


Abstract
ENP1 is an essential Saccharomyces cerevisiae gene encoding a 483 amino acid polypeptide. Enp1 protein is localized in the nucleus and concentrated in the nucleolus. An enp1-1 temperature-sensitive mutant inhibited 35S pre-rRNA early processing at sites A0, A1 and A2 as shown by northern analysis of steady state levels of rRNA precursors. Pulse-chase analysis further revealed that the enp1-1 strain was defective in the synthesis of 20S pre-rRNA and hence 18S rRNA, which led to reduced formation of 40S ribosomal subunits. Co-precipitation analysis revealed that Enp1 was associated with Nop1 protein, as well as with U3 and U14 RNAs, two snoRNAs implicated in early pre-rRNA processing steps. These results suggest a direct role for Enp1 in the early steps of rRNA processing.
INTRODUCTION
Ribosome biogenesis is one of the major cellular activities in eukaryotic cells. It takes place primarily in a specialized subnuclear compartment, the nucleolus (1,2). In the yeast Saccharomyces cerevisiae, each rRNA gene is transcribed by RNA polymerase I into a 35S rRNA precursor, consisting of 18S, 5.8S and 25S rRNA sequences flanked by two external transcribed spacers (ETS) and separated by two internal transcribed spacers (ITS) (Fig. 1). This 35S precursor goes through a series of modifications and processing steps to generate the mature 18S, 5.8S and 25S rRNAs. The processing occurs first at sites A0, A1 and A2, resulting in the 20S pre-rRNA and 27SA2 pre-rRNA. The 20S pre-rRNA is then cleaved, leading to the mature 18S rRNA found in the 40S ribosomal subunit. The 27SA2 pre-rRNA is processed through two alternative pathways. The majority of 27SA2 pre-rRNA is cleaved at sites A3 and B2 to form the 27SA3 pre-rRNA, which is subsequently processed to produce 27SBS. Alternatively, 27SA2 pre-rRNA can be processed at sites B1L and B2 to generate 27SBL pre-rRNA. Both 27SBS and 27SBL pre-rRNAs are then cleaved at sites C1 and C2 to generate the mature 25S rRNA, and 7SS or 7SL intermediates, which are then processed to mature 5.8SS or 5.8SL rRNAs (Fig. 1). The 25S rRNA and 5.8S rRNA are the RNA components of the 60S ribosomal subunit (3). During the course of rRNA modification and processing, many of the ribosomal proteins are assembled onto the rRNA molecules to form the ribosome complex.
Figure 1.

Pre-rRNA processing pathways in S.cerevisiae. The 35S pre-rRNA contains the sequences of mature 18S, 5.8S and 25S rRNA flanked by the external transcribed spacers (5′ ETS and 3′ ETS) and separated by the internal spacers (ITS1 and ITS2). Letters A0 to E indicate the processing sites. The processing of pre-rRNA occurs first at sites A0, A1 and A2, generating the 20S pre-rRNA and 27SA2 pre-rRNA. The 20S pre-rRNA is then cleaved at site D, leading to the mature 18S rRNA found in the 40S ribosomal subunit. The 27SA2 pre-rRNA is processed by two alternative pathways. The majority of 27SA2 pre-rRNA is cleaved at sites A3 and B2 to form the 27SA3 pre-rRNA, which is subsequently processed to B1S by exonucleases to produce 27SBS. Alternatively, 27SA2 pre-rRNA can be processed at sites B1L and B2 to generate 27SBL pre-rRNA. Both 27SBS and 27SBL pre-rRNAs are then cleaved at sites C1 and C2 to generate the mature 25S rRNA, and 7SS or 7SL intermediates, which are then converted to mature 5.8SS or 5.8SL rRNAs.
Ribosome biogenesis needs a large number of trans-acting factors, including small nucleolar RNAs (snoRNAs), protein components of the snoRNP complexes, rRNA modifying enzymes, endo- and exonucleases, putative RNA helicases and other protein factors (3,4). In yeast cells there are more than 100 different snoRNAs playing important roles in rRNA modification and processing. On the basis of their structure, the snoRNAs can be divided into two groups: the box C/D family and box H/ACA family. Only one snoRNA, MRP RNA, belongs to neither family (5–7). While the majority of the snoRNAs participate in RNA pseudouridylation and 2′-O-ribose methylation, a few of them, including MRP snoRNA, box C/D snoRNAs U3 and U14, and box H/ACA snoRNAs snR10 and snR30, are required for processing of the pre-rRNA (8–12). Not only are U3, U14, snR10 and snR30 essential for early cleavages of 35S pre-rRNA to 18S rRNA, the proteins associated with them, including Nop1, Nop5, Gar1 and Nop10, have also been shown to be required for 18S rRNA synthesis (13–16).
The ENP1 (Essential Nuclear Protein 1) gene was identified in a genetic screen for suppressors of an ost4 mutation (oligosaccharide transferase 4) (17). However, in subsequent studies it was found not to be involved in OST4 function (18). ENP1 is an essential gene encoding a 483 amino acid polypeptide. The Enp1 protein is highly conserved and homologs are found in all eukaryotes. The yeast protein was localized to the nucleus in a previous study (18). On the other hand, an Enp1 human homolog, called bystin, was reported to localize to the cytoplasm and was proposed to be involved in cell adhesion (19).
In this study, we report that the Enp1 protein not only is nuclear but is enriched in the nucleolus. We also found that Enp1 is required for the synthesis of 40S ribosomal subunits, and its presence is necessary for the pre-rRNA processing to form 20S pre-rRNA and 18S rRNA. An association between Enp1 and U3 and U14 snoRNAs, and with the nucleolar protein Nop1, was established. We also found that human Enp1, expressed in yeast, was located in the nucleus and the nucleolus, suggesting that the function of this protein is conserved.
