

Abstract
Heterotrimeric G-proteins are molecular switches that regulate numerous signaling pathways involved in cellular physiology. This characteristic is achieved by the adoption of two principal states: an inactive, GDP-bound and an active, GTP-bound state. Under basal conditions G-proteins exist in the inactive GDP-bound state, thus nucleotide exchange is crucial to the onset of signaling. Despite our understanding of G-protein signaling pathways, the mechanism of nucleotide exchange remains elusive. We employed phage display technology to identify nucleotide-state-dependent Gα binding peptides. Herein, we report a GDP-selective Gα-binding peptide, KB-752, that enhances spontaneous nucleotide exchange of Gαi subunits. Structural determination of the Gαi1/peptide complex reveals unique changes in the Gα switch regions predicted to enhance nucleotide exchange by creating a GDP dissociation route. Our results cast light onto a potential mechanism by which Gα subunits adopt a conformation suitable for nucleotide exchange.
Introduction
Heterotrimeric G-proteins are crucial intracellular mediators of a diverse array of extracellular signals including hormones, photons, odorants, and small molecules (Cabrera-Vera et al., 2003; McCudden et al., 2005). In the standard model of G-protein signaling, seven transmembrane domain G protein-coupled receptors (GPCRs) are associated with inactive, membrane-tethered G-protein heterotrimers consisting of Gα·GDP bound to Gβγ. Gβγ facilitates the receptor coupling of Gα·GDP, stabilizes its GDP-bound state and prevents spontaneous nucleotide exchange, thus serving as a guanine nucleotide dissociation inhibitor (GDI) (Higashijima et al., 1987). Ligand-activated GPCRs serve as guanine nucleotide exchange factors (GEFs), catalyzing exchange of GDP for GTP on Gα. GTP binding alters the conformation of three flexible “switch” regions within Gα, leading to Gβγ dissociation. Both Gα·GTP and Gβγ subsequently regulate several downstream effectors including adenylyl cyclases, phospholipases, kinases, and ion channels (Cabrera-Vera et al., 2003; McCudden et al., 2005). Based on sequence similarity and functional differences in effector regulation, G proteins are grouped into four distinct families: Gαi/o, Gαs, Gαq/11, and Gα12/13 (Cabrera-Vera et al., 2003; McCudden et al., 2005). Signal termination is achieved by the intrinsic GTP hydrolysis activity of Gα and accelerated by “regulators of G-protein signaling” (RGS proteins; Neubig and Siderovski, 2002). Formation of Gα·GDP causes heterotrimer reassociation, thereby preventing further effector interactions by either Gα or Gβγ. Accordingly, the duration of G-protein signaling is determined by the lifetime of Gα in the GTP-bound state (Sprang, 1997). Thus, G-proteins serve as temporal regulators of signaling pathways and understanding the molecular determinants of their guanine nucleotide cycle is of particular interest.
Structures of Gα subunits, including Gαi1, in both inactive and activated states have revealed critical conformational changes that occur during GTP binding and hydrolysis (Sprang, 1997). Gα consists of a Ras-like domain, a structural fold present in many GTPases, and a unique all-helical domain. Bound nucleotide resides in a cleft between these two domains. Although flexibility between these domains is thought to govern the rate of spontaneous nucleotide exchange (Remmers et al., 1999), the mechanism whereby Gα GEFs induce nucleotide exchange is not yet clear. Two distinct types of Gα GEFs are now known: membrane-bound GPCRs and the soluble, cytoplasmic RIC-8 proteins. The structure of the prototypical GPCR rhodopsin provided the first structural glimpse of the most prominent class of Gα GEFs (Palczewski et al., 2000); however, the receptor was in an inactive form and not bound to heterotrimer, and thus little direct information was gained about the mechanism for G-protein activation. The non-receptor Gα GEF RIC-8 is widely conserved across metazoa as a critical determinant (along with Gαi subunits) in mitotic spindle force generation during mitosis (reviewed in McCudden et al., 2005). Unlike the GEFs for Ras-superfamily GTPases, such as the RhoGEF family (Rossman et al., 2005), that have no preference for nucleotide state (GDP- or GTP-bound), RIC-8 exhibits selective interaction with the GDP-bound state of Gα subunits and does not bind nor act as a GEF toward Gα·GTP (Afshar et al., 2004; Tall et al., 2003). As RIC-8 proteins have only been recently discovered, structural studies of these proteins have yet to be reported. Thus, the structural determinants of Gα activation by GEFs remain largely unknown.
Phage display is a powerful technique to identify small peptides capable of binding desired targets in an unbiased manner. Identified peptides can then serve as tools to study target protein binding surfaces, protein-protein interaction sites, and protein function and regulation (reviewed in Rodi et al., 2002). This technology has identified peptide modulators of a variety of enzyme classes and signaling molecules (e.g., Ashraf et al., 2003; Hyde-DeRuyscher et al., 2000). In particular, phage display and similar approaches have been used to investigate G-protein binding interfaces on GPCRs (Gilchrist et al., 1998; Martin et al., 1996) and effector binding regions on Gβγ subunits (Scott et al., 2001), as well as to identify peptides with G-protein regulatory properties, including both GEF and GDI activities (Hessling et al., 2003; Ja and Roberts, 2004). In the present study, we have identified guanine nucleotide-dependent Gα binding peptides from a phage display peptide library. In particular, we describe the guanine nucleotide exchange factor activity of a GDP-selective peptide, termed KB-752. To understand the mechanism of KB-752 GEF activity, we determined the crystal structure of the peptide bound to Gαi1. These studies are the first to describe the structure of a Gα subunit in complex with a GEF and provide direct structural evidence in support of a previously proposed mechanism for the GPCR-catalyzed nucleotide exchange reaction.
