

Abstract
We have previously demonstrated that phospholemman (PLM), a 15 kDa integral sarcolemmal phosphoprotein, inhibits the cardiac Na+/Ca2+ exchanger (NCX1). In addition, p rotein kinase A phosphorylates serine68 while protein kinase C phosphorylates both serine63 and serine68 of PLM. Using HEK293 cells that are devoid of both endogenous PLM and NCX1, we first demonstrated that the exogenous NCX1 current (INaCa) was increased by phorbol 12-myristate 13-acetate (PMA) but not by forskolin. When co-expressed with NCX1, PLM resulted in: (i) decreases in INaCa; (ii) attenuation of the increase in INaCa by PMA; and (iii) additional reduction in INaCa in cells treated with forskolin. Mutating serine63 to alanine (S63A) preserved PLM’s sensitivity to forskolin in terms of suppression of INaCa, whereas mutating serine68 to alanine (S68A) abolished PLM’s inhibitory effect on INaCa. Mutating serine68 to glutamic acid (phosphomimetic) resulted in additional suppression of I NaCa as compared to wild-type PLM. These results suggest that PLM phosphorylated at serine68 inhibited INaCa. The physiological significance of inhibition of NCX1 by phosphorylated PLM was evaluated in PLM-knockout (KO) mice. When compared to wild-type myocytes, INaCa was significant larger in PLM-KO myocytes. In addition, PMA-induced increase in INaCa was significantly higher in PLM-KO myocytes. By contrast, forskolin had no effect on INaCa in wild-type myocytes. We conclude that PLM, when phosphorylated at serine68, inhibits Na+/Ca2+ exchange in the heart.
Abbreviations: The abbreviations used are: ANOVA, analysis of variance; 8-Br-cAMP, 8-bromoadenosine 3′, 5′ cyclic monophosphate; [Ca2+]o, extracellular Ca2+ concentration; Cm, whole cell membrane capacitance; CMV, cytomegalovirus; DMEM, Dulbecco’s modified Eagle’s medium; DMSO, dimethylsulfoxide; EGTA, ethylene glycol-bis-(β-aminoethyl ether)N,N,N’,N’-tetraacetic acid; Em, membrane potential; em., emission; ex., excitation; ENaCa, equilibrium potential for Na+, Ca2+ exchange current; FBS, fetal bovine serum; GFP, green fluorescent protein; HEK, human embryonic kidney; HEPES, N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid; INaCa, Na+, Ca2+ exchange current; KO, knock-out; MEM, minimal essential media; NCX1, Na+, Ca2+ exchanger; NIMA, never in mitosis A; PKA, protein kinase A; PKC, protein kinase C; PLM, phospholemman; PMA, phorbol 12-myristate 13-acetate; PMSF, phenylmethylsulfonyl fluoride; PVDF, polyvinylidene difluoride; SE, standard error; SERCA2, sarco(endo)plasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum; SDS-PAGE, sodium dodecyl sulfate- polyacrylamide gel electrophoresis; Vmax, maximum velocity; WT, wild-type
Introduction
Phospholemman (PLM), a 72-amino acid membrane phosphoprotein with a single transmembrane domain (1), belongs to the FXYD gene family of small ion transport regulators (2). With the exception of γ-subunit of Na+-K+-ATPase (FXYD2), all other known members of the FXYD gene family have at least one serine or threonine within the cytoplasmic tail (2), indicating potentia l phosphorylation sites. In particular, PLM (FXYD1) is the only FXYD family member to have a consensus sequence for phosphorylation by PKA (RRXS), PKC (RXXSXR), and NIMA kinase (FRXS/T). Indeed, PLM has been shown to be phosphorylated by PKA at serine68 and PKC at both serine63 and serine68 (3).
To-date, PLM has been demonstrated to modulate ion fluxes through both the Na+-K+-ATPase (4–8) and the cardiac Na+/Ca2+ exchanger (NCX1)(9–11). Based on analogy of phospholamban inhibition of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA2)(12), and experimental observation on the effects of PLMS (a 15-kDa homologue of PLM isolated from shark rectal glands) on shark Na+-K+-ATPase (13,14), the current working hypothesis is that the Na+ pump is inhibited by unphosphorylated PLM. On phosphorylation of PLM, inhibition of Na+-K+-ATPase is relieved. This hypothesis has been given strong support by the observation that the Vmax of sarcolemmal Na+-K+-ATPase was increased 3-fold after acute cardiac ischemia, in association with increased PLM phosphorylation by >300% (5). In addition, Na+ pump current has been demonstrated to directly increase in association with PLM phosphorylation in response to forskolin (6). More recently, comparison of β-adrenergic effects on Na+ pump function between wild-type and PLM-knockout (KO) myocytes supports the notion that the inhibitory effects of PLM on Na+-K+-ATPase is relieved by phosphorylation (8). It is at present not clear whether dissociation of the phosphorylated PLM from Na+-K+-ATPase is required to relieve its inhibition on the Na+ pump (5,6,8,13,14). With respect to the cardiac Na+/Ca2+ exchanger, previous studies demonstrated that overexpression of PLM inhibited Na+/Ca2+ exchange activity (9,10) while downregulation of PLM enhanced NCX1 current (INaCa)(11). The importance of PLM phosphorylation in mediating its modulatory effects on NCX1 was not addressed in these early studies except that serine68 in PLM was found to be important (15).
Here, we demonstrated that PKC but not PKA activation enhanced INaCa when NCX1 was expressed alone in HEK293 cells. Co-expression of PLM with NCX1 resulted in decreased INaCa in the basal state, additional decrease in I NaCa when stimulated with forskolin, and attenuation of the magnitude of increase in INaCa by PKC activation. Mutating serine68 to glutamic acid (S68E) enhanced while substituting serine68 with alanine (S68A) abolished PLM’s inhibitory effect on INaCa. Mutating serine63 to alanine (S63A) preserved PLM’s sensitivity to forskolin in terms of additional inhibition of INaCa. Using a fundamentally different model system of murine cardiac myocytes, we first showed that endogenous INaCa was larger in PLM-KO myocytes when compared to wild-type (WT) myocytes, despite similar NCX1 protein levels. PKC but not PKA activation increased INaCa in WT myocytes. PLM-KO myocytes exhibited significantly larger increases in INaCa when stimulated with phorbol 12-myristate 13-acetate (PMA) as compared to WT myocytes. We conclude that PLM, when phosphorylated at serine68, inhibits cardiac Na+/Ca2+ exchanger.
Experimental Procedures
Construction Of PLM Mutants and NCX1 Clones
PLM serine mutants (S63A, S68A and S68E) were constructed with PLM in pAlter-1, using Altered Sites II in vitro Mutagenesis System (Promega; Madison, WI) as described previously (15). PLM and its serine mutants were authenticated by DNA sequencing, and subcloned into the mammalian expression vector pAdTrack-CMV (16). Rat cardiac NCX1 clone in pcDNA3.1(+) was a generous gift from Dr. J. Lytton and subcloned into pAdTrack-CMV as previously described (17). We chose the pAdTrack shuttle vector since it allowed us to identify successfully transfected HEK293 cells through a separate cytomegalovirus (CMV) promoter present on the vector backbone that drives the expression of green fluorescent protein (GFP).
Transfection of HEK293 cells
HEK293 cells (American Type Culture Collection, ATCC; Manassas, VA) were cultured and transfected with various NCX1 and PLM or its mutant clones as previously described (10). Briefly, cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 containing 10% heat-inactivated fetal bovine serum (FBS) at a density of 1.2 x 106 cells per 100 mm dish. After 24 h, medium was changed and cells were transfected with 25 μl Lipofectamine and total of 3μg plasmid DNA per dish: either pAdTrack-CMV alone (3μg), pAdTrack-CMV-NCX1 (1μg) + pAdTrack-CMV (2μg), pAdTrack-CMV-NCX1 (1μg) + pAdTrack-CMV-PLM (1μg) + pAdTrack-CMV (1μg), or pAdTrack-CMV-NCX1 (1μg) + pAdTrack-CMV-PLM serine mutant (1μg) + pAdTrack-CMV (1 μg). The lipid-DNA complex was left on cells for 5 h at 37ºC/5% CO2. Medium was then replaced with DMEM/Ham’s F12 + 10% FBS and cells were cultured for additional 48h before experiments. For patch-clamp applications, cells were trypsinized at 24 h post-transfection using Trypsin-EDTA, transferred to 35 mm dishes containing sterile glass coverslips and incubated a further 24 h prior to experiments. Transfection according to this protocol routinely yielded 30–50% transfection efficiency.