MATERIALS AND METHODS
Yeast strains and media
The S.cerevisiae strains used in this study are all derivatives of a wild-type diploid strain W303 (MATa/MATα ura3-1/ura3-1 leu2-3,112/leu2-3,112 trp1-1/trp1-1 his3-11,15/his3-11,15 ade2-1/ade2-1 can1-100/can1-100) except for strain RS1938. Strain JBY45 (MATa/MATα ENP1/Δenp1::his5+) was constructed by replacing one copy of the ENP1 open reading frame (ORF) with the Schizosaccharomyces pombe his5+ gene (18). Strain JBY46 [MATa Δenp1::his5+/pJB23 (ENP1, URA3, CEN6)] is a haploid strain derived from JBY45 with wild-type ENP1 on a low copy number plasmid. Strain JBY48 [MATa Δenp1::his5+/pJB19 (enp1-1, TRP1, CEN6)] and strain JBY49 [MATa Δenp1::his5+/pJB39 (enp1-2, TRP1, CEN6)] have plasmids with enp1 temperature-sensitive (ts) mutations. Strain JBY51 (MATa Δenp1::his5+ TRP1::enp1-1) is an enp1 ts strain generated by integrating the enp1-1 gene at the chromosomal trp1-1 locus. Strain CWY13 [MATa Δenp1::his5+/pJB24 (pMET25-GFP-ENP1, URA3, CEN6)] has GFP-ENP1 under control of the MET25 promoter. Strain CWY14 (MATa ENP1-TAP) was created by fusing sequences encoding a Tandem Affinity Purification (TAP) tag (20,21) to the 3′ end of ENP1. Strain YRH39 (MATα HST2-TAP) was created by fusing sequences encoding a TAP tag to the 3′end of HST2. Strain RS1938 (Δnop1::URA3 pUN100-ProtA-NOP1) was created by transforming RS1935 (MATa/α leu2/leu2 ura3/ura3 lys2/lys2 ade2/ade2 Δnop1::URA3/NOP1) with pUN100-ProtA-NOP1 (strain and plasmid supplied by T. Schafer), followed by tetrad dissection. Strain CWY15 [MATa/pCW109 (pMET25-GFP-hENP1, URA3, CEN6)] has a human homolog of Enp1 expressed in W303-1a. The media used were prepared as described (22).
Cloning of ENP1
pRS426-MEG1 (The original name of ENP1) was a gift from Dr William J. Lennarz. It contains the ENP1 gene within a 2.6 kb EcoRI genomic fragment cloned into pRS426 (18). The EcoRI fragment was subsequently cloned into pRS314 (TRP1, CEN6), pRS316 (URA3, CEN6) and pRS424 (TRP1, 2µ) to create pJB20, pJB23 and pJB21, respectively. pGFP-N-FUS is a centromeric plasmid for fusing green fluorescence protein (GFP) to a polypeptide’s N-terminus under control of the MET25 promoter (23). Cloning the ENP1 ORF into pGFP-N-FUS via XbaI and SalI sites generated plasmid pJB24. pJB19 (enp1-1, TRP1, CEN6) contains the enp1-1 mutant gene. The human homolog of yeast Enp1 was PCR amplified from a human cDNA clone BC007340 (Research Genetics), then cloned into pGFP-N-FUS, p415-ADH, p415-GPD and p415-TEF (24) via XbaI and SalI sites to generate pCW109, pCW113, pCW115 and pCW117, respectively. The fragment of the human Enp1 homolog (amino acids 152–437) was also cloned into these vectors to generate pCW110, pCW114, pCW116 and pCW118.
Random mutagenesis of ENP1 to generate enp1 ts mutants
The enp1 ts mutants were generated with a protocol introducing random mutations by PCR (25). The PCR was performed with oligonucleotides ENP1-MUT5′ (5′-GGTGGTGTCAGT AGGGGA-3′), ENP1-MUT3′ (5′-CAGTCTGCAATATA TGGAC-3′) and plasmid template pJB20 (see above). The nucleotide concentrations in the reaction were 1 mM each for dATP, dGTP, dCTP and dTTP. After strain JBY46 was transformed with the PCR product and with pJB20 gapped by NheI and NsiI, the transformants were replica-plated onto two plates containing 5-FOA synthetic medium without tryptophan; one replica was incubated at 23°C and the other at 37°C. Colonies growing at 23°C but not at 37°C were picked as candidates. Since approximately 10% of the colonies were inviable at both temperatures, we knew that 10% of the ENP1 PCR products lost their function after the PCR mutagenesis. This confirmed the effectiveness of the mutagenesis procedure. Two candidates showed good growth at 23°C but no growth at 37°C. They were named strains JBY48 and JBY49 and contained plasmids with the enp1-1 and enp1-2 mutations, respectively. The mutated enp1-1 gene on pJB19 was cloned into an integration vector, pRS304, as an EcoRI fragment. The plasmid was subsequently linearized with SnaBI within the TRP1 marker and was integrated at the chromosomal trp1-1 locus of strain JBY46. Selection on 5-FOA was used to remove plasmid pJB23, creating strain JBY51.
Sucrose gradient analysis
Polyribosome preparation and analysis were carried out essentially as described (26). Cells grown in YPD were collected at mid-log phase (OD600 0.8–1.0) and were broken with glass beads. The lysate was frozen immediately in liquid N2 and was stored at –80°C. Lysate (30 U of absorbance at OD260) was layered over a 7–47% (w/v) sucrose gradient, which was centrifuged at 28 000 r.p.m. for 5 h at 4°C in a SW28 rotor and was analyzed with an ISCO UA-5 gradient UV detection system on absorbency at 254 nm.
Immunofluorescence
Immunofluorescence analysis was carried out essentially as described (27). Cells were fixed with 3.7% formaldehyde at room temperature for 1.5 h. Antibodies included a mouse monoclonal anti-Nop1 (a gift from John P. Aris, University of Florida, Gainesville, Florida) and a Texas-red-conjugated donkey-anti-mouse antibody (Jackson Lab), both used at 1:500 dilution. Images were taken on a Zeiss Axioplan2 microscope equipped with a Zeiss AxioCam camera.
Pulse-chase labeling analysis of rRNA
For [methyl-3H]methionine pulse-chase analysis, cells were grown in synthetic medium without methionine at room temperature or 37°C for 2 h. When the OD600 reached 1.0, 6 ml of the culture were pulse-labeled with 250 µCi [methyl-3H]methionine (Amersham Pharmacia) for 3 min and chased with cold methionine (500 µg/ml) for 2, 4 or 12 min. For each time point of the chase 1.25 ml of culture was mixed with ice and collected. The pellets were frozen immediately in liquid N2 and stored at –80°C before total RNA was purified using a hot phenol method (28). The RNAs were separated on a 1.2% agarose formaldehyde gel and transferred onto Hybond-N+ nylon membranes (Amersham Pharmacia). After being sprayed with EN3HANCE (Du Pont), the membranes were exposed to film at –80°C (29).