Results
Identification of nucleotide-dependent Gα binding peptides
We used phage display to obtain peptides that recognize the distinct conformations of Gα when bound to GDP versus GTPγS (Cabrera-Vera et al., 2003; Sprang, 1997). Biotinylated Gαi1·GDP and Gαi1·GTPγS were independently immobilized onto streptavidin-coated microtiter plates for selection of phage-displayed peptides. Phage selectivity was monitored by comparing phage ELISA signals between wells containing Gαi1 and wells blocked with albumin. After four iterative rounds of selection, clonal bacteriophage isolates were purified, amplified and screened for selective binding to Gαi1 in GDP- or GTPγS-bound states (e.g., Figure 1A). In total, we isolated 51 GDP-dependent, 12 GTPγS-dependent, and 5 nucleotide-state-independent phage-displayed peptides. Extensive database searches suggest that none of these peptides has sequence similarity to known Gα interacting proteins (data not shown).
Figure 1.
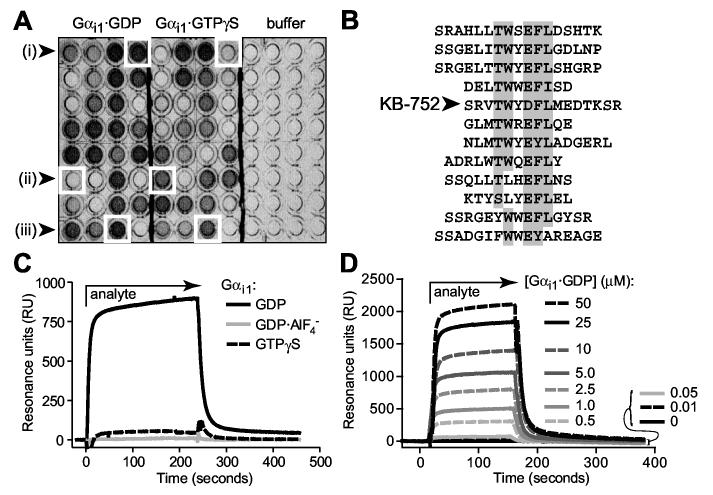
Identification of nucleotide-dependent Gαi-binding peptides via phage display. (A) Representative phage ELISA results indicating the identification of (i) GDP-selective, (ii) GTPγS-selective, and (iii) nucleotide state-independent Gαi1-binding peptides. (B) Sequences of 12 isolated peptides with GDP-selective binding to Gαi1, sharing a consensus TWXE/DFL motif with the particular peptide used in this study: KB-752. (C) Nucleotide-dependent binding of KB-752 as measured by surface plasmon resonance (SPR). 5 μM Gαi1 protein (“analyte”), in each of three nucleotide bound states, was injected over immobilized, biotinylated KB-752. Non-specific binding to a control peptide was subtracted from each curve. (D) GDP-bound Gαi1 was injected at each indicated concentration over immobilized KB-752 to determine the dissociation constant (Kd) for this interaction pair. SPR-derived dissociation constants for the interaction of KB-752 with Gαi1 and Gαo, in their ground state (GDP-bound), transition state-mimetic (GDP·AlF4- bound), and activated state (GTPγS-bound) forms, were obtained from analyses (n = 4-6 for each state) similar to that shown in panel D. Dissociation constants of >1000 μM were obtained for both Gα subunits in their GDP·AlF4--bound form, and for Gαo bound to GTPγS.
A representative group of GDP-dependent phage (Figure 1B) showed strong sequence similarity around the motif TWXE/DFL. Of these GDP-selective peptides, we focused initially on KB-752. Nucleotide-dependent Gα binding was quantitated by surface plasmon resonance (SPR) measurements on a streptavidin biosensor chip coated with biotinylated KB-752 (e.g., Figure 1C for Gαi1). Dissociation constants (Kd values) were obtained by simultaneous kinetic analysis of on (ka) and off (kd) rates obtained by injecting increasing concentrations of Gα in GDP-, GDP·AlF4-, and GTPγS-bound states (e.g., Figure 1D for Gαi1·GDP). In agreement with the phage selection, KB-752 displayed highest affinity binding to Gαi1 in its GDP-bound form (Kd of 3.9 ± 0.6 μM). No appreciable binding was observed to the transition-state-mimetic form of Gαi1·GDP·AlF4-, although measurable (albeit low affinity) binding was observed for Gαi1·GTPγS (Kd of 28.0 ± 3.2 μM); given the slow rate of spontaneous nucleotide exchange of Gαi1 (Fields and Casey, 1997), this observed binding may be due to residual GDP-bound protein. KB-752 demonstrated lower affinity for the closely-related Gαo, with a Kd of 18.2 ± 3.0 μM for Gαo·GDP, but no measurable interaction to GDP·AlF4- nor GTPγS-bound forms of Gαo (data not shown).
KB-752 binding affects guanine nucleotide exchange
To examine the effects of KB-752 on nucleotide exchange by Gα binding partners, [35S]GTPγS binding to purified Gα was quantified in the absence or presence of peptide. KB-752 enhanced the nucleotide exchange rate of Gαi1 (Figure 2A); the effective concentration for 50% maximal response (EC50) for KB-752 GEF activity on Gαi1 was 5.6 ± 1.1 μM (Figure 2B), comparable to its observed Kd. Equipotent GEF activity was found for both Gαi2 and Gαi3 (Figure 2B). KB-752 did not affect the nucleotide exchange rate of Gαi2β1γ2 (Figure 2C), suggesting that KB-752 cannot disrupt a native heterotrimer and interacts solely with free Gα.