For brevity, HEK293 cells expressing NCX1 alone are referred in the text as NCX1 cells, while cells co-expressing NCX1 and PLM or its serine mutants are referred as PLM cells or SNNX cells (where NN is either 63 or 68, and X is either A or E).
Na+/Ca2+ Exchange Current (INaCa) Measurements
Whole cell patch-clamp recordings were performed at 30ºC as described previously (10,11,18,19). Briefly, fire-polished pipettes (tip-diameter 2–3 μm) were filled with a buffered Ca2+ solution containing (in mM): 100 Cs+ glutamate, 7.25 Na+ HEPES, 1 MgCl2, 12.75 HEPES, 2.5 Na2ATP, 10 EGTA, and 6 CaCl2, pH 7.2. Free Ca2+ in the pipette solution was 205 nM, measured fluorimetrically with fura 2. Cells were bathed in an external solution containing (in mM): 130 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 CaCl2, 10 HEPES, 10 Na+ HEPES, and 10 glucose, pH 7.4. Verapamil (1 μM), ouabain (1mM), and niflumic acid (30 μM) were used to block Ca2+, Na+-K+-ATPase, and Cl− currents, respectively. K+ currents were minimized by Cs+ substitution for K+ in both pipette and external solutions. Only cells that fluoresced green (ex. 380nm, em. 510 nm), indicating successful pAdTrack transfection, were selected for current measurements. Membrane potential (Em) was held at the calculated reversal potential of INaCa (−73 mV) for 5 min before stimulation. A descending voltage ramp (from +100 to −120 mV; 500 mV/s) was immediately followed by an ascending voltage ramp (from –120 to +100 mV; 500 mV/s)(Fig. 1A). Membrane currents were measured both before and after addition of 1 mM CdCl2 to the external solution (Fig. 1B). INaCa was defined as the difference current measured during the descending voltage ramp in the absence and presence of Cd2+ (Fig. 1C). To facilitate comparison of NCX1 currents, INaCa of each cell was divided by its whole cell capacitance (Cm) to account for variations in cell sizes. Except as otherwise stated, all results were obtained using these standard solutions.
Fig. 1. Measurement of Na+/Ca2+ exchange current (INaCa) in transfected HEK293 cells.

HEK293 cells were transfected with NCX1. At 48h post-transfection, INaCa was measured at 5 mM [Ca2+]o and 30oC with a descending-ascending voltage ramp protocol (A) described in Experimental Procedures. Free [Ca2+] in the Ca2+ buffered pipette solution was 205 nM. Holding potential was at the calculated reversal potential of INaCa (−73 mV) under our experimental conditions. Ca2+, Na+-K+-ATPase, Cl− and K+ currents were blocked by appropriate inhibitors. (B) Membrane currents recorded in a transfected cell during the descending-ascending voltage-ramp from +100 to −120 and back to +100 mV, in the absence and presence of 1 mM Cd2+. (C) Derived Cd2+-sensitive current in the transfected cell shown in B.
When indicated, PMA (0.1 μM) or forskolin (1 μM)(both dissolved in DMSO) was added to cells after baseline INaCa was obtained. Repeat INaCa was measured ~3–5 min. after drug addition.
In a 2nd series of experiments, the effects of PMA on INaCa were measured under Cl−-free conditions. Pipette solutions consisted of (in mM): 100 Cs+ glutamate, 7.25 Na+ HEPES, 1 MgSO4, 12.75 HEPES, 2.5 Na2ATP, 10 EGTA, and 6 Ca(OH)2, pH 7.2. External solutions contained (in mM): 130 Na+ aspartate, 5 Cs+ glutamate, 1.2 MgSO4, 1.2 NaH2PO4, 5 Ca(OH)2, 10 HEPES, 10 Na+ HEPES, and 10 glucose, pH 7.4. Verapamil, ouabain and niflumic acid were added to the bath as before. Holding potential was −73 mV. INaCa was defined as the difference current measured during the descending voltage ramp in the absence and presence of Cd2+ (1 mM) or Ni2+ (5 mM).
In a 3rd series of experiments, the effects of PMA on INaCa were measured under high [Na+]i conditions. Pipette solutions contained (in mM): 60 Cs+ glutamate, 40 Na+ glutamate, 7.25 Na+ HEPES, 1 MgCl2, 12.75 HEPES, 2.5 Na2ATP, 10 EGTA, and 6 CaCl2, pH 7.2. External solution consisted of 130 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 0.2 CaCl2, 10 HEPES, 10 Na+ HEPES, and 10 glucose, pH 7.4; and the usual inhibitors. [Ca2+]o was deliberately lowered to 0.2 mM so that the calculated reversal potential of INaCa (−103 mV), and thus the holding potential, was closer to the holding potential of −73 mV used in other experiments. Keeping [Ca2+]o at 5 mM would have resulted in a very negative holding potential of −188 mV. INaCa was defined as the difference current measured during the descending voltage ramp in the absence and presence of Ni2+ (5 mM).
Generation of PLM-KO Mice
A mouse line deficient in PLM was generated by replacing exons 3 to 5 of the PLM gene with lacZ and neomycin resistance genes, as described in detail previously (20). These mice grow to adulthood and are fertile. Studies were performed using mice backcrossed to a pure congenic C57BL/6 background. Homozygous adult littermates 3–6 months old were used in the experiments. Mice were housed in ventilated racks in a barrier facility supervised by the Department of Comparative Medicine at the Pennsylvania State University College of Medicine. Standard care was provided to all mice used for experiments.
PLM, NCX1 and calsequestrin immunoblotting
Mouse left ventricles were excised, rinsed in ice-cold PBS, and cut into small pieces. Approximately 60 mg of tissue were suspended in 700 μl of ice-cold lysis buffer containing (in mM): 50 Tris (pH 8.0), 150 NaCl, 1 Na+ orthovanadate, 1 PMSF, 100 NaF, 1 EGTA, and 0.5% NP40. A Complete Mini protease inhibitor cocktail tablet (Roche, Penzberg, Germany) was also added to 10 ml of lysis buffer. The tissue was homogenized with a glass dounce homogenizer (15–20 strokes), placed on ice for 15 min, before centrifugation at 20,800g for 10 min at 4°C. The supernatant was snap-frozen with dry ice-ethanol and stored at −80°C.
Protein in heart homogenates were subjected to 7.5% (NCX1 and calsequestrin) or 12% (PLM) SDS-PAGE under either non-reducing (10 mM N-ethylmaleimide for NCX1 and calsequestrin) or reducing (5% β-mercaptoethanol for PLM) conditions. The fractionated proteins were transferred to ImmunBlot PVDF membranes. Primary antibodies used were polyclonal antibody C2Ab (1:10,000) for PLM (21), polyclonal antibody π11–13 (1:500; Swant, Bellinzona, Switzerland) for NCX1, and rabbit anti-calsequestrin antibody (1:5,000; Swant). The secondary antibodies used were donkey anti-rabbit IgG (Amersham, Piscataway, NJ). Immunoreactive proteins were detected with an enhanced chemiluminescence Western blotting system. Protein band signal intensities were quantitated by scanning autoradiograms of the blots with a phosphorimager (Molecular Dynamics; Sunnyvale, CA). Because calsequestrin expression has been shown to be unchanged during ontogenic development, aging, cardiac hypertrophy, and failing human myocardium (22), we used calsequestrin as an internal control for protein loading.
Isolation of Murine Myocytes and Measurement of INaCa
Cardiac myocytes were isolated from the septum and left ventricular free wall of WT and PLM-KO mice (25–37g) according to the protocol of Zhou et al. (23). Briefly, mice were heparinized (1500 u/kg ip) and anesthetized (pentobarbital sodium, 50 mg/kg ip). The heart was excised, mounted on a steel cannula and retrograde perfused (100 cm H2O, 37ºC) with Ca2+-free bicarbonate buffer followed by enzymatic digestion (collagenases B and D, protease XIV) as described (23). Isolated myocytes were plated on laminin -coated glass coverslips in a petri dish, and the Ca2+ concentration of the buffer was progressively increased from 0.05 to 0.125 to 0.25 to 0.5 mM in 3 steps (10 min interval each). The 0.5 mM Ca2+ buffer was then aspirated and replaced with minimal essential medium (MEM, Sigma M1018) containing 1.2 mM Ca2+, 2.5% FBS and antibiotics (1% penicillin/streptomycin). After 1 h (5% CO2, 37º), media was replaced with FBS-free MEM. Myocytes were used within 2–8 h of isolation. The protocol for heart excision for myocyte isolation was approved by the Institutional Animal Care and Usage Committee.