Northern analysis
Cells were grown inYPD at room temperature or 37°C for 2–4 h. When the OD600 reached 1.0, 10 ml of cells were collected and frozen immediately. Total RNA was extracted as described (28). Five micrograms of RNA were separated on 1.2% agarose formaldehyde gels (for high molecular weight RNA) or on 6% polyacrylamide 7 M urea denaturing gels (for low molecular weight RNA) for each sample. The RNA was then transferred to Zeta-Probe GT nylon membranes (Bio-Rad). Probes for hybridyzation were specific oligonucleotides end-labeled using [γ-32P]ATP. After hybridization and washes, membranes were exposed to phosphorimager screens or X-ray films (29). Oligonucleotides specific for various regions of 35S pre-rRNA are: 1, GGTCTCTCTGCTGCCG GAAATG; 3, AATGAGCCATTCGCAGTTTCACTG; 4, GCTCTCATGCTCTTGCCAAAAC; 5, TGTTTGTTACCT CTGGGCCCCG; 6, TCCAGTTACGAAAATTCTTG; 7, CGTATCGCATTTCGCTGCGTTC; 8, GTTCGCCTAGAC GCTCTCTCTTC; 9, GCGAGATTCCCCTACCCAC. The regions complementary to these oligonucleotides are shown in Figure 6A. The oligonucleotides probing box C/D snoRNAs are: U3, TTCGGTTTCTCACTCACTCTGGGGTAC; U14, GGAACCAGTCTTTCATCACCGTG. The oligonucleotides probing box H/ACA snoRNAs are: snoRNA10, CCTTG CAACGGTCCTCATCCGGG; snoRNA30, GTCCGAAGC GCCATCTAGATGA.
Figure 6.

Effect of enp1-1 on steady state levels of pre-rRNAs and snoRNAs. Cells of W303-1a (ENP1) and JBY51 (enp1-1) were grown in YPD at 23 or 37°C for 2 or 4 h. Total RNA was separated on either 1.2% formaldehyde agarose gels (B–H) or 6% polyacrylmide 7 M urea denaturing gels (I) and transferred onto nylon membranes. Oligonucleotide probes specific for various regions of the pre-rRNA are shown in (A) and numbered 1–9. Northern blots with specific probes, labeled p3, p9, p5, p1, p4, p6, p8 and p7, are shown in panels B–H. Results with probes specific for various snoRNAs are shown in panel I.
RNA and protein precipitation analysis
Cells were grown in YPD (1 l) to OD600 1.0, then collected, washed once in ice-cold PBS and broken using glass beads in 10 ml IPP150 buffer (10 mM Tris pH 8.0, 150 mM NaCl and 0.1% NP-40) with protease inhibitors. The lysates were mixed with 200 µl IgG agarose beads for 2 h at 4°C. After several washes with 50 ml IPP150 buffer, the IgG beads were collected and the RNA associated with the beads extracted by the hot phenol method (28). One-tenth of the precipitated RNA was then separated on 6% polyacrylamide 7 M urea denaturing gels and electro-transferred to Zeta-Probe GT nylon membranes. They were probed with [γ-32P]ATP labeled oligonucleotides. For the lanes showing total RNA, 2 µg RNA prepared from exponentially growing cultures were loaded. For co-precipitation analysis of proteins, the IgG beads were resuspended in 2× Laemmli sample buffer, boiled for 5 min and separated by SDS–polyacrylamide gel electrophoresis. Approximately 1% of the precipitate was loaded onto each lane. For the total protein, 0.1% of the extract prior to precipitation was loaded onto each lane. Following electrophoresis, the proteins were transferred to nitrocellulose membranes and detected using anti-Nop1 antibody at 1:3000 dilution (provided by J. Aris) and anti-L3 antibody (provided by J. Warner) at 1:3000 dilution followed by peroxidase-conjugated anti-mouse secondary antibody at 1:5000 dilution. The antibody complexes were detected using ECL-Plus reagents (Amersham Pharmacia) as specified by the manufacturer.
RESULTS
Construction and analysis of ENP1 temperature-sensitive alleles
ENP1 is an essential yeast gene conserved among eukaryotes (18). To study its functions, we first created a diploid strain, JBY45, heterozygous for the Δenp1 mutation. Then two enp1 ts mutant alleles (enp1-1 and enp1-2) were generated by random PCR mutagenesis, as described in Materials and Methods. Both mutant alleles were recessive to the wild-type ENP1 gene. The enp1-1 gene was integrated into the chromosomal trp1-1 locus to create strain JBY51 and all subsequent experiments were done with this enp1 allele. At 23°C the growth of JBY51 was comparable to that of W303-1a (ENP1), but at 37°C it did not grow (Fig. 2). Sequence analysis revealed that enp1-1 contains two point mutations, resulting in substitutions of two amino acids: W242→G and V415→A. No attempt was made to determine whether both mutations were required for the ts phenotype. Flow cytometry of strain JBY51 showed DNA content profiles similar to those of the wild-type strain at the non-permissive temperature (data not shown), suggesting that ENP1 is not involved in cell cycle regulation.
Figure 2.
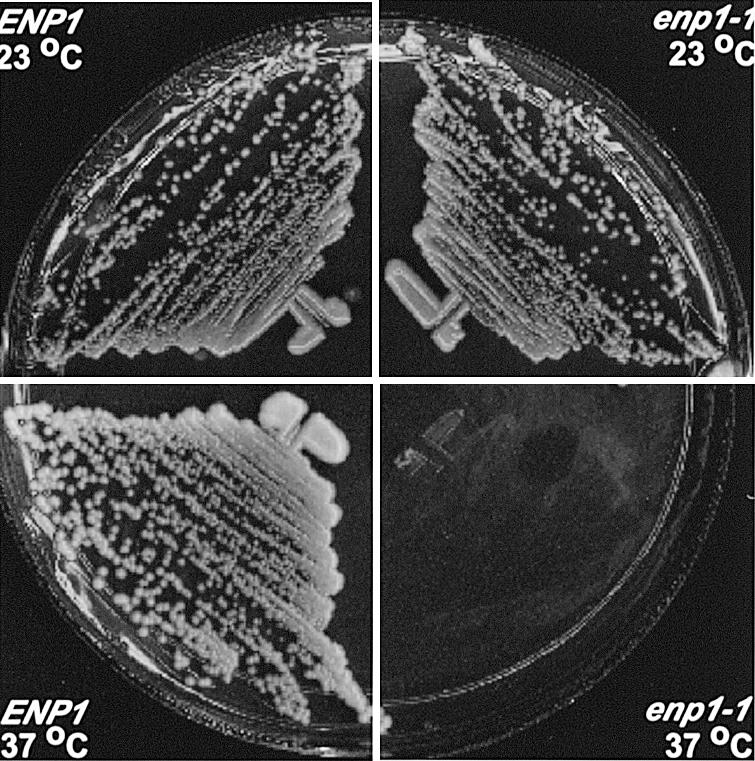
Growth of wild-type and enp1-1 ts mutant strains. Strain W303-1a (ENP1) and enp1 mutant strain JBY51 (enp1-1) were incubated for 3 days at 23 or 37°C on YPD medium.