Figure 2.
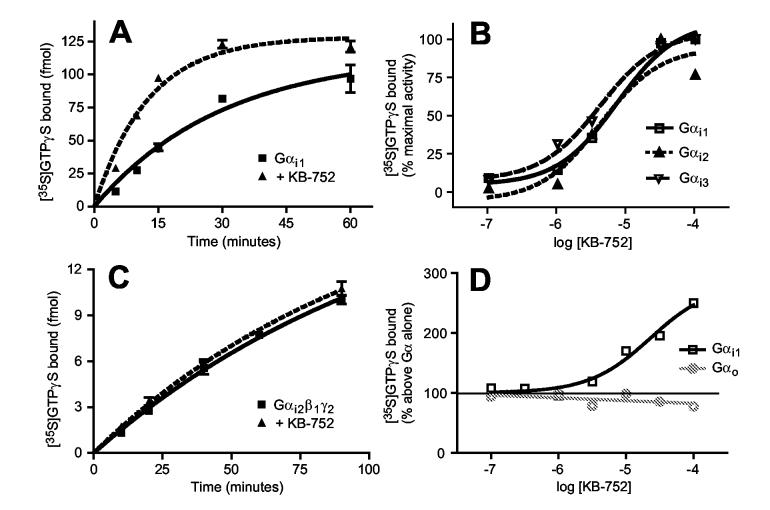
KB-752 is a selective guanine nucleotide exchange factor for Gαi subunits. (A) KB-752 (10 μM) enhances the GTPγS binding rate of Gαi1·GDP (50 nM); rate constants at 30 °C: Gαi1 alone = 0.029 ± 0.006 min-1, Gαi1 + KB-752 = 0.086 ± 0.008 min-1. (B) KB-752 is equipotent as a GEF on all three Gαi members. 50 nM Gαi1, Gαi2, or Gαi3 was incubated with indicated concentrations of KB-752 and the amount of [35S]GTPγS binding was measured after 10 min at 30°C, expressed as percent of maximal GTPγS binding. KB-752 does not alter the rate of GTPγS binding by (C) Gαi-heterotrimer Gαi2·GDP/Gβ1γ2 (peptide and protein amounts as in panel A), nor (D) isolated Gαo·GDP. For the dose-response curve of panel (D), 50 nM Gαi1 or Gαo was incubated in the presence of the indicated concentrations of KB-752 and the amount of [35S]GTPγS binding was measured (after 10 min at 30 °C for Gαi1; after 5 min at 20 °C for Gαo) as described in Experimental Procedures and is expressed as the percent of GTPγS bound in the absence of KB-752. The EC50 value for GEF activity on Gαi1 was 5.6 ± 1.1 μM. Data shown are from a representative experiment of 3-5 independent experiments.
Despite binding to Gαo·GDP, KB-752 did not affect nucleotide exchange even at saturating concentrations (Figure 2D). We hypothesized that the higher intrinsic rate of spontaneous nucleotide exchange of Gαo vs Gαi contributes to the lack of KB-752 activity on Gαo. To test this, we purified Gαi1 containing an arginine-144 to alanine (R144A) mutation that disrupts an interaction between the all-helical and Ras-like domains, and thus renders the spontaneous exchange rate equivalent to that of Gαo (Remmers et al., 1999); confirmed in Figure 3A). KB-752 did not enhance the exchange rate of Gαi1 (R144A) (Figure 3B), highlighting the mechanism of KB-752 as enhancing GDP release from the intrinsically slow exchanger Gαi.
Figure 3.

KB-752 GEF activity increases steady-state GTP hydrolysis by Gαi1, but does not act on a Gαi1 point mutant (R144A) with accelerated spontaneous nucleotide release. (A) 200 nM of wild-type (wt) Gαi1, R144A Gαi1, or wild-type Gαo was added to cuvettes containing 1 μM BODIPY-FL-GTPγS. Real-time nucleotide binding (Kimple et al., 2004) was measured at 25 °C as an increase in fluorescence response (λex = 485 nm; λem = 530 nm; slit widths of 3.0 nm) upon binding BODIPY-FL-GTPγS. Mutation of arginine 144 to alanine (R144A) resulted in Gαi1 nucleotide binding kinetics indistinguishable from that of Gαo, as previously reported by Remmers and colleagues (Remmers et al., 1999). (B) KB-752 does not alter the rate of GTPγS binding by the mutant Gαi1 subunit (R144A) with accelerated spontaneous nucleotide exchange comparable to that of wildtype Gαo (experiment performed as in Figure 2A except conducted at 20 °C). (C) Confirming the GEF activity of KB-752 on wild type Gαi1, addition of KB-752 (100 μM) to Gα (200 nM) enhances the steady-state hydrolysis of [γ32-P]GTP by Gαi1, but has no effect on Gαo. Note that the rate-limiting step in steady-state hydrolysis of GTP by Gα subunits is release of product (i.e., GDP) and not the hydrolysis of GTP per se (Ross, 2002).
To validate these results, we employed steady-state GTPase assays. Given that GDP release is the rate-limiting step of the Gα guanine nucleotide cycle, any alteration of GDP release, either positively (i.e., GEF activity) or negatively (i.e., GDI activity), will be reflected in the overall steady-state rate of GTP hydrolysis (Ross, 2002). KB-752 enhanced steady-state GTP hydrolysis by Gαi1 (Figure 3C), further indicating that it has GEF activity for Gαi subunits. No effect of KB-752 was seen on Gαo. These results support the conclusion that KB-752 possesses Gαi-selective GEF activity.