INaCa was measured in isolated murine myocytes with the same protocol and standard solutions used for transfected HEK293 cells except that pipette tip diameter was increased to 4–6 μm and niflumic acid was decreased to 10 μM.
Statistical Analysis
All results are expressed as means ± _SE. For analysis of a parameter (e.g., INaCa) as a function of group (e.g., NCX1 vs. PLM) and voltage, two-way ANOVA was used to determine statistical significance. For analysis of Cm, Student’s t-test was used. A commercial software package (JMP version 4.0.5, SAS Institute; Cary, NC) was used. In all analyses, p<0.05 was taken to be statistically significant.
Results
Effects of PMA or Forskolin on INaCa in HEK293 Cells Expressing NCX1 alone
We have previously shown that HEK293 cells did not express NCX1 or demonstrate measurable INaCa or Na+-dependent Ca2+ uptake (10). When transfected with rat cardiac NCX1, HEK293 cells exhibited characteristic INaCa demonstrating both forward (inward current, 3 Na+ in: 1 Ca2+ out) and reverse (outward current, 3 Na+ out: 1 Ca2+ in) Na+/Ca2+ exchange (Fig. 2A). In addition, the reversal potential of INaCa was between −70 to −60 mV, close to its theoretical equilibrium potential of −73 mV under our experimental conditions (Fig. 2A). There were no significant (p<0.76) differences in baseline INaCa measured with either Cd2+ or Ni2+ (data not shown).Treatment with PMA which activates PKC resulted in a large increase in INaCa in NCX1 cells (Fig. 2A; p<0.0001). For example, at +100 mV, PKC stimulation resulted in ~120% increase in INaCa. Control experiments performed in Cl−-free solutions demonstrated that the PMA-induced current increase was not due to increase in Cl− currents (Fig. 2B). In addition, PMA induced large increases in currents whether Cd2+ (~122% at +100 mV)(Fig. 2B) or Ni2+ (~81% at +100 mV) (data not shown) was used to define INaCa under Cl− -free conditions. To control for the possibility that the observed PMA-induced INaCa increase was due to small changes in [Na+]i rather than enhancing intrinsic NCX1 activity, experiments were performed in high [Na+]i conditions such that INaCa would not be so sensitive to small changes in [Na+]i. Fig. 2C shows that baseline INaCa was significantly (p<0.0001) smaller in high [Na+]i and low [Ca2+]o (0.2 mM) when compared to normal [Na+]i and high [Ca2+]o (5 mM) conditions (Fig, 2A), likely due to the 25-fold reduction of [Ca2+]o. However, addition of PMA increased INaCa (~84% at +100mV) under high [Na+]i conditions, similar to the observations obtained under lower but more physiological [Na+]i conditions.
Fig. 2. Effects of PMA on INaCa in transfected HEK293 cells.
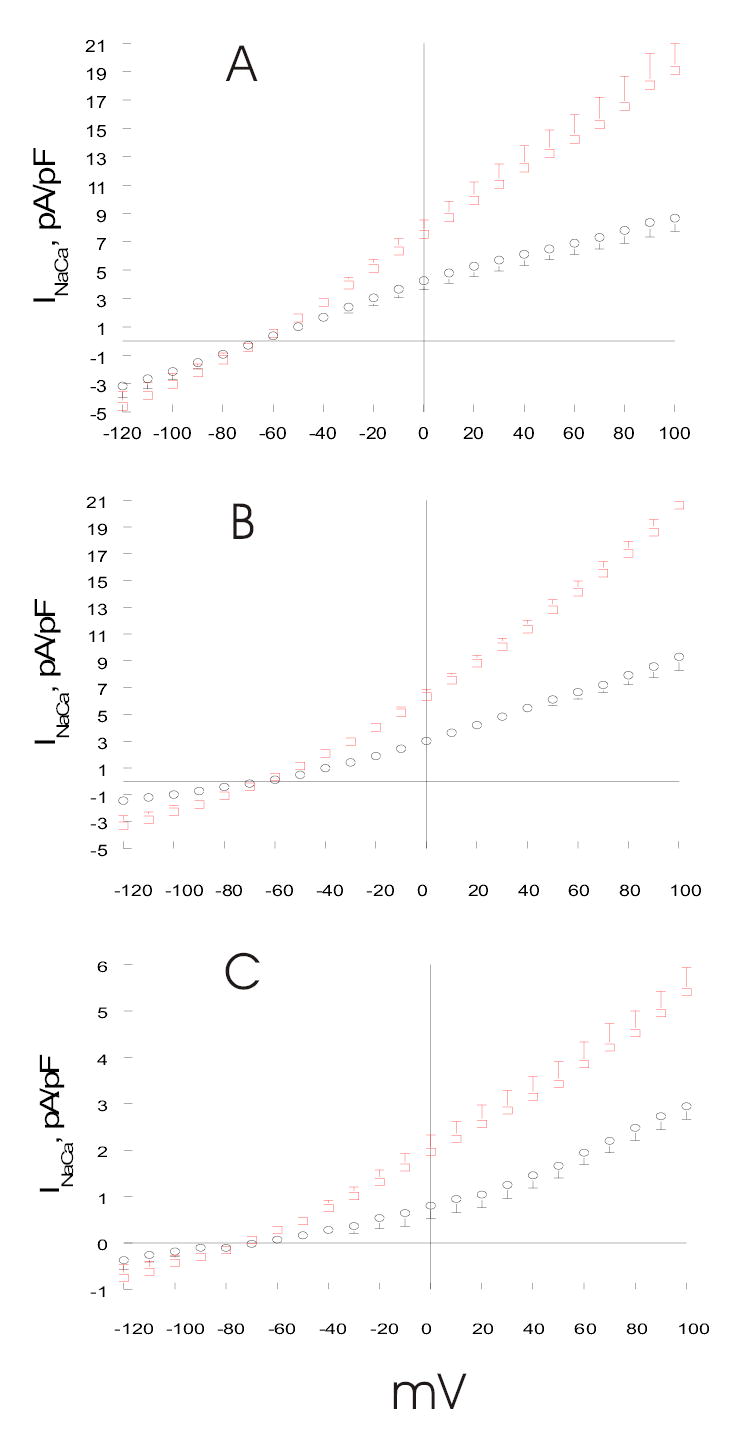
(A). HEK293 cells were transfected with NCX1 (open circles, n=14). At 48h post-transfection, INaCa was measured at 30°C using standard solutions and Cd2+ as described in Experimental Procedures and Fig. 1. After baseline INaCa was obtained, PMA (0.1 μM) was added and INaCa was measured 3 to 5 min after drug addition (open squares; n=8). (B). INaCa was measured in HEK293 cells transfected with NCX1 under Cl−-free conditions (Experimental Procedures), both before (open circles, n=4) and after (open squares, n=4) addition of PMA. Cd2+ was used to define INaCa. (C). INaCa was measured in HEK293 cells transfected with NCX1 under high [Na+]i conditions (Experimental Procedures), both before (open circles, n=6) and after (open squares, n=7) addition of PMA. [Ca2+]o was 0.2 rather than 5.0 mM so that the calculated reversal potential for INaCa was −103 mV, as compared to the holding potential of −73 mV used in other experiments. Ni2+ was used to define INaCa. Error bars are not shown if they fall within boundaries of the symbols.
In contrast to results obtained with PMA stimulation, forskolin treatment did not affect INaCa in NCX1 expressing cells (Fig. 3B; p<0.64).
Fig. 3. Effects of PMA and forskolin on INaCa in transfected HEK293 cells.