Enp1 is enriched in the nucleolus
Enp1 tagged at the C-terminus with the myc epitope was previously found to localize to the nucleus (18). To expand this analysis, we fused GFP to the N-terminus of Enp1, and expressed the tagged Enp1 protein under control of the MET25 promoter as the sole source of Enp1 protein in the cells. Cells of strain CWY13 (pMET25-GFP-ENP1) were grown in media with methionine (transcription partially repressed) or without methionine (transcription not repressed). Growth of CWY13 was comparable to that of wild-type cells in both media (data not shown), indicating that the GFP tagged Enp1 protein was functional. In cells cultured in medium without methionine, in which the ENP transcription was not repressed, the GFP-Enp1 protein was expressed at a high level and showed strong green fluorescence distributed throughout the nucleus (data not shown), consistent with the original observation (18). With methionine added to the medium, however, the fluorescent signal of GFP-Enp1 was weaker and surprisingly showed crescent or cap-like patterns typical of nucleolar proteins (Fig. 3A). This nucleolar enrichment was confirmed by its co-localization with the nucleolar protein Nop1 (Fig. 3B).
Figure 3.

Enp1 is localized to the nucleolus. Strain CWY13 (GFP-Enp1) was grown at 30°C and then processed for immunofluorescence. The GFP-Enp1 was detected by its own green fluorescence (A), the nucleolus was visualized with anti-Nop1 antibody (B) and the nuclear DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (C).
ENP1 mutation leads to reduced levels of 40S ribosomal subunits
The nucleolar enrichment suggested that Enp1 might play some role in ribosome synthesis. To test this possibility, cells of W303-1a (ENP1), JBY51 (enp1-1) and a top2 ts strain RS191 (30) were grown in YPD at 23 or 37°C and their ribosome profiles analyzed after separation on sucrose gradients. At 23°C, enp1-1 cells showed polysome profiles similar to those of the wild-type and top2-1 strains (Fig. 4A–C). In contrast, after incubation at the non-permissive temperature (37°C) for 40 min (data not shown) and 2 h, enp1-1 cells had reduced levels of 40S ribosomal subunits, 80S monosomes and polysomes, along with a dramatic increase in the free 60S subunit peak (Fig. 4F), while the wild-type and top2-1 cells showed little change in polysome profile at 37°C (Fig. 4D and E). These results demonstrate that the changes of the enp1-1 polysome profile were due to specific defects caused by the enp1-1 mutation, and not due simply to the shift to 37°C for a wild-type or ts strain.
Figure 4.

The level of free 40S ribosomal subunits is reduced in enp1-1 mutants at 37°C but not in the wild-type or top2-1 strains. Cells of W303-1a (ENP1, A and D), RS191 (top2-1, B and E) and JBY51 (enp1-1, C and F) were grown in rich medium at 23 or 37°C for 2 h. The extracts were prepared and the ribosomes separated on 7–47% sucrose gradients.
The processing of pre-rRNA for 18S rRNA is impaired in enp1 mutants
In most cases reductions in ribosomal subunit levels are the results of defects in pre-rRNA processing or ribosome assembly or both (3). To study the mechanism by which Enp1 affects the 40S subunit, we analyzed the effects of enp1 mutations on processing of the pre-rRNA using pulse-chase labeling. [methyl-3H]methionine is preferred for labeling rRNAs in pulse-chase analysis because rRNAs are specifically methylated during the early steps of the processing. Cells of W303-1a (ENP1) and JBY51 (enp1-1) were grown at 23 or 37°C for 2 h before they were labeled with [methyl-3H]methionine for 3 min and chased with cold methionine for 2, 4 and 12 min. In wild-type cells, the labeled 35S rRNA precursor, 27S and 20S rRNA intermediates were rapidly chased into mature 25S and 18S rRNAs (Fig. 5), at 23 or 37°C. In contrast, enp1-1 cells showed dramatic changes after incubation at 37°C. Although at 23°C the synthesis and processing were comparable to those of wild-type cells, cells cultured at 37°C for 2 h had neither 20S pre-rRNA nor 18S rRNA, while 25S rRNA was generated at normal levels. The 37°C grown enp1-1 cells also had low levels of aberrant 23S and possibly 21S intermediates (Fig. 5). The defect in 18S rRNA synthesis was further supported by pulse-chase experiments carried out using [5,6-3H]uracil. The results obtained were essentially the same; no 20S pre-rRNA or 18S rRNA was produced, while processing to 25S rRNA was not affected (data not shown).
Figure 5.

Pulse-chase analysis shows reduced 18 rRNA synthesis in enp1 mutants. Strains W303-1a (ENP1) and JBY51 (enp1-1) were grown at 23 or 37°C for 2 h, labeled with [methyl-3H]methionine for 3 min, and subsequently chased for 2, 4 or 12 min. Total RNA was purified, samples with 20 000 c.p.m. were loaded on each lane of a 1.2% agarose formaldehyde gel.
Taken together, these results suggested that the enp1 mutation leads to specific defects in the processing pathway for 20S pre-rRNA and 18S rRNA, resulting in reduced 40S subunit synthesis and a lowered level of the 40S subunit.