Structure of KB-752 bound to αi1
To ascertain the molecular mechanism of KB-752 GEF activity, we determined the structure of KB-752-bound Gαi1·GDP (PDB ID 1Y3A; Figure 4 and Table 1). KB-752 assumes a partial α-helical structure and binds Gαi1 between switch II and the α3 helix of the Ras-like domain (Figures 4 and 5A). The repositioning of switch II affords the binding groove for KB-752, as the α3 helix is not significantly altered in conformation compared to other structures of Gαi1. Indeed, the ability to reposition switch II likely defines the nucleotide specificity of KB-752 binding, given predicted steric hindrance between the N-terminus of KB-752 and switch II within Gαi1·GTPγS and Gαi1·GDP·AlF4- (Figure 5B). In particular, the positioning of tryptophan-211 of switch II would not accommodate tryptophan-5 of KB-752 (Figure 5B); however, tryptophan-211 is repositioned in the Gαi1·GDP/KB-752 structure and creates part of a critical hydrophobic pocket used by KB-752 for binding (see below).
Figure 4.
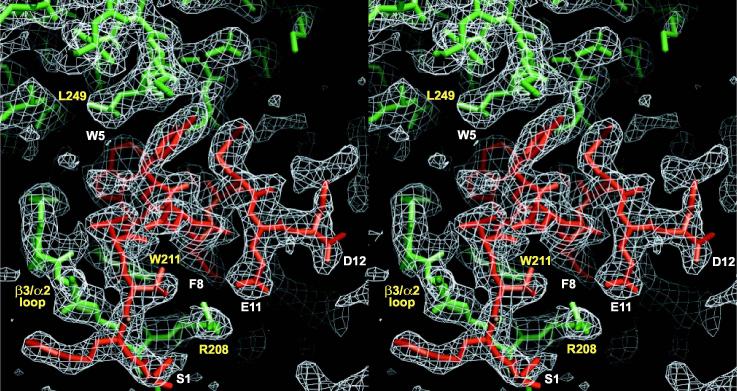
Stereoview of experimental electron density for the KB-752 peptide bound to Gαi1. The region highlighted is the entire peptide density (model in red; labels in white) found between switch II (α2 helix) and the α3 helix of Gαi1 (model in green; labels in yellow). Shown is a 2Fo-Fc simulated annealing composite omit map (generated with 5% overall model omitted) contoured at 1 s with electron density shown in white cage.
Table 1.
Data collection and refinement statistics
| Data collectiona | |
|---|---|
| Space group | P21 |
| No. of molecules per asymmetric unit | 4 |
| Unit cell dimensions | |
| a, b, c (Å) | 72.9, 112.8, 109.5 |
| α, β, γ (°) | 90, 93.8, 90 |
| Wavelength (Å) | 1.0093 |
| Resolution (Å) | 50-2.5 (2.59-2.5) |
| Rsymm (%) | 26.6 |
| Linear R-factorb | 0.072 (0.266) |
| Square R-factorc | 0.065 (0.232) |
| <IσI>d | 24 (3.6) |
| Completeness (%) | 96.4 (89.6) |
| Redundancy | 3.5 |
| Refinement | |
| Resolution (Å) | 20-2.5 (2.53-2.5) |
| No. reflections (working/test) | 29795/1561 |
| Rwork/Rfree (%)e | 24.9/28.1 |
| No. of nonhydrogen protein atoms | 10584 |
| GDP molecules | 4 |
| Water molecules | 136 |
| R.m.s. deviations | |
| Bonds (Å) | 0.062 |
| Angles (°) | 1.9 |
| Overall B-factors (chain B:chain F dimer) | |
| G-alpha | 41.9 (38.2) |
| KB-752 peptide | 52.4 (43.9) |
| GDP | 35.6 (32.6) |
| Water | 32.5 |
| Ramachandran plot (% in region) | |
| Most favored | 88.6 |
| Allowed | 9.0 |
| Generously allowed | 2.4 |
| Disallowed | 0.0 |
Numbers in parentheses pertain to the highest resolution shell.
Linear R-factor = Σ(|I - <I>|)/Σ(I)
Square R-factor = Σ(|I - <I>|)2 /Σ(I)2
<I/σI>, Mean signal-to-noise, where I is the integrated intensity of a measured reflection and σI is the estimated error in measurement.
Rwork = Σ(|Fp - Fp(calc)|)/ΣFp, where Fp and Fp(calc) are the observed and calculated structure factor amplitudes. Rfree is calculated similarly using test set reflections never used during refinement.
Figure 5.