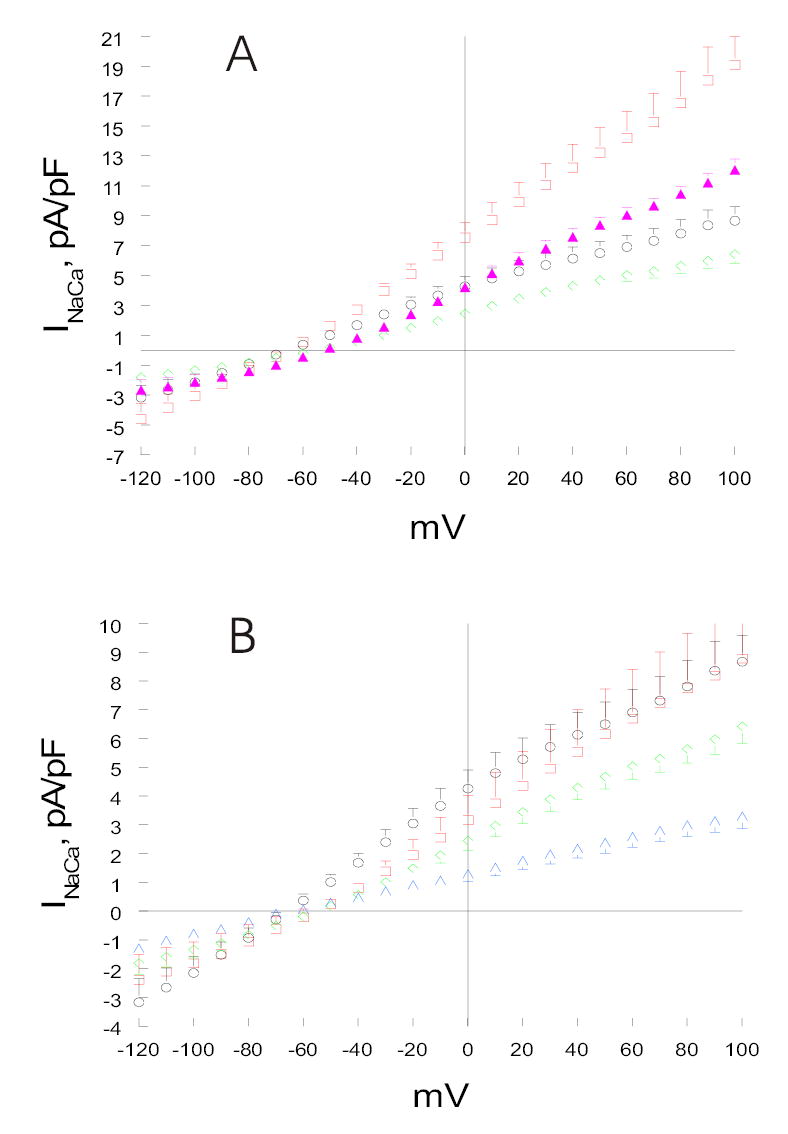
(A). HEK293 cells were transfected with either NCX1 alone (open circles, n=14) or PLM+NCX1 (open diamonds, n=15). At 48h post-transfection, INaCa was measured at 5 mM [Ca2+]o and 30°C as described in Fig. 1. After baseline INaCa was obtained, PMA (0.1 μM) was added to NCX1 (open squares; n=8) and PLM+NCX1 (filled triangles; n=9) cells. Measurement of INaCa was repeated ~3 to 5 min after drug addition. (B). HEK293 cells were transfected with either NCX1 alone (open circles, n=14) or PLM+NCX1 (open diamonds, n=15). At 48h post-transfection, baseline INaCa was obtained. Forskolin (1 μM) was then added to NCX1 (open squares; n=4) and PLM+NCX1 (open triangles; n=7) cells. Measurement of INaCa was repeated ~3 to 5 min after drug addition. Error bars are not shown if they fall within boundaries of the symbols.
Effects of PMA or Forskolin on INaCa in Cells Expressing both NCX1 and PLM
Co-expression of PLM with NCX1 in HEK293 cells resulted in significant decrease in INaCa compared to cells expressing NCX1 alone (Figs. 3A & 3B; p<0.0005), consistent with our previous observations (10). At +100 mV, PLM inhibited INaCa by ~26%. PMA treatment of PLM cells resulted in significant increase in INaCa when compared to unstimulated NCX1 or PLM cells (Fig. 3A; p<0.0001). However, the magnitude of INaCa increase by PMA was much smaller in PLM cells when compared to NCX1 cells (39 vs. 120% at +100mV).
Despite absence of forskolin’s effect on INaCa in cells expressing NCX1 alone, PKA stimulation in PLM cells resulted in significant decrease in INaCa compared to unstimulated PLM cells (Fig. 3B; p<0.0001). For example, at +100 mV, forskolin effected ~49% decrease in INaCa in PLM cells (Fig. 3B).
Effects of PLM Serine68 Mutants on INaCa in Transfected HEK293 Cells
Because serine68 in PLM is the common phosphorylation target for both PKA and PKC, we next investigated the effects of serine68 mutants on INaCa in cells co-expressing NCX1 and PLM serine68 mutants. Mutating serine68 to alanine (S68A) resulted in abolition of WT PLM’s effect on INaCa (Fig. 4A; p<0.08), consistent with our previous observations (10). Treating S68A cells with PMA, instead of increasing INaCa as observed in PLM cells (Fig. 3A), resulted in a modest but significant suppression of in INaCa when compared to unstimulated NCX1 cells (Fig. 4A; p<0.0004).
Fig. 4. Effects of serine68 mutants of PLM on INaCa in transfected HEK293 cells.

(A). HEK293 cells were transfected with either NCX1 alone (open circles, n=14) or S68A+NCX1 (open squares, n=7). At 48 h post-transfection, INaCa was measured at 5 mM [Ca2+]o and 30°C as described in Fig. 1. After baseline INaCa was obtained, PMA (0.1 μM) was added to S68A+NCX1 cells (open triangles; n=7) and INaCa was again measured. (B). HEK293 cells were transfected with NCX1 alone (open circles, n=10), PLM+NCX1 (open diamonds, n=8), or S68E+NCX1 (open squares, n=6). INaCa was measured 48h post-transfection. In S68E+NCX1 cells, INaCa was measured both before (open squares) and after (open triangles) addition of PMA (0.1 μM). Error bars are not shown if they fall within boundaries of the symbols.
Mutating serine68 to glutamic acid (S68E) resulted in greater suppression of INaCa when compared to WT PLM (Fig. 4B; p<0.0001). Stimulating S68E cells with PMA, in contrast to increases in INaCa in PLM cells (Fig. 3A), did not result in appreciable changes in INaCa when compared to unstimulated S68E cells (Fig. 4B; p<0.70). The lack of INaCa stimulation by PMA in both PLM serine68 mutants (Figs. 4A & 4B) as compared to WT PLM (Fig. 3A) suggests altered PLM interaction with NCX1 by serine68 mutants may somehow interfere with PMA’s stimulatory effects on NCX1.
Effects of PLM Serine63 Mutant on INaCa in Transfected HEK293 Cells
Unlike PKA which phosphorylates serine68 only, PKC phosphorylates both serine63 and serine68 in PLM (3). Co-expressing PLM serine63 to alanine mutant (S63A) with NCX1 resulted in inhibition of INaCa compared to cells expressing NCX1 alone (Fig. 5A; p<0.03). The magnitude of inhibition by S63A was quite modest (~8% at +100mV) when compared to that by WT PLM (~26% at +100mV; Fig. 3A). Treating S63A cells with forskolin resulted in additional inhibition of INaCa (~37% at +100mV) when compared to unstimulated S63A cells or NCX1 cells (Fig. 5A; p<0.0001).
Fig. 5. Effects of serine63 mutants of PLM on INaCa in transfected HEK293 cells.
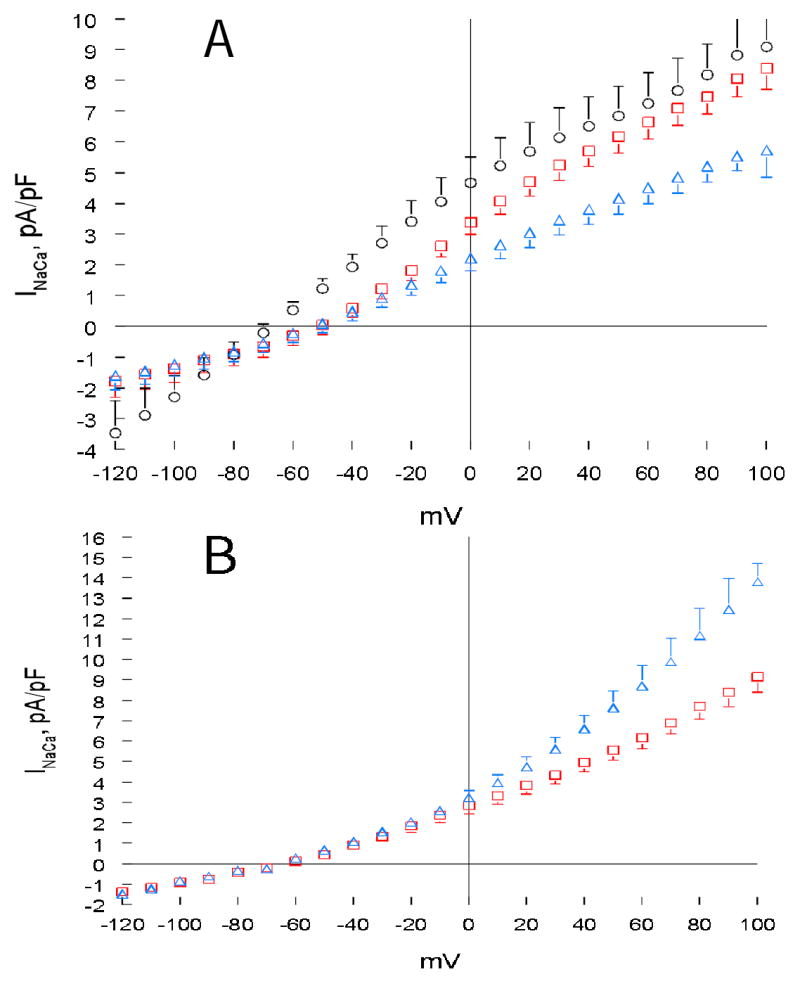
(A). HEK293 cells were transfected with either NCX1 alone (open circles, n=10) or S63A+NCX1 (open squares, n=5). At 48 h post-transfection, INaCa was measured at 5 mM [Ca2+]o and 30°C as described in Fig. 1. After baseline INaCa was obtained, forskolin (1 μM) was added to S63A+NCX1 cells (open triangles; n=5) and INaCa was again measured. (B). HEK293 cells were transfected with S63A+NCX1 (open squares, n=6). INaCa was measured 48h post-transfection, both before (open squares) and after (open triangles) addition of PMA (0.1 μM). Error bars are not shown if they fall within boundaries of the symbols.