Enp1 is required for pre-rRNA processing at A0, A1 and A2 sites
To define the processing steps affected by the enp1 mutation, the steady state levels of rRNA precursors and mature RNAs were analyzed by northern blotting. Total RNAs were isolated from cells of W303-1a (ENP1) and JBY51 (enp1-1) grown at 37°C for 2 or 4 h. After separation on formaldehyde agarose, RNAs were transferred onto nylon membranes and probed with radiolabeled oligonucleotides specific to various regions of the 35S pre-rRNA (Fig. 6A). After incubation at 37°C, enp1-1 cells had wild-type levels of 25S rRNA, but reduced levels of 18S rRNA (Fig. 6B). A probe specific to ITS1 (P5) further revealed that enp1-1 cells incubated at the non-permissive temperature accumulated two aberrant rRNAs, 23S and 21S (Fig. 6C). Aberrant rRNA processing products of similar sizes have been described in numerous studies (14,31–34) and result from defects in processing at A0, A1 and A2. Additional probes were used to verify the origins of the 23S and 21S RNAs in the enp1-1 strain. The 23S product was detected by oligos 1, 2, 4 and 5, but not by oligos 6, 7 or 8 (Fig. 6D, E, C, F, G and data not shown). Thus this RNA extends from the 5′ end of the 35S rRNA to the A3 site. The 21S product was detected by oligos 4 and 5, but not by oligos 1, 2, 6, 7 and 8 (Fig. 6E, C, D, F, G and data not shown), indicating that it extends from the A1 to the A3 site. At 37°C the enp1-1 cells also had less 32S and 20S rRNA intermediates (Fig. 6B and E). These results suggest that the enp1 mutation led to complete or nearly complete inhibition of processing at site A0 and A2, and partial inhibition of processing at site A1. As a consequence, the 35S pre-rRNA was cleaved at site A3 instead, producing 23S and 21S rRNA products. Consistent with this theory, the 27SA2 rRNA intermediate level was greatly reduced in enp1 cells at 37°C (Fig. 6C) due to the inhibition of processing at A2, while the 27SB pre-rRNA level was similar to wild-type cells (Fig. 6G). Moreover, no difference in the 5.8 rRNA level was observed between the wild-type cells and enp1 cells (Fig. 6H).
Enp1 is associated with U3 and U14 snoRNAs and with Nop1
It has previously been shown that mutations in the U3, U14, snR10 and snR30 snoRNAs and in their associated proteins affect processing of 35S pre-rRNA at A0, A1 and A2. Because of the similarity with the processing defects of enp1-1 strains, we tested the association between Enp1 and snoRNAs. To do this, we created a strain in which a TAP tag was fused to the C-terminus of Enp1. The TAP tag contains Staphylococcus aureus Protein A as well as calmodulin-binding peptide sequences, so the tagged Enp1 binds IgG beads with high specificity. Cells grew well with the TAP-tagged Enp1 as the only source of Enp1 protein, showing that it was functional. As a control, we used a TAP tag on an unrelated cytoplasmic protein, Hst2. Strain CWY14 (ENP1-TAP) and the Hst2-TAP tagged strain were grown in YPD to mid-exponential phase before being harvested. Extracts were prepared and mixed with IgG agarose beads to precipitate Enp1-TAP and associated RNAs. Total RNAs were extracted from washed IgG beads, separated on 6% polyacrylamide denaturing gels, transferred to nylon membranes and probed with radiolabeled oligonucleotides. Northern analysis revealed that Enp1-TAP precipitates were enriched with U3 and U14 snoRNAs relative to precipitates from the Hst2-tagged strain (Fig. 7A). The Enp1-TAP samples were not enriched with snR30 snoRNA (Fig. 7A), nor with U24 or U18 snoRNAs (data not shown). We found no change in the levels of U3, U14, snR10 and snR30 snoRNAs in the enp1 mutant, indicating that the mutation did not affect snoRNAs levels (Fig. 6I). The results indicate that Enp1 associates in vivo with U3 and U14 snoRNAs. Much less U3 RNA co-immunoprecipitated with Enp1-TAP than with Protein A-tagged Nop1, which is known to associate with U3 and U14 snoRNAs (Fig. 7A). However, Protein A-Nop1 was much more efficiently precipitated than was the Enp1-TAP protein (data not shown). Therefore, it is likely that similar amounts of RNAs are associated with the two proteins and that the proteins are part of the same complex. To address this further, we checked for the presence of Nop1 in the Enp1-TAP immunoprecipitates. Figure 7B shows that Nop1 indeed is in the Enp1 precipitate but not in the Hst2 control, whereas an abundant ribosomal protein, L3, is in neither precipitate.
Figure 7.
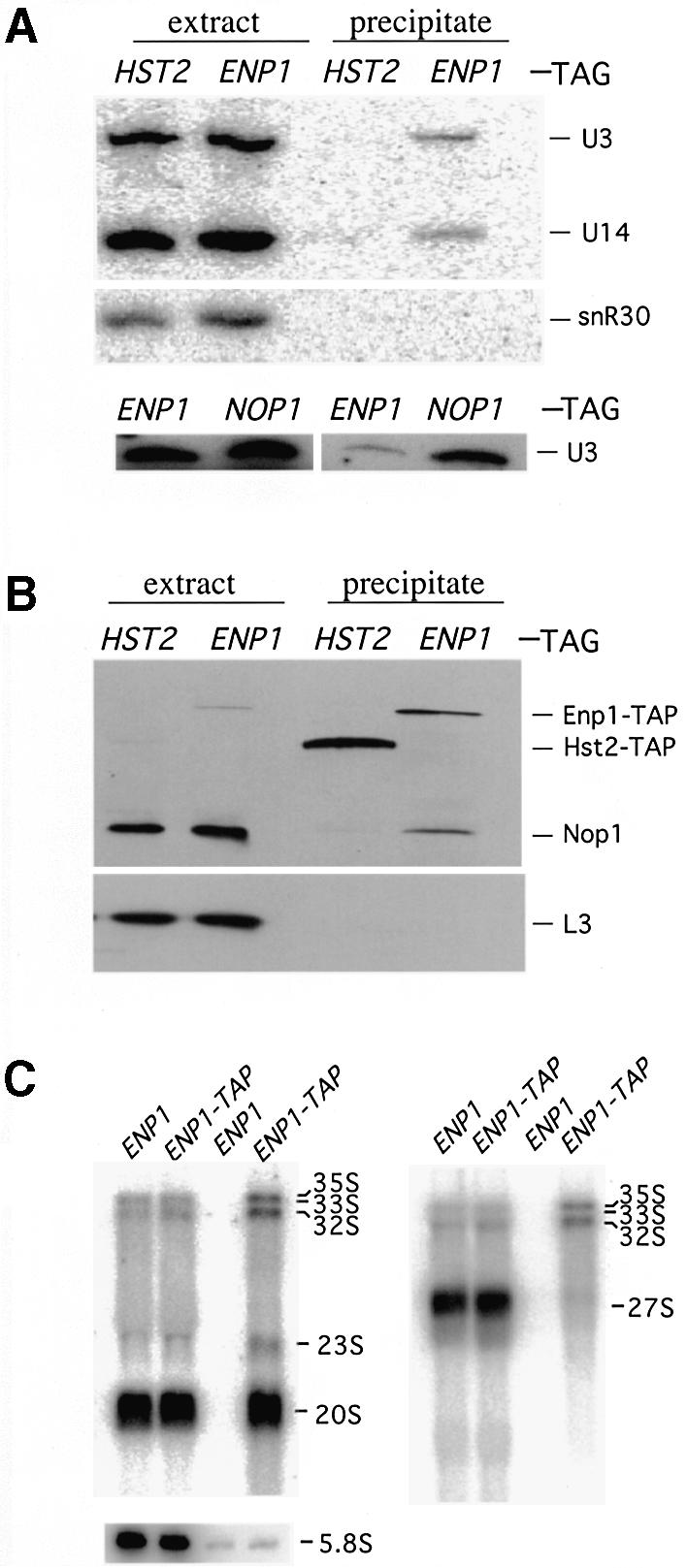
Enp1 co-immunoprecipitates with components of the 18S rRNA processing machinery. Cell extracts from YRH39 (HST2-TAP), CWY14 (ENP1-TAP) and RS1938 (Protein A-NOP1) were prepared and mixed with IgG agarose beads. The beads were washed and analysed for associated RNAs or proteins. (A) Northern analysis of snoRNAs associated with Enp1-TAP. Total RNA was isolated from cell extracts or IgG bead precipitates. RNA was then separated on a 6% polyacrylamide 7 M urea denaturing gel and probed with oligonucleotides specific for U3, U14 and snR30 snoRNAs. (B) Western analysis of proteins associated with Enp1-TAP. The proteins bound to the IgG agarose beads were separated on a 10% SDS–polyacrylamide gel. Subsequently the proteins were transferred onto nitrocellulose membranes and probed with anti-Nop1 antibody or anti-L3 antibody. (C) Enp1 associates with 18S rRNA precursors. RNAs isolated from extracts (lanes 1, 2, 5 and 6) and precipitates (lanes 3, 4, 7 and 8) were separated on either 1.2% formaldehyde agarose gels or 6% polyacrylamide 7 M urea denaturing gels (for 5.8S) and probed with radioactive oligonucleotides described in Figure 6: P4 (left panel), P8 (right panel) or specific for 5.8S RNA.