Biochemical confirmation of the overall structural features of the Gαi1/KB-752 interaction. (A) Ribbon trace of KB-752 (red) bound between the α2 (“switch II”) and α3 helices of the Gαi1 Ras-like domain (blue). No contacts are made between KB-752 and the all-helical domain (yellow) or bound GDP (magenta). Switch regions are denoted in green. (B) Structural basis for nucleotide selective binding of KB-752 to Gαi1. KB-752 peptide (red, translucent) binds Gαi1 between switch II and the α3 helix; the conformations of these two helices are shown for Gαi1·GDP/KB-752 (green), Gαi1·GTPγS (yellow), and Gαi1·GDP·AlF4- (magenta). Whereas the α3 helix is not significantly altered, switch II is displaced to accommodate KB-752 binding. Switch II in both Gαi1·GTPγS and Gαi1·GDP·AlF4- assumes an extended α-helical conformation that is stabilized relative to Gαi1·GDP (Mixon et al., 1995; Sprang, 1997). This conformation of switch II is not permissive to KB-752 binding as it creates extensive steric hindrance. In particular, W211 of switch II (shown in space filling) is in a restrictive position relative to W5 of KB-752. (C) The GoLoco motif of RGS14 (orange) is also seen to bind, in an alpha-helical conformation, between switch II and the α3 helix of Gαi1 (PDB ID 1KJY); highlighted within the C α-carbon ribbon trace of the GoLoco peptide. Other features are colored as in panel A. (D) KB-752 GEF activity does not rely on the all-helical domain. 100 nM of Gαi1 or a chimeric α containing the Ras-like domain of Gαi1 and the all-helical domain of Gαo (“Gαioi”; ref. (Remmers et al., 1999) was incubated in the absence or presence of 50 μM KB-752 and [35S]GTPγS binding after 10 min at 30 °C was measured as described in Experimental Procedures. Data are expressed as percent GTPγS bound relative to Gα protein in the absence of KB-752 (“Control”) and are the average ± SEM of 4 independent experiments. (E) The KB-752 binding site on Gαi1 overlaps that of GoLoco motif peptides. Gαi1 (50 nM) was incubated in the absence or presence of the indicated concentrations of a peptide representing the GoLoco motif of RGS12 (R12GL) (Kimple et al., 2002). GTPγS binding was then measured in the presence of the indicated concentrations of KB-752. Data are expressed as fmol of GTPγS bound above that measured in the absence of KB-752 and are from a representative experiment of 3 independent experiments. (F) The binding of KB-752 has no effect on the kinetics of Gαi1 activation by AlF4-, unlike the slowed activation rate seen upon GoLoco peptide binding. Gαi1-CFP (200 nM) and YFP-RGS4 (280 nM) fusion proteins, previously shown to generate increased fluorescence resonance energy transfer (FRET) upon Gαi1 activation by AlF4- and subsequent RGS-box binding (Willard et al., 2004), were mixed together and pre-incubated with either 10 μM KB-752 peptide or 5 μM GoLoco consensus peptide (AGS3Con; Kimple et al., 2002), prior to the addition of NaF and AlCl 3 to final concentrations of 20 mM and 30 μM, respectively, at the 150-second mark.
The switch II/α3 helix binding pocket for KB-752 is similar to that of the N-terminal alpha-helix of the RGS14 GoLoco motif, a short polypeptide that displays GDI activity toward Gαi1 (PDB ID 1KJY) (Kimple et al., 2002); however, the GoLoco motif binding site extends into the all-helical domain (Figure 5C), whereas KB-752 makes no contacts with this region of Gαi1 (Figure 5A). The lack of functional contacts made between KB-752 and the all-helical domain was validated using a chimera (“Gαioi”) with the Ras-like domain of Gαi1 but the all-helical domain of Gαo (Remmers et al., 1999); KB-752 displays GEF activity on this chimera equal to that on wildtype Gαi1 (Figure 5D), suggesting that interactions with the Ras-like domain are sufficient for GEF activity. The use of a switch II/α3 helix binding pocket for both KB-752 and GoLoco motif peptides was also validated biochemically. The GEF activity of KB-752 on Gαi1 was found to be competitively antagonized by the GoLoco motif of the RGS14 paralogue, RGS12 (Figure 5E). Unlike the GoLoco motif, which lies over the GDP-binding pocket and uses an arginine finger to stabilize GDP (Kimple et al., 2002), KB-752 does not occlude nor make contact with GDP (Figure 5A vs 5C), suggesting that its GEF activity relies on conformational changes induced within Gαi1. In support of these distinct modes of interaction about the GDP-binding pocket, KB-752 binding has almost no effect on the rate by which Gαi1 is activated by aluminum tetrafluoride (Fig. 5F), unlike the inhibitory effect of GoLoco motif peptides (Willard et al., 2004).
Structural basis for the conserved TWXE/DFL binding motif
Figure 6 shows specific contacts between KB-752 and Gαi1. Glutamate-11 (E11) of KB-752 forms a salt bridge with R208 of Gαi1. Tryptophan-5 (W5) is found within a hydrophobic pocket formed by F215, L249, and I253 of Gαi1. Phenylalanine-8 (F8) is also placed within a hydrophobic environment established by W211, I212, and F215 of Gαi1. The burial of large hydrophobic residues within the hydrophobic groove between switch II and the α3 helix is common among several known Gα binding partners: p115RhoGEF-RGS inserts a methionine (M165) into the Gαi/13 chimera (Chen et al., 2005), the C2 domain of adenylyl cyclase inserts a phenylalanine (F991) into Gαs (Tesmer et al., 1997b), and the gamma subunit of cGMPphosphodiesterase inserts a tryptophan (W70) into Gαt (Slep et al., 2001). Burial of the peptide residues W5 and F8 within Gαi1 validates the results of the phage selection, as these two hydrophobic residues figure prominently within the TWXE/DFL binding motif (Figure 1B). An intramolecular hydrogen bond network between threonine-4 (T4) and both the side-chain carboxylate and peptide-bond nitrogen of aspartate-7 (D7) (Figure 6B) underscores the conservation of threonine and acidic residues within the TWXE/DFL motif. Specifically, the side-chain hydroxyl of T4 forms a hydrogen bond with both the side-chain carboxylate and main-chain amide nitrogen of D7, and the main-chain carbonyl oxygen of T4 forms a hydrogen bond with the main-chain amide nitrogen of D7. Additionally, this hydrogen bonding network within the α-helical portion of KB-752 serve to orient both W5 and F8 side chains toward the α binding face of the peptide.