In another series of experiments, the effects of PMA on INaCa in S63A cells were evaluated. Unlike cells co-expressing NCX1 and PLM serine68 mutants in which INaCa was not stimulated at all by PMA (Figs. 4A & 4B), S63A cells demonstrated significant PMA-induced enhancement of INaCa (Fig. 5B; p<0.0001). The effects of PMA on INaCa in cells co-expressing NCX1 and S68A, S68E or S63A, when considered together, are consistent with the notion that retaining normal serine68 in PLM is absolutely essential for PMA’s stimulatory effect on INaCa in cells co-expressing PLM and NCX1.
Effects of PMA on INaCa in ventricular myocytes isolated from wild-type and PLM-KO mice
Results from transfected HEK293 cells strongly suggest that PLM, when phosphorylated at serine68, inhibits cardiac Na+/Ca2+ exchanger. To put the findings in physiological perspective, we examined the effects of PMA on INaCa in myocytes isolated from WT and PLM-KO mice. Western blots confirmed the absence of PLM in PLM-KO myocytes (Fig. 6). Importantly, NCX1 protein levels (normalized to calsequestrin) were not significantly different (p<0.52) between wild-type (99.8 ± 11.7 arbitrary units; n=8) and PLM-KO myocytes (90.7 ± 7.3 arbitrary units; n=8)(Fig. 6). Wild-type and PLM-KO myocytes had similar cell sizes, as evidenced by no differences (p <0.97) in whole cell capacitance (a measure of membrane surface area) between WT (184 ± 7 pF, n=13) and PLM-KO myocytes (184 ± 8 pF, n=13). Unlike HEK293 cells expressing exogenous NCX1 (Fig. 1B), in cardiac myocytes a contaminant inward Na+ current was evident during the ascending voltage ramp (Fig. 7B). For this reason, the Cd2+-sensitive current (INaCa) was quantitated during the descending portion of the voltage ramp (Fig. 7C). INaCa was significantly larger in PLM-KO myocytes when compared to WT myocytes (Fig. 8; p<0.0001). In another series of experiments, PKC stimulation resulted in increases in INaCa in both WT (p<0.0001) and PLM-KO (p<0.0001) myocytes when compared to their respective unstimulated controls (Fig. 9A). However, PMA-induced increase in INaCa was significantly (p<0.002) higher in PLM-KO (~132% increase at +100mV) than wild-type myocytes (~91% at +100mV).
Fig. 6. Immunoblots of Na+/Ca2+ exchanger (NCX1), calsequestrin and phospholemman (PLM) from murine hearts.
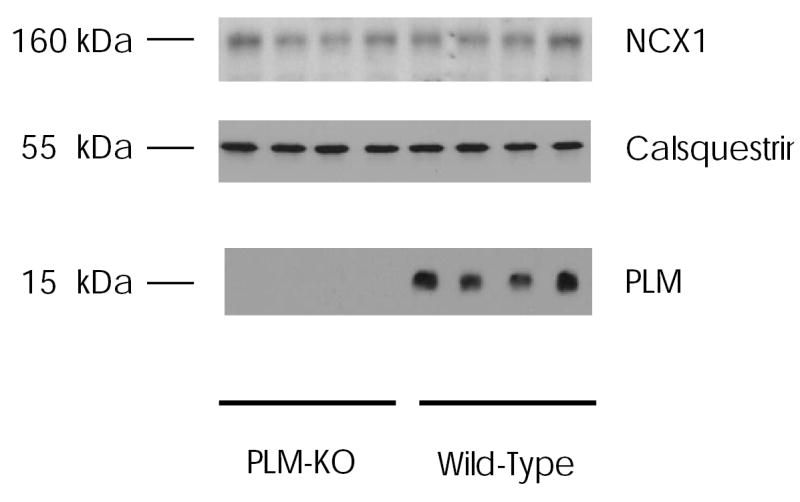
Left ventricular homogenates were prepared from wild-type and PLM-KO mice of congenic C57BL/6 background, as described in Experimental Procedures. Proteins were separated by gel electrophoresis under non-reducing conditions for NCX1 (50 μg/lane) and calsequestrin (100 μg/lane), and reducing conditions for PLM (5 μg/lane). After transfer to PVDF membranes, immunoblotting were performed as described in Experimental Procedures. Numbers on the left refer to apparent molecular mass.
Fig. 7. Measurement of Na+/Ca2+ exchange current (INaCa) in murine cardiac myocytes.

INaCa was measured in ventricular myocytes isolated from adult mouse hearts at 5 mM [Ca2+]o and 30°C with a descending-ascending voltage ramp protocol (A) as described in Experimental Procedures. Free Ca2+ in the Ca2+-buffered pipette solution was 205 nM. Holding potential was at the calculated reversal potential of INaCa (−73 mV) under our experimental conditions. Ca2+, Na+-K+-ATPase, Cl− and K+ currents were blocked by appropriate inhibitors. (B) Membrane currents recorded in a wild -type myocyte during the descending-ascending voltage-ramp from +100 to −120 and back to +100 mV, in the absence and presence of 1 mM Cd2+. (C) Derived Cd2+-sensitive current in the wild-type myocyte shown in B.
Fig. 8. INaCa is larger in PLM-KO when compared to wild-type cardiac myocytes.

INaCa was measured in ventricular myocytes isolated from wild-type and PLM-KO mouse hearts at 5 mM [Ca2+]o and 30°C as described in Fig. 7. Shown are current density-voltage relationships of INaCa (means ± SE) from wild-type (open circles; n=20) and PLM-KO (filled circles; n=23) myocytes. Error bars are not shown if they fall within the boundaries of symbols.
Fig. 9. Effects of PMA and forskolin on INaCa in wild-type and PLM-KO cardiac myocytes.