U3 and U14 snoRNAs have been shown to interact with rRNA precursors and these interactions are required for pre-rRNA processing (35–37). Using an in vitro system, U3 was also shown to be associated with its rRNA substrate and processing product (38). This raised the possibility that Enp1 might also be associated with rRNAs since its function in rRNA processing appeared linked to those of U3 and U14 snoRNAs. To test this possibility, we further analyzed the RNAs that co-precipiate with TAP-tagged Enp1. The RNAs were separated on 1.2% agarose formaldehyde gels or on polyacrylamide denaturing gels, transferred to nylon membranes and probed with radiolabeled oligonucleotides for pre-rRNAs. A probe (P4; see Fig. 6) specific to 20s and its precursors revealed that pre-rRNAs 35S, 33S, 32S and 20S specifically co-precipitated with Enp1-TAP (Fig. 7C). No such enrichment was found using a probe (P8; see Fig. 6) specific for 27S RNAs or a probe for 5.8S RNA (Fig. 7C). These results clearly demonstrated that Enp1 is associated with substrates and products of the early steps of 18S rRNA processing.
Comparison of Enp1 with homologs in other organisms
Homologs of Enp1 protein in human, Drosophila and Caenorhabditis elegans have been reported (18). A previously described human homolog, bystin, was only 306 amino acids in length, and lacked sequences corresponding to the N-terminal 163 amino acids of yeast Enp1 (19). In contrast, we identified a human expressed sequence tag (EST), BC007340 in a Blast search that revealed an ORF of 1311 nucleotides encoding a 437 amino acid polypeptide (39). Comparison of this 437 amino acid polypeptide with human bystin (19) showed that they were encoded by the same gene on human chromosome 6. The reported sequence for human bystin is a fragment of the 437 amino acid human Enp1 protein, due to a truncated cDNA sequence. Also, a sequence discrepancy at the C-terminus is due to inaccurate DNA sequence of the human bystin, as confirmed by available human genome and EST sequences. Searches of the database of other organisms identified Enp1 homologs in S.pombe, Arabidopsis thaliana, C.elegans, Drosophila melanogaster and mouse. The alignment of the homologs showed that they shared homology from the N-terminus to the C-terminus, with the C-terminal half extremely well conserved, with close to 90% similarity (Fig. 8). One interesting observation is that the two amino acids changed in the enp1-1 mutant, W242 and V415, are conserved among all homologs.
Figure 8.

Alignment of Enp1 homologs. Enp1 homologs were found in seven organisms, including S.cerevisiae, S.pombe, A.thaliana, C.elegans, D.melanogaster, Mus musculus and Homo sapiens. The amino acid sequences were analyzed and aligned with MacVector 6.5.3. Identical amino acids are in shaded boxes and similar amino acids are in unshaded boxes. Amino acid numbers are marked on both sides of each sequence. Accession numbers for the sequences are as follows: S.cerevisiae, P38333; S.pombe, CAA18662; A.thaliana, NP_174447; C.elegans, Q20932; D.melanogaster, P51406; M.musculus, BG977156; and H.sapiens, BC007340.
The human Enp1 homolog was cloned and expressed in yeast and tested for function. Expressed under the control of ADH, TEF or GPD promoter, the human Enp1 homolog did not complement a yeast enp1 null mutant. However, a GFP fusion to the N-terminus of human Enp1 homolog localized to the nucleus and was enriched in the nucleolus (data not shown).
DISCUSSION
ENP1 is a yeast gene first identified in a genetic screen for complementation of mutations in ost4, which encodes a subunit of oligosaccharide transferase (17), although subsequent work showed that it is unlikely that Enp1 has any connection to oligosaccharide transferase (18). ENP1 was shown to be essential for viability and the Enp1 protein localized to the nucleus (18).
When we re-examined the localization of Enp1, we observed that the protein was enriched in the nucleolus. This finding led us to test for a role of Enp1 in ribosome synthesis. Using an enp1 ts mutant, we found that depletion of Enp1 caused a 40S ribosomal subunit deficiency. Further analysis revealed that this deficiency was not due to a change of the subunit’s stability, but to a defect in the synthesis of the subunit’s 18S rRNA component. Pulse-chase analysis of RNA synthesis in an enp1-1 strain revealed that no 20S pre-rRNA or 18S rRNA was made at the non-permissive temperature, while levels of 25S rRNA appeared normal. Low levels of precursors to 18S rRNA were detected in the mutants, and they were extremely unstable. Northern analysis of steady state RNA levels demonstrated that the enp1 mutation specifically inhibited the pre-rRNA early cleavages at sites A0, A1 and A2, which are required for the production of the 20S pre-rRNA and the 18S rRNA. These defects of the enp1 mutation on rRNA processing are very similar to those caused by mutation of KRR1, another essential nucleolar gene required for synthesis of the 40S, but not the 60S subunit (40).