Figure 6.
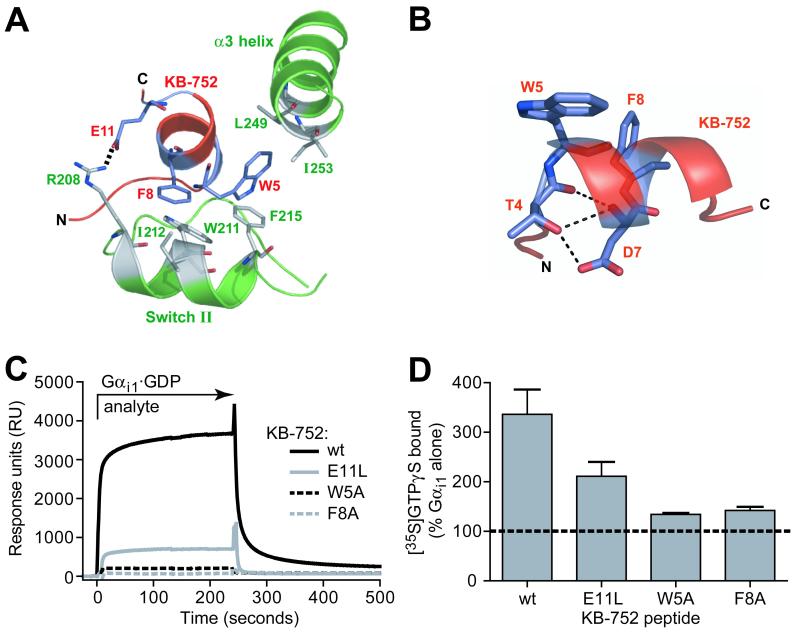
Biochemical confirmation of specific interactions between KB-752 and Gαi1. (A) Positions of KB-752 residues W5, F8, and E11 relative to residues in the switch II and α3 helices of Gαi1. W5 and F8 are placed within hydrophobic pockets formed by Gαi1 residues F215, L249, and I253, and W211, I212, and F215, respectively. E11 forms a salt bridge with R208 of Gαi1. (B) Peptide residues T4 and D7 of the conserved TWXE/DFL binding motif form an intrapeptide hydrogen bond network that helps to orient W5 and F8. (C, D) Effects of W5A, F8A, and E11L mutations to KB-752 activity. (C) Indicated KB-752 mutant or wildtype (wt) peptides were each immobilized to a density of ∼1000 RUs on separate streptavidin-coated flow cells and 50 μM GDP-bound Gαi1 (“analyte”) was injected simultaneously over all four surfaces. (D) Compared to the increase in GTPγS binding observed by addition of 50 μM wildtype KB-752 to 100 nM Gαi1·GDP, substantial reduction of GEF activity is seen upon mutation to the W5, F8, or E11 residue of KB-752.
Based on contacts between KB-752 and Gαi1, we generated three KB-752 variants to validate biochemically the structural model. E11 was replaced with leucine to eliminate the ionic interaction with R208. W5 and F8 of the TWXE/DFL motif were each independently replaced with alanine to reduce the potential for burial within hydrophobic environments created by switch II and the α3 helix. We first confirmed by SPR that each mutation abrogated Gαi1 binding. Gαi1·GDP was capable of interacting with the E11L peptide, but binding was significantly attenuated compared to wild-type (Figure 6C). Both W5A and F8A peptides displayed a near complete loss of binding to Gαi1·GDP. We then tested the ability of each peptide to enhance nucleotide exchange by Gαi1·GDP. Wild-type KB-752 resulted in an approximately 3-fold increase in the rate of [35S]GTPγS binding. The E11L peptide had diminished GEF activity compared to wild-type (Figure 6D), while W5A and F8A peptides lacked significant GEF activity. These results corroborate the critical contacts made between Gαi1 and these residues of KB-752 in the structural model.
Structural basis for KB-752 GEF activity
Exchange of GDP for GTP results in movement of the three switch regions to stabilize bound GTP and adopt the conformation responsible for effector binding (Sprang, 1997). Gαi1·GDP/KB-752 possesses significant alterations in each switch region compared to Gα·GDP/Gβ1γ2 (Figure 7A). Most apparent is switch II, which is displaced down and outward compared to the Gαi1·GDP/Gβ1γ2 structure (Wall et al., 1995). This movement results in the lip of switch II, normally ordered and helical in the GTPγS-bound state (Coleman et al., 1994; Sunahara et al., 1997), being displaced away from the nucleotide binding pocket and GDP. This conformation in Gαi1·GDP/KB-752 contrasts with the movement of switch II towards the nucleotide pocket when GTPγS is bound. Switch III is also slightly displaced from GDP within KB-752-bound Gαi1 compared to the heterotrimer (Figure 7A). However, of the four Gαi1/KB-752 dimers in the asymmetric unit (Table 1), only one Gαi1 molecule (chain B of PDB ID 1Y3A) had sufficient electron density to accurately model the switch III loop, suggesting that this region of Gαi1 is inherently flexible even when bound to KB-752. Similar alterations to both switch regions II and III are seen in GoLoco-bound Gαi1 (Gαi1·GDP/R14GL) (Kimple et al., 2002); however, switch II is more dramatically displaced in Gαi1·GDP/KB-752 (Figure 7B). Interestingly, despite movement in switch II, the β3/α2 loop at the entry to switch II is not significantly displaced in the Gαi1·GDP/R14GL structure compared to the Gαi1·GDP/Gβ1γ2 structure (Figure 7A vs. B). In contrast, this β3/α2 loop is removed from the guanine nucleotide pocket along with switch II in the Gαi1·GDP/KB-752 structure (Figure 7B). Displacement of the β3α2 loop is stabilized through several interactions with KB-752, including hydrogen bonding between the carbonyl oxygen of glycine-202 of the β3/α2 loop and the indole nitrogen of tryptophan-5 in KB-752 (Figure 7C), indicating an additional role for this key peptide residue. The displacement of switch II positions the catalytic glutamine-204 residue far from the nucleotide binding pocket compared to structures of Gαi1·GTPγS and Gαi1·GDP·AlF4- (Figure 8), and this residue makes an intramolecular bond with valine-201 (Figure 7C).