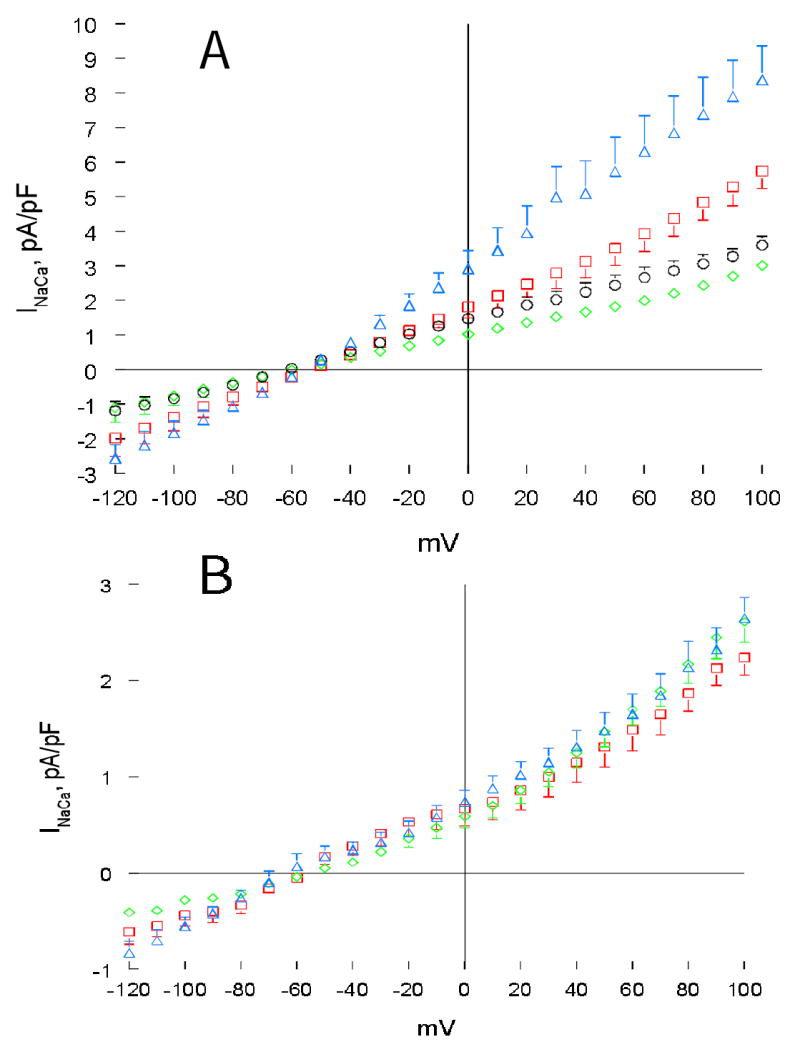
(A). INaCa was measured in a second group of ventricular myocytes isolated from wild-type (diamonds, n=7) and PLM-KO (circles, n=7) mouse hearts at 5 mM [Ca2+]o and 30°C as described in Fig. 6. After baseline INaCa was obtained, PMA (1 μM) was added to both wild-type (squares, n=7) and PLM-KO (triangles, n=7) and INaCa measurement was repeated. (B). INaCa was measured in a third group of wild-type myocytes, both before (diamonds, n=6) and after (squares, n=6) addition of forskolin (1 μM). Similarly, INaCa was measured in PLM-KO myocytes. For clarity of presentation, only data from PLM-KO myocytes treated with forskolin (triangles, n=6) are shown. Error bars are not shown if they fall within boundaries of the symbols.
Effects of Forskolin on INaCa in wild-type and PLM-KO ventricular myocytes
In a third series of experiments, we measured the effects of forskolin on INaCa in murine cardiac myocytes. Baseline INaCa was again significantly (p<0.0001) higher in PLM-KO than WT myocytes (for clarity, KO data not shown in Fig. 9B). PKA stimulation did not result in appreciable changes in INaCa in WT myocytes (Fig. 9B; p<0.11). In addition, there were no differences in INaCa between WT and PLM-KO myocytes after forskolin treatment (Fig. 9B, p<0.15).
Discussion
We have previously demonstrated in both rat cardiac myocytes (9,11,15) and transfected HEK293 cells (10) that PLM, in addition to its well-known modulatory effects on the Na+ pump (4,6,8,24), inhibited cardiac Na+/Ca2+ exchanger. Specifically, PLM co-localized and co-immunoprecipitated with NCX1, and functionally decreased INaCa and Na+-dependent Ca2+ uptake (9,10). Whereas PLM phosphorylation during ischemia (5) or by β-adrenergic stimulation (8) was associated with relief of its inhibition of Na+-K+-ATPase, it is not clear whether inhibition of Na+/Ca2+ exchanger is mediated by phosphorylated or unphosphorylated PLM.
Incorporation of 32P into PLM in intact guinea pig ventricles was enhanced ~2.6 fold with isoproterenol treatment, suggesting WT PLM was partially phosphorylated in the unstimulated state (25). Based on C68P Ab and C2 Ab which are antibodies specific for phosphorylated (at serine68) and unphosphorylated PLM, respectively (24,26), it has been estimated that ~41% of PLM in adult rat myocytes (24) and ~25% of PLM in guinea pig myocytes (6) were phosphorylated at serine68 under the basal state. Using another approach of comparing the effects of WT PLM and its serine68 and serine63 mutants on INaCa in adult rat myocytes, ~46% of serine68 and ~16% of serine63 were estimated to be phosphorylated in the resting state (15). The results from these 3 fundamentally different experimental approaches strongly indicate that PLM was only partially phosphorylated in cardiac myocytes. Overexpression of PLM did not grossly distort the relative level of phosphorylation on serine68 of PLM in adult rat cardiac myocytes (24). Therefore it is difficult to ascertain which form of PLM (phosphorylated or unphosphorylated) mediated the inhibition of Na+/Ca2+ exchange in studies employing PLM overexpression strategies (9,15).
Because PLM is known to regulate Na+-K+-ATPase (4–6,8,24), it is tempting to explain the effects of PLM on NCX1 as indirect, i.e., changes in [Na+]i due to alterations in Na+ pump activity by PLM would change the driving force of NCX1 and hence INaCa magnitude. The conditions used in our INaCa measurements were carefully designed to avoid this ambiguity in that Na+ pump activity was eliminated by exclusion of K+ in pipette and bathing solutions as well as by the inclusion of ouabain. In addition, the measured and theoretical equilibrium potentials for INaCa (ENaCa) were in reasonable agreement, suggesting that under the heavily buffered [Ca2+]i conditions used in our INaCa measurements, the [Na+]i sensed by NCX1 could be approximated by [Na+]pip. Finally, the measured ENaCa between NCX1 and PLM cells were in close agreement, indicating that the [Na+]i sensed by NCX1 were similar in both types of cells. Therefore, the thermodynamic parameters ([Ca2+]i, [Ca2+]o, [Na+]i, [Na+]o ) that determine ENaCa, and hence the driving force for INaCa (Em- ENaCa), were identical between NCX1 and PLM cells. In addition, we have previously demonstrated that the protein levels of NCX1 in HEK293 cells were similar in the absence or presence of co-transfected PLM (10). The observed differences in INaCa between NCX1 and PLM cells can thus be unambiguously assigned to the direct inhibitory effects of PLM on NCX1. Similar arguments can be advanced that the observed differences in INaCa between wild-type and PLM-KO myocytes (with similar NCX1 protein levels) were due to direct inhibition of NCX1 by PLM.
NCX1 is known to be modulated by α-adrenergic stimulation (27), presumably mediated via PKC (28). Our finding that in HEK293 cells expressing NCX1 alone, PKC activation by PMA resulted in large increase in Na+/Ca2+ exchange activity is similar to that observed in CCL39 fibroblasts expressing NCX1 (28). In our experiments on HEK293 cells expressing NCX1, the increase in current by PMA was not due to activation of Cl− current since similar current increases were observed under Cl−-free conditions. Another potential concern is that although Ca2+ was heavily buffered under our experimental conditions, small changes in [Na+]i by PMA may have large effects in INaCa (proportional to 3rd power of [Na+]i) with only small effects on ENaCa (proportional to 3rd root of the Na+ gradient). Under conditions of high [Na+]i in which I NaCa would not be expected to be so sensitive to small changes in cytoplasmic Na+, PKC stimulation still effected a large increase in INaCa. Our control experiments with Cl−-free solutions and high [Na+]i conditions indicate that the observed increase in currents by PMA was due to PKC’s enhancement of intrinsic NCX1 activity, rather than an artifactual increase in Cl− currents or changes in driving force for the exchanger.
PKC activation was associated with increased NCX1 phosphorylation at serine249, serine250 and serine357 (29). In normal cardiac myocytes, however, NCX1 is associated with PLM (10,11). Therefore the physiologically more relevant model system is one which co-expresses both NCX1 and PLM. In HEK293 cells co-expressing both NCX1 and PLM, PMA treatment also resulted in enhancement of Na+/Ca2+ exchange activity, similar to that observed in rat sarcolemmal vesicles (27). The magnitude of I NaCa increase, however, was much smaller in cells co-expressing NCX1 and PLM when compared to cells expressing NCX1 alone. These results suggest that the stimulatory effects of PMA on NCX1 were attenuated by increased PLM phosphorylation. The implication on Na+/Ca2+ exchange in intact myocytes exposed to PKC activators is that the direct stimulatory effects on NCX1 are somewhat opposed by an indirect inhibitory effect by increased phosphorylated PLM.
