkina TM, Borisy GG. Methods Enzymol. 1998;298:570–592. doi: 10.1016/s0076-6879(98)98045-4. [DOI] [PubMed] [Google Scholar]
- 18.Vaiskunaite R, Kozasa T, Voyno-Yasenetskaya TA. J Biol Chem. 2001;276:46088–46093. doi: 10.1074/jbc.M108711200. [DOI] [PubMed] [Google Scholar]
- 19.Voyno-Yasenetskaya TA, Faure MP, Ahn NG, Bourne HR. J Biol Chem. 1996;271:21081–21087. doi: 10.1074/jbc.271.35.21081. [DOI] [PubMed] [Google Scholar]
- 20.Preissner KT, Nawroth PP, Kanse SM. J Pathol. 2000;190:360–372. doi: 10.1002/(SICI)1096-9896(200002)190:3<360::AID-PATH574>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 21.Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JG, Verin AD. FASEB J. 2004;18:1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- 22.Amos LA, Lowe J. Chem Biol. 1999;6:R65–69. doi: 10.1016/s1074-5521(99)89002-4. [DOI] [PubMed] [Google Scholar]
- 23.Ohashi K, Nagata K, Maekawa M, shizaki T, Narumiya S, Mizuno K. J Biol Chem. 2000;275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 24.Foletta VC, Moussi N, Sarmiere PD, Bamburg JR, Bernard O. Exp Cell Res. 2004;294:392–405. doi: 10.1016/j.yexcr.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 25.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. J Biol Chem. 1998;273:21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 26.Ohashi K, Nagata K, Maekawa M, shizaki T, Narumiya S, Mizuno K. J Biol Chem. 2000;275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 27.Verin AD, Birukova A, Wang P, Liu F, Becker P, Birukov K, Garcia JG. Am J Physiol Lung Cell Mol Physiol. 2001;281:L565–574. doi: 10.1152/ajplung.2001.281.3.L565. [DOI] [PubMed] [Google Scholar]
- 28.Lee TY, Gotlieb AI. Microsc Res Tech. 2003;60:115–127. doi: 10.1002/jemt.10250. [DOI] [PubMed] [Google Scholar]
- 29.Harris BZ, Lim WA. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 30.Retaux S, Bachy I. Mol Neurobiol. 2002;26:269–281. doi: 10.1385/MN:26:2-3:269. [DOI] [PubMed] [Google Scholar]
- 31.Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Gill GN, Kikkawa U. J Biol Chem. 1996;271:31029–31032. doi: 10.1074/jbc.271.49.31029. [DOI] [PubMed] [Google Scholar]
- 32.Wang JY, Frenzel KE, Wen D, Falls DL. J Biol Chem. 1998;273:20525–20534. doi: 10.1074/jbc.273.32.20525. [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Yu K, Hao Y, Li DM, Stewart R, nsogna KL, Xu T. Nat Cell Biol. 2004;6:609–617. doi: 10.1038/ncb1140. [DOI] [PubMed] [Google Scholar]
- 34.Hirokawa N. Curr Opin Cell Biol. 1994;6:74–81. doi: 10.1016/0955-0674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 35.Belmont L, Mitchison TJ. Cell. 1996;84:623–631. doi: 10.1016/s0092-8674(00)81037-5. [DOI] [PubMed] [Google Scholar]
- 36.Riederer BM, Pellier V, Antonsson B, Di Paolo G, Stimpson SA, Lutjens R, Catsicas S, Grenningloh G. Proc Natl Acad Sci U S A. 1997;94:741–745. doi: 10.1073/pnas.94.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roychowdhury S, Panda D, Wilson L, Rasenick MM. J Biol Chem. 1999;274:13485–13490. doi: 10.1074/jbc.274.19.13485. [DOI] [PubMed] [Google Scholar]
- 38.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez OC, Schaefer AW, Mandato CA, Forscher P, Bement WM, Waterman-Storer C. Nat Cell Biol. 2003;5:599–609. doi: 10.1038/ncb0703-599. [DOI] [PubMed] [Google Scholar]
- 40.Ren XD, Kiosses WB, Schwartz MA. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krendel M, Zenke FT, Bokoch GM. Nat Cell Biol. 2002;4:294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]