Figure 7.

Conformational changes in Gα switch regions induced by KB-752 binding. (A,B) Relative orientations of the three switch regions (SI — III) in heterotrimeric (PDB code 1GP2; blue in panel A), Gαi1·GDP/R14GL (PDB code 1KJY; orange in panel B), and Gαi1·GDP/KB-752 (green). Movement of switch II (α2 helix) and the connected β3/α2 loop away from the nucleotide binding pocket in Gαi1·GDP/KB-752 is thought to contribute to GEF activity by creating a route for GDP release. The RGS14 GoLoco motif peptide (R14GL) displaces switch II but does not significantly alter the position of the β3/α2 loop. (C) Binding of KB-752 displaces switch II resulting in a reorientation of the β3/α2 loop away from the guanine nucleotide pocket. KB-752 (red) stabilizes the β3/α2 loop (green) via several hydrogen bonds (indicated as yellow dashes), including the carbonyl oxygen of G202 (Gα) with the indole nitrogen of W5 (KB-752), and the side chain hydroxyl and main chain amide nitrogen of S206 (Gα) with the main chain amide nitrogen and carbonyl oxygen of V3 (KB-752), respectively. A water molecule (magenta ball) is coordinated by both Gα and KB-752 contacts.
Figure 8.
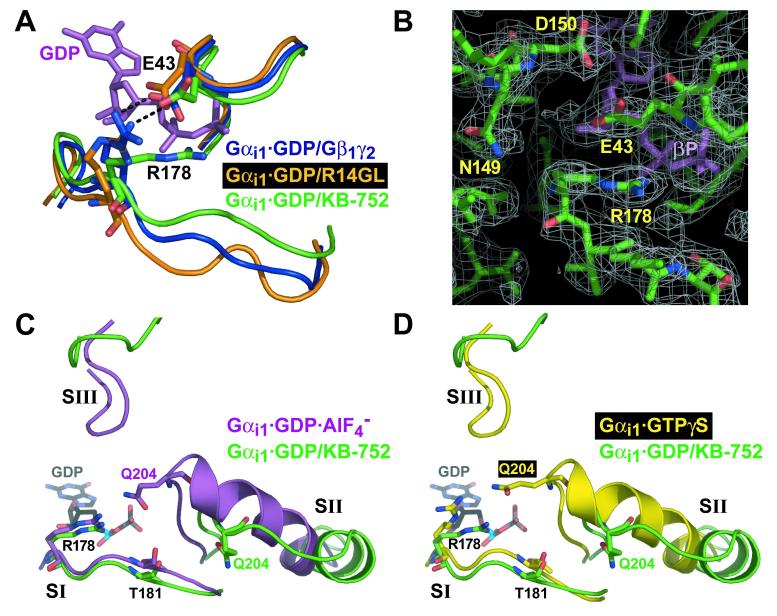
Comparison of switch regions and core catalytic residues of KB-752-bound Gαi1 with other states of Gαi1. (A) Movement of switch I in the Gαi1·GDP/KB-752 complex (green), versus its position in the Gαi1β1γ2 heterotrimer (blue) and the Gαi1·GDP/R14GL complex (orange), results in disruption of a salt-bridge (black dotted line) between R178 and E43 that normally stabilizes bound GDP (magenta) within Gαi1 when complexed to a GDI (Gβγ or GoLoco peptide). (B) Electron density of the R178 side-chain in the Gαi1·GDP/KB-752 complex (from a 2Fo-Fc simulated annealing composite omit map contoured to a level of 1 σ) is denoted by white mesh. In the background is the beta-phosphate of the bound GDP (βP). (C,D) Switch region comparisons with activated Gαi1 states. Switch regions of Gαi1·GDP/KB-752 (green), Gαi1·GDP·AlF4- (PDB code 1GFI; magenta; panel C), and Gαi1·GTPγS (PDB code 1GIA; yellow; panel D), are shown along with the residues critical for GTP hydrolysis (R178 and T181 within switch I and Q204 within switch II). GDP from the Gαi1·GDP/KB-752 structure is shown for reference in each case. Overall conformation of the switch regions of Gαi1·GDP·AlF4- and Gαi1·GTPγS are very similar, save for key changes in the position of catalytic residue side chains (Wall et al., 1998). Whereas switch I of Gαi1·GDP/KB-752 is very similar to that of the activated forms, both switch II and III are dramatically removed from the guanine nucleotide to allow for GDP release. The catalytic Q204 residue within switch II is far removed from the bound nucleotide and active site for GTP hydrolysis in the Gαi1·GDP/KB-752 structure. However, R178 and T181 of switch I are in a strikingly similar position to that of the Gαi1·GDP·AlF4- structure.