Since PKC induces phosphorylation at both serine63 and serine68 of PLM (3), we next activated PKA to evaluate the effects of PLM phosphorylated only at serine68 on NCX1. The effects of PKA on the cardiac Na+/Ca2+ exchanger are quite controversial. For example, PKA activation did not enhance phosphorylation of NCX1 expressed in CCL39 fibroblasts (29) but the catalytic subunit of PKA was quite capable of in vitro phosphorylation of NCX1 immunoprecipitated from xenopus oocytes expressing the Na+/Ca2+ exchanger (30). It is at present equally contentious as to whether the mammalian cardiac Na+/Ca2+ exchange activity is affected by PKA activation. For example, no enhancement of INaCa by 8-bromoadenosine 3′, 5′ cyclic monophosphate (8-Br-cAMP) was observed in HEK cells expressing dog NCX1 (31). Likewise, 8-Br-cAMP had no effect on Na+-dependent Ca2+ uptake in CCL39 fibroblasts expressing dog heart NCX1 (29). In giant membrane patches excised from blebs of guinea pig ventricular cells, no stimulatory effect of β-adrenergic stimulation or PKA on Na+/Ca2+ exchange activity was observed (32). In isolated rat sarcolemmal vesicles, isoproterenol had no effect on Na+/Ca2+ exchange activity (27). In intact cardiac myocytes, isoproterenol was reported to increase INaCa in guinea pig (33) and pig (34) but not in rabbit myocytes (35). Recently, an elegant study shed light on the confusing literature concerning the effects of PKA activation on mammalian cardiac Na+/Ca2+ exchange activity (36). The apparent augmentation of I NaCa by isoproterenol in guinea pig myocytes was due to the activation of a cAMP-dependent and Ni2+-sensitive Cl− current (36). In rat and mouse ventricular cells in which cAMP did not activate this cAMP-dependent Cl− current (37), isoproterenol treatment did not increase the amplitude of INaCa (36). Therefore to-date, the weight of current evidence suggests that β-adrenergic stimulation with subsequent PKA activation had no discernible effects on mammalian cardiac Na+/Ca2+ exchange activity. Our observations that forskolin had no stimulatory effects on INaCa in transfected HEK293 cells expressing NCX1 alone and in wild-type mouse myocytes are thus consistent with this view. However, in HEK293 cells expressing both NCX1 and PLM, forskolin resulted in additional suppression of I NaCa. This observation suggests that PLM, when phosphorylated at serine68, inhibited cardiac Na+/Ca2+ exchange in a heterologous expression system. The importance of phosphorylated serine68 in mediating PLM’s inhibition of INaCa is supported by the experimental results with serine68 mutants. S68A which cannot be phosphorylated resulted in loss of function while S68E which mimicked 100% phosphorylation resulted in additional suppression of INaCa when compared to WT PLM; both in transfected HEK293 cells (current study) and in adult rat cardiac myocytes overexpressing PLM or its serine68 mutants (15).
The results of S63A mutant on INaCa are interesting in 3 respects. First, leaving serine68 intact but prohibiting phosphorylation at serine63 resulted in much more modest inhibition of INaCa when compared to wild-type PLM. This suggests that phosphorylation at serine63 may also contribute to PLM’s inhibitory effect on INaCa. However, the lack of effects on I NaCa by S68A mutant (with or without PMA stimulation) indicates that serine68 phosphorylation is of primary importance in PLM’s inhibition of NCX1. Second, treating S63A cells with forskolin resulted in a more substantial suppression of INaCa, again indicating the primacy of serine68 phosphorylation in mediating PLM’s inhibitory effect on INaCa. Third and perhaps the most intriguing is that while PMA resulted in large INaCa increases in cells expressing NCX1 alone or NCX1+PLM, cells which expressed NCX1 and S68A or S68E mutants showed no increases in INaCa when stimulated with PMA. Cells which expressed NCX1 and S63A mutant (in which serine68 is intact), on the other hand, were able to increase INaCa with PKC activation - similar to cells expressing both NCX1 and wild-type PLM. Our results on the serine63 and serine68 mutants suggest that changes in conformation in PLM by mutating serine68 may alter its interaction with NCX1, resulting in NCX1 not accessible to PKC action perhaps due to steric hindrance.
The relative lack of effects by S68A and S63A mutants on INaCa in transfected HEK293 cells may be due to loss of interaction between these PLM mutants and NCX1. This is unlikely, however, as we have previously demonstrated that both S68A and S63A mutants, similar to WT PLM, were able to co-immunoprecipitate NCX1 in HEK293 cells co-expressing NCX1 and PLM or its serine mutants (15).
The physiological relevance of serine68 phosphorylation in PLM on NCX1 function was examined in WT and PLM-KO myocytes. There are many similarities between the results obtained in transfected HEK293 cells and murine myocytes. For example, similar to the observation that INaCa was higher in HEK293 cells expressing NCX1 alone as compared to cells co-expressing NCX1 and PLM, baseline I NaCa was higher in PLM-KO than WT myocytes. PMA treatment resulted in enhancement of I NaCa in both WT and PLM-KO myocytes, although the increase in INaCa was much higher in PLM-KO myocytes. This is also similar to our findings in the heterologous expression system. On the other hand, there are some differences between the effects of PKA on INaCa in HEK293 cells and murine myocytes. For example, forskolin treatment resulted in suppression of INaCa in HEK293 cells co-expressing NCX1 and PLM. By contrast, PKA stimulation in WT myocytes did not result in any detectable changes in I NaCa, in agreement with observations by Ginsburg and Bers (35) and Lin et al. (36). The differences between the results obtained in HEK293 cells and murine myocytes with respect to PKA effects on INaCa are not intuitively obvious but may relate to association of NCX1 with the catalytic subunit of PKA and protein phosphatase 1 (PP1) in rat hearts (30). It is known that NCX1 exhibited significant basal phosphorylation in cardiac myocytes (28). In addition, dephosphorylation of NCX1 by PP1 resulted in reduction of INaCa (34) whereas increased NCX1 phosphorylation was associated with enhancement of Na+/Ca2+ exchange activity (28). PKA stimulation of intact cardiac myocytes would be expected to simultaneously increase phosphorylation in both NCX1 (stimulatory)(30) and PLM (inhibitory), plus or minus other unknown effects on PP1 such that the net effect would be no measurable changes in I NaCa. In NCX1 expressed heterologously in HEK293 cells, there may not be such close association of PKA with NCX1 in an assembled “macromolecular complex” (38) so that PKA can exert its effects on NCX1. On the other hand, in our simplified heterologous expression system, phosphorylation of PLM by ubiquitous PKA present in these cells (BA Ahlers, unpublished observations) or the phosphomimetic S68E mutant would be expected to suppress INaCa.
In the intact heart, β-adrenergic stimulation increases Na+ influx into the myocytes because of the chronotropic effect (more frequent depolarizations). In addition, L-type Ca2+ current and SERCA2 activity are also increased in response to β-adrenergic stimulation, resulting in increased Ca2+ entry and Ca2+ loading of the sarcoplasmic reticulum. Increased SR Ca2+ available for release largely accounts for the increased inotropy of β-adrenergic agonists. To maintain steady-state Ca2+ balance, the increased myocyte Ca2+ entry must necessitate increased Ca2+ efflux mediated by forward Na+/Ca2+ exchange, thereby bringing more Na+ into the cell. Therefore, enhanced Na+-K+-ATPase activity (by PLM phosphorylation) during β-adrenergic stimulation is necessary to prevent cellular Na+ overload. On the other hand, unchecked stimulation of Na+-K+-ATPase would decrease intracellular Na+ concentration, thereby increasing the thermodynamic driving force of forward Na+/Ca2+ exchange, resulting in Ca2+ depletion. The ensuing decreased inotropy is clearly not desirable under the circumstances of fight or flight. Our presented evidence suggests a coordinated paradigm in which PLM, on phosphorylation at serine68, enhances Na+-K+-ATPase (5,8) but inhibits Na+/Ca2+ exchange activities in cardiac myocytes. The consequences of Na+-K+-ATPase stimulation on the one hand, and Na+/Ca2+ exchange inhibition on the other, on cellular Ca2+ homeostasis and contractility are complex and difficult to predict or model and clearly requires further study.
Finally, it should be pointed out that the magnitude of inhibition of I NaCa by WT PLM in HEK293 cells was ~26% at +100 mV in our current experiments, much more modest than our previous results of ~80% inhibition at +100 mV (10). This is because we deliberately decreased the amount of plasmid DNA encoding PLM used in the transfection (from 1.5 μg to 1.0 μg per dish) so that we would better be able to detect additional inhibition of INaCa when PLM was phosphorylated or when a phosphomimetic PLM mutant was used.
In summary, we have demonstrated that phospholemman phosphorylated at serine68 inhibited Na+/Ca2+ exchange in both transfected HEK293 cells and mouse myocytes. We conclude that in intact cardiac myocytes, phosphorylation of phospholemman results in relief of inhibition of Na+-K+-ATPase and inhibition of Na+/Ca2+ exchange.