Co-precipitation analyses provided strong evidence that Enp1 is directly involved in pre-rRNA processing. In these experiments, TAP-tagged Enp1 bound to IgG beads specifically co-precipitated with two snoRNAs, U3 and U14, and with the 35S, 33S, 32S and 20S pre-rRNAs. Among the more than 100 snoRNAs in yeast cells, U3, U14, snR10, snR30 and MRP RNA are the only ones required for rRNA processing. It has been shown that mutations in U3, U14, snR10 and snR30 snoRNAs and protein components of the snoRNPs affect processing at sites A0, A1 or A2 (15,16,41,42). Enp1’s interaction with U3 and U14 snoRNAs suggests that Enp1’s function in rRNA processing involves these two snoRNAs. The fact that the nucleolar protein, Nop1, known to bind to U3 and U14 RNAs, also was found in the Enp1 precipitate, provides additional evidence that Enp1 is part of a complex involved in processing of rRNA. Recently, a genome-wide study of yeast protein complexes, using a TAP tag method similar to ours, reported a number of proteins that co-immunoprecipitated with Enp1 (43). These proteins included Imp4, Kre31, Kre33, Mpp10, Nop1, Nop14 and Sof1, all of which have been implicated in 18S RNA processing or 40S biogenesis. Very recently (after the studies in this manuscript were concluded) Grandi et al. published an analysis of components of the 90s preribosomal particle, which include the 35S pre-rRNA, U3 snoRNP and rRNA processing factors for the 40S subunit (44). In agreement with our findings, they found that Enp1 is a component of this 90s complex and also of a smaller complex containing the 20S pre-rRNA. Surprisingly, none of the other proteins involved in processing of pre-rRNAs that were tested associated with the 20S rRNA, suggesting that those proteins, but not Enp1, are released prior to 20S pre-rRNA formation (44). The authors also showed co-precipitation of Enp1 with dimethylated 20S pre-rRNA, which is formed after export of 20S to the cytoplasm. These results of Grandi et al. suggest that Enp1 may also be involved in later steps of processing or nuclear export.
A previous study on the Enp1 human homolog, bystin, reported that the protein was localized in the cytoplasm of mammalian cells and might be involved in cell adhesion (19). Our discovery of the 437 amino acid human Enp1 homolog revealed that the bystin studied previously was from a truncated library cDNA encoding only the C-terminal 306 amino acids. Although the human homolog of Enp1 did not complement a yeast Δenp1 mutant, we did show that it was localized to the nucleus and enriched in the nucleolus (data not shown). This strongly suggests that the conserved function of Enp1 is in rRNA processing. The cytoplasmic localization of bystin and its proposed function in cell adhesion are unlikely to reflect the actual function of the human Enp1 homolog.
It will be important to study the nature of the associations between Enp1 and U3 and U14 snoRNPs, and to learn more about the exact role of Enp1 in ribosomal RNA processing.
Acknowledgments
ACKNOWLEDGEMENTS
We thank J. Aris, J. Warner, T. Schafer and P. Silver for reagents, and J. Warner and A. Neiman for valuable advice. This work was supported by NIH grant GM28220.
REFERENCES
- 1.Shaw P.J. and Jordan,E.G. (1995) The nucleolus. Annu. Rev. Cell Dev. Biol., 11, 93–121. [DOI] [PubMed] [Google Scholar]
- 2.Scheer U. and Hock,R. (1999) Structure and function of the nucleolus. Curr. Opin. Cell Biol., 11, 385–390. [DOI] [PubMed] [Google Scholar]
- 3.Venema J. and Tollervey,D. (1999) Ribosome synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet., 33, 261–311. [DOI] [PubMed] [Google Scholar]
- 4.Kressler D., Linder,P. and de La Cruz,J. (1999) Protein trans-acting factors involved in ribosome biogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 7897–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maden T. (1996) Ribosomal RNA. Click here for methylation. Nature, 383, 675–676. [DOI] [PubMed] [Google Scholar]
- 6.Maxwell E.S. and Fournier,M.J. (1995) The small nucleolar RNAs. Annu. Rev. Biochem., 64, 897–934. [DOI] [PubMed] [Google Scholar]
- 7.Tollervey D. (1996) Small nucleolar RNAs guide ribosomal RNA methylation. Science, 273, 1056–1057. [DOI] [PubMed] [Google Scholar]
- 8.Hughes J.M. and Ares,M.,Jr (1991) Depletion of U3 small nucleolar RNA inhibits cleavage in the 5′ external transcribed spacer of yeast pre-ribosomal RNA and impairs formation of 18S ribosomal RNA. EMBO J., 10, 4231–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H.D., Zagorski,J. and Fournier,M.J. (1990) Depletion of U14 small nuclear RNA (snR128) disrupts production of 18S rRNA in Saccharomyces cerevisiae. Mol. Cell. Biol., 10, 1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tollervey D. (1987) A yeast small nuclear RNA is required for normal processing of pre-ribosomal RNA. EMBO J., 6, 4169–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrissey J.P. and Tollervey,D. (1993) Yeast snR30 is a small nucleolar RNA required for 18S rRNA synthesis. Mol. Cell. Biol., 13, 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shuai K. and Warner,J.R. (1991) A temperature sensitive mutant of Saccharomyces cerevisiae defective in pre-rRNA processing. Nucleic Acids Res., 19, 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tollervey D., Lehtonen,H., Carmo-Fonseca,M. and Hurt,E.C. (1991) The small nucleolar RNP protein NOP1 (fibrillarin) is required for pre-rRNA processing in yeast. EMBO J., 10, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu P., Brockenbrough,J.S., Metcalfe,A.C., Chen,S. and Aris,J.P. (1998) Nop5p is a small nucleolar ribonucleoprotein component required for pre-18 S rRNA processing in yeast. J. Biol. Chem., 273, 16453–16463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Girard J.P., Lehtonen,H., Caizergues-Ferrer,M., Amalric,F., Tollervey,D. and Lapeyre,B. (1992) GAR1 is an essential small nucleolar RNP protein required for pre-rRNA processing in yeast. EMBO J., 11, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henras A., Henry,Y., Bousquet-Antonelli,C., Noaillac-Depeyre,J., Gelugne,J.P. and Caizergues-Ferrer,M. (1998) Nhp2p and Nop10p are essential for the function of H/ACA snoRNPs. EMBO J., 17, 7078–7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roos J., Sternglanz,R. and Lennarz,W.J. (1994) A screen for yeast mutants with defects in the dolichol-mediated pathway for N-glycosylation. Proc. Natl Acad. Sci. USA, 91, 1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roos J., Luz,J.M., Centoducati,S., Sternglanz,R. and Lennarz,W.J. (1997) ENP1, an essential gene encoding a nuclear protein that is highly conserved from yeast to humans. Gene, 185, 137–146. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki N., Zara,J., Sato,T., Ong,E., Bakhiet,N., Oshima,R.G., Watson,K.L. and Fukuda,M.N. (1998) A cytoplasmic protein, bystin, interacts with trophinin, tastin and cytokeratin and may be involved in trophinin-mediated cell adhesion between trophoblast and endometrial epithelial cells. Proc. Natl Acad. Sci. USA, 95, 5027–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puig O., Caspary,F., Rigaut,G., Rutz,B., Bouveret,E., Bragado-Nilsson,E., Wilm,M. and Seraphin,B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods, 24, 218–229. [DOI] [PubMed] [Google Scholar]
- 21.Rigaut G., Shevchenko,A., Rutz,B., Wilm,M., Mann,M. and Seraphin,B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol., 17, 1030–1032. [DOI] [PubMed] [Google Scholar]
- 22.Kaiser C., Michaelis,S. and Mitchell,A. (1994) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Niedenthal R.K., Riles,L., Johnston,M. and Hegemann,J.H. (1996) Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast, 12, 773–786. [DOI] [PubMed] [Google Scholar]
- 24.Mumberg D., Muller,R. and Funk,M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene, 156, 119–122. [DOI] [PubMed] [Google Scholar]
- 25.Muhlrad D., Hunter,R. and Parker,R. (1992) A rapid method for localized mutagenesis of yeast genes. Yeast, 8, 79–82. [DOI] [PubMed] [Google Scholar]
- 26.Baim S.B., Pietras,D.F., Eustice,D.C. and Sherman,F. (1985) A mutation allowing an mRNA secondary structure diminishes translation of Saccharomyces cerevisiae iso-1-cytochrome c. Mol. Cell. Biol., 5, 1839–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pringle J.R., Adams,A.E., Drubin,D.G. and Haarer,B.K. (1991) Immunofluorescence methods for yeast. Methods Enzymol., 194, 565–602. [DOI] [PubMed] [Google Scholar]
- 28.Kohrer K. and Domdey,H. (1991) Preparation of high molecular weight RNA. Methods Enzymol., 194, 398–405. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 30.Thomas W., Spell,R.M., Ming,M.E. and Holm,C. (1991) Genetic analysis of the gyrase A-like domain of DNA topoisomerase II of Saccharomyces cerevisiae. Genetics, 128, 703–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zanchin N.I., Roberts,P., DeSilva,A., Sherman,F. and Goldfarb,D.S. (1997) Saccharomyces cerevisiae Nip7p is required for efficient 60S ribosome subunit biogenesis. Mol. Cell. Biol., 17, 5001–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong B., Brockenbrough,J.S., Wu,P. and Aris,J.P. (1997) Nop2p is required for pre-rRNA processing and 60S ribosome subunit synthesis in yeast. Mol. Cell. Biol., 17, 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baudin-Baillieu A., Tollervey,D., Cullin,C. and Lacroute,F. (1997) Functional analysis of Rrp7p, an essential yeast protein involved in pre-rRNA processing and ribosome assembly. Mol. Cell. Biol., 17, 5023–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torchet C. and Hermann-Le Denmat,S. (2000) Bypassing the rRNA processing endonucleolytic cleavage at site A2 in Saccharomyces cerevisiae. RNA, 6, 1498–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma K. and Tollervey,D. (1999) Base pairing between U3 small nucleolar RNA and the 5′ end of 18S rRNA is required for pre-rRNA processing. Mol. Cell. Biol., 19, 6012–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang W.Q. and Fournier,M.J. (1995) U14 base-pairs with 18S rRNA: a novel snoRNA interaction required for rRNA processing. Genes Dev., 9, 2433–2443. [DOI] [PubMed] [Google Scholar]
- 37.Maser R.L. and Calvet,J.P. (1989) U3 small nuclear RNA can be psoralen-cross-linked in vivo to the 5′ external transcribed spacer of pre-ribosomal-RNA. Proc. Natl Acad. Sci. USA, 86, 6523–6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kass S., Tyc,K., Steitz,J.A. and Sollner-Webb,B. (1990) The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell, 60, 897–908. [DOI] [PubMed] [Google Scholar]
- 39.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 40.Sasaki T., Toh,E.A. and Kikuchi,Y. (2000) Yeast Krr1p physically and functionally interacts with a novel essential Kri1p and both proteins are required for 40S ribosome biogenesis in the nucleolus. Mol. Cell. Biol., 20, 7971–7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colley A., Beggs,J.D., Tollervey,D. and Lafontaine,D.L. (2000) Dhr1p, a putative DEAH-box RNA helicase, is associated with the box C+D snoRNP U3. Mol. Cell. Biol., 20, 7238–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunbar D.A., Wormsley,S., Agentis,T.M. and Baserga,S.J. (1997) Mpp10p, a U3 small nucleolar ribonucleoprotein component required for pre-18S rRNA processing in yeast. Mol. Cell. Biol., 17, 5803–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gavin A.C., Bosche,M., Krause,R., Grandi,P., Marzioch,M., Bauer,A., Schultz,J., Rick,J.M., Michon,A.M., Cruciat,C.M., Remor,M., Hofert,C., Schelder,M., Brajenovic,M., Ruffner,H., Merino,A., Klein,K., Hudak,M., Dickson,D., Rudi,T., Gnau,V., Bauch,A., Bastuck,S., Huhse,B., Leutwein,C., Heurtier,M.A., Copley,R.R., Edelmann,A., Querfurth,E., Rybin,V., Drewes,G., Raida,M., Bouwmeester,T., Bork,P., Seraphin,B., Kuster,B., Neubauer,G. and Superti-Furga,G. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature, 415, 141–147. [DOI] [PubMed] [Google Scholar]
- 44.Grandi P., Rybin,V., Bassler,J., Petfalski,E., Strauss,D., Marzioch,M., Schafer,T., Kuster,B., Tschochner,H., Tollervey,D., Gavin,A.C. and Hurt,E. (2002) 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol. Cell, 10, 105–115. [DOI] [PubMed] [Google Scholar]