Acknowledgments
This study was supported in part by the National Institutes of Health Grants HL-58672 & HL-74854 (J.Y. Cheung), DK-46678 (J.Y. Cheung, co-investigator), GM-69841 (L.I. Rothblum), HL-70548 and GM-64640 (J.R. Moorman), and HL-69074 (A.L. Tucker); American Heart Association Pennsylvania Affiliate Grants-in-Aid 0265426U (X. Zhang) and 0355744U (J.Y.Cheung); American Heart Association Pennsylvania Affiliate Post-Doctoral Fellowship 0425319U (B.A. Ahlers); and by grants from the Geisinger Foundation (J.Y. Cheung and L.I. Rothblum).
References
- 1.Palmer CJ, Scott BT, Jones LR. J Biol Chem. 1991;266:11126–11130. [PubMed] [Google Scholar]
- 2.Sweadner KJ, Rael E. Genomics. 2000;68:41–56. doi: 10.1006/geno.2000.6274. [DOI] [PubMed] [Google Scholar]
- 3.Waalas SI, Czernik AJ, Olstad OK, Sletten K, Walaas O. Biochem J. 1994;304 ( Pt 2):635–640. doi: 10.1042/bj3040635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Proc Natl Acad Sci U S A. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuller W, Eaton P, Bell JR, Shattock MJ. Faseb J. 2004;18:197–199. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- 6.Silverman BZ, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY, James AF, Shattock MJ. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Bossuyt, J., Ai, X., Moorman, J. R., Pogwizd, S. M., and Bers, D. M. (2005) Circ Res [DOI] [PubMed]
- 8.Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, Tucker AL, Bers DM. Circ Res. 2005;97:252–259. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- 9.Zhang XQ, Qureshi A, Song J, Carl LL, Tian Q, Stahl RC, Carey DJ, Rothblum LI, Cheung JY. Am J Physiol Heart Circ Physiol. 2003;284:H225–233. doi: 10.1152/ajpheart.00698.2002. [DOI] [PubMed] [Google Scholar]
- 10.Ahlers BA, Zhang XQ, Moorman JR, Rothblum LI, Carl LL, Song J, Wang J, Geddis LM, Tucker AL, Mounsey JP, Cheung JY. J Biol Chem. 2005;280:19875–19882. doi: 10.1074/jbc.M414703200. [DOI] [PubMed] [Google Scholar]
- 11.Mirza MA, Zhang XQ, Ahlers BA, Qureshi A, Carl LL, Song J, Tucker AL, Mounsey JP, Moorman JR, Rothblum LI, Zhang TS, Cheung JY. Am J Physiol Heart Circ Physiol. 2004;286:H1322–1330. doi: 10.1152/ajpheart.00997.2003. [DOI] [PubMed] [Google Scholar]
- 12.Simmerman HK, Jones LR. Physiol Rev. 1998;78:921–947. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- 13.Mahmmoud YA, Vorum H, Cornelius F. J Biol Chem. 2000;275:35969–35977. doi: 10.1074/jbc.M005168200. [DOI] [PubMed] [Google Scholar]
- 14.Mahmmoud YA, Cramb G, Maunsbach AB, Cutler CP, Meischke L, Cornelius F. J Biol Chem. 2003;278:37427–37438. doi: 10.1074/jbc.M305126200. [DOI] [PubMed] [Google Scholar]
- 15.Song J, Zhang XQ, Ahlers BA, Carl LL, Wang J, Rothblum LI, Stahl RC, Mounsey JP, Tucker AL, Moorman JR, Cheung JY. Am J Physiol Heart Circ Physiol. 2005;288:H2342–2354. doi: 10.1152/ajpheart.01133.2004. [DOI] [PubMed] [Google Scholar]
- 16.He T, Zhou S, da Costa L, Yu L, Kinzler K, Vogelstein B. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang XQ, Song J, Rothblum LI, Lun M, Wang X, Ding F, Dunn J, Lytton J, McDermott PJ, Cheung JY. Am J Physiol Heart Circ Physiol. 2001;281:H2079–2088. doi: 10.1152/ajpheart.2001.281.5.H2079. [DOI] [PubMed] [Google Scholar]
- 18.Tadros GM, Zhang XQ, Song J, Carl LL, Rothblum LI, Tian Q, Dunn J, Lytton J, Cheung JY. Am J Physiol Heart Circ Physiol. 2002;283:H1616–1626. doi: 10.1152/ajpheart.00186.2002. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Tillotson D, Moore R, Zelis R, Cheung J. Am Journal of Physiol. 1996;271:C1800–C1807. doi: 10.1152/ajpcell.1996.271.6.C1800. [DOI] [PubMed] [Google Scholar]
- 20.Jia LG, Donnet C, Bogaev RC, Blatt RJ, McKinney CE, Day KH, Berr SS, Jones LR, Moorman JR, Sweadner KJ, Tucker AL. Am J Physiol Heart Circ Physiol. 2005;288:H1982–1988. doi: 10.1152/ajpheart.00142.2004. [DOI] [PubMed] [Google Scholar]
- 21.Song J, Zhang X, Carl L, Qureshi A, Rothblum L, Cheung J. Am Journal of Physiol Heart Circ Physiol. 2002;283:H576–H583. doi: 10.1152/ajpheart.00197.2002. [DOI] [PubMed] [Google Scholar]
- 22.Hasenfuss G. Cardiovasc Res. 1998;37:279–289. doi: 10.1016/s0008-6363(97)00277-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Am J Physiol Heart Circ Physiol. 2000;279:H429–436. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]
- 24.Zhang XQ, Moorman JR, Ahlers BA, Carl LL, Lake DE, Song J, Mounsey JP, Tucker AL, Chan YM, Rothblum LI, Stahl RC, Carey DJ, Cheung JY. J Appl Physiol. 2006;100:212–220. doi: 10.1152/japplphysiol.00757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Presti CF, Jones LR, Lindemann JP. J Biol Chem. 1985;260:3860–3867. [PubMed] [Google Scholar]
- 26.Rembold CM, Ripley ML, Meeks MK, Geddis LM, Kutchai HC, Marassi FM, Cheung JY, Moorman JR. J Vasc Res. 2005;42:483–491. doi: 10.1159/000088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ballard C, Schaffer S. J Mol Cell Cardiol. 1996;28:11–17. doi: 10.1006/jmcc.1996.0002. [DOI] [PubMed] [Google Scholar]
- 28.Iwamoto T, Pan Y, Wakabayashi S, Imagawa T, Yamanaka HI, Shigekawa M. J Biol Chem. 1996;271:13609–13615. doi: 10.1074/jbc.271.23.13609. [DOI] [PubMed] [Google Scholar]
- 29.Iwamoto T, Pan Y, Nakamura TY, Wakabayashi S, Shigekawa M. Biochemistry. 1998;37:17230–17238. doi: 10.1021/bi981521q. [DOI] [PubMed] [Google Scholar]
- 30.Ruknudin A, He S, Lederer WJ, Schulze DH. J Physiol 529 Pt. 2000;3:599–610. doi: 10.1111/j.1469-7793.2000.00599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He LP, Cleemann L, Soldatov NM, Morad M. J Physiol. 2003;548:677–689. doi: 10.1113/jphysiol.2002.036426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins A, Somlyo AV, Hilgemann DW. J Physiol. 1992;454:27–57. doi: 10.1113/jphysiol.1992.sp019253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perchenet L, Hinde AK, Patel KC, Hancox JC, Levi AJ. Pflugers Arch. 2000;439:822–828. doi: 10.1007/s004249900218. [DOI] [PubMed] [Google Scholar]
- 34.Wei SK, Ruknudin A, Hanlon SU, McCurley JM, Schulze DH, Haigney MC. Circ Res. 2003;92:897–903. doi: 10.1161/01.RES.0000069701.19660.14. [DOI] [PubMed] [Google Scholar]
- 35.Ginsburg K, DM B. Biophys J. 2004;86:26a. [Google Scholar]
- 36.Lin, X., Jo, H., Sakakibara, Y., Tambara, K., Kim, B., Komeda, M., and Matsuoka, S. (2005) Am J Physiol Cell Physiol [DOI] [PubMed]
- 37.Levesque PC, Hume JR. Cardiovasc Res. 1995;29:336–343. [PubMed] [Google Scholar]
- 38.Schulze DH, Muqhal M, Lederer WJ, Ruknudin AM. J Biol Chem. 2003;278:28849–28855. doi: 10.1074/jbc.M300754200. [DOI] [PubMed] [Google Scholar]