Switch I within Gαi1·GDP/KB-752 adopts a conformation more similar to the activated states of Gαi1·GTPγS and Gαi1·GDP·AlF4- (Figure 8), moving in closer proximity to GDP (compared to Gβγ- and GoLoco-bound states) and affecting the position of arginine-178 (R178) (Figure 8A). In the Gαi1·GDP/Gβ1γ2 heterotrimer (PDB 1GP2) (Wall et al., 1995), R178 of switch I forms a salt-bridge interaction with glutamate-43 (E43) across the bound GDP (Figure 8A). This “seatbelt” conformation, resulting from reoriented coordinating residues N149 and D150 due to Gβ1γ2 binding (Wall et al., 1998), is proposed to stabilize bound GDP (Lambright et al., 1996; Wall et al., 1995). This same interaction occurs in Gαi1·GDP/R14GL (Kimple et al., 2002) (Figure 8A), but not in the structures of free Gαi1·GDP (Wall et al., 1998) suggesting that the formation of this R178/E43 salt-bridge represents a common mechanism used by GDIs for Gαi1. Interestingly, in the KB-752-bound structure, the seatbelt interaction is not present (Figure 8A, B); the conformation of R178 is nearly identical to that of the Gαi1·GDP·AlF4- transition state (PDB 1GFI) (Figure 8C) (Coleman et al., 1994) in which R178 is oriented to participate in GTP hydrolysis by stabilization of the leaving γ-phosphate group as mimicked by the aluminum tetrafluoride anion. These findings suggest that the R178/E43 interaction is broken during nucleotide exchange and that an “unbuckled seatbeat” conformation may be essential for GDP release in addition to GTP hydrolysis. Thus, KB-752 appears to alter switch I and II to create a feasible exit route for GDP (see Discussion below). Magnesium was not observed in the nucleotide binding pocket of Gαi1·GDP/KB-752 (although its coordinating residue T181 is unaltered; Figure 8C,D), consistent with studies showing that Mg2+ has no effect on GDP binding to Gα (e.g., Higashijima et al., 1987).
Discussion
Despite many biochemical and structural studies of the guanine nucleotide cycle, the mechanism of heterotrimeric G-protein activation remains elusive. Mutagenesis studies have highlighted several determinants governing the G-protein coupling and nucleotide exchange properties of GPCRs (Bourne, 1997; Hamm, 2001), but precisely how a Gα subunit is induced to exchange GDP for GTP has remained unanswered, given the inherent difficulty in obtaining structural information on the GPCR:G-protein complex. Structural determinants of the recently described GEF activity of RIC-8 (Afshar et al., 2004; Tall et al., 2003) are also not known.
An alternative approach has been the use of small peptides that possess nucleotide-dependent binding and biochemical properties akin to known G-protein regulators. Mastoparan, a 14 aa peptide found in wasp venom, is a GEF for Gαi and Gαo (Higashijima et al., 1990). The solution structure of mastoparan bound to Gαi indicates a helical conformation for this peptide; however, biochemical studies suggest its binding interface resides at the extended N-terminus of Gα (Sukumar and Higashijima, 1992). Moreover, mastoparan (INLKALAALAKKIL) shows no similarity to the TWXE/DFL motif found in KB-752 and other Gα·GDP binding peptides from our screen. Synthetic peptides from the third intracellular loop of several GPCRs, a region involved in G-protein coupling and activation by receptor, have also been used to study G protein activation. A solution structure of a peptide from the third intracellular loop of the CB1 cannabinoid receptor suggests the necessity for a helical conformation (Ulfers et al., 2002). Finally, phage display has identified short hepta-peptides with biochemical activity at specific G-protein subunits (Hessling et al., 2003), although no structural information has been reported. Our results further highlight the power of phage display as a useful technique to identify conformation-dependent binding peptides that can be useful tools to investigate protein function. The structure of the Gαi1·GDP/KB-752 complex clearly demonstrates the basis of nucleotide specificity of this peptide.
Furthermore, our structural determination of KB-752 bound to Gαi1 represents the first glimpse of a Gα subunit bound to a GEF. As with other Gα regulators, KB-752 modulates the conformation of the switch regions critical to the guanine nucleotide cycle (Sprang, 1997). Previous structures of uncomplexed Gαi1·GDP have revealed structural disorder in these switch regions, particularly switch II and III (Coleman and Sprang, 1998). However, structures in which Gαi1 is bound to regulators (Gβγ, RGS4, GoLoco motif) or in the activated state (GTPγS- or GDP·AlF4--bound), the switch regions become ordered in specific, defined conformations (Coleman et al., 1994; Kimple et al., 2002; Tesmer et al., 1997a; Wall et al., 1995). Similarly, our structure of Gαi1·GDP/KB-752 reveals order in the switch regions, suggesting that the peptide stabilizes this conformation resulting in its GEF activity -- specifically by creating a stabilized route for GDP egress.
Since the Gα nucleotide binding pocket is buried far from the proposed Gα/receptor interacting surface, it is thought that GPCRs use Gβγ as a lever to “pull open” Gα, creating a GDP exit route. By modeling onto Gα the structural changes in EF-Tu induced by EF-Ts during nucleotide exchange, Bourne and colleagues have pointed to the β3/α2 loop as a potential "lip" that occludes GDP release (Iiri et al., 1998). Gβγ makes several contacts with this region and has been proposed to use additional contacts within the α2 helix (switch II), namely D228 of Gβ1 contacting K210 in Gαi1 (K206 in Gαs), to lever open the lip to induce GDP release (Rondard et al., 2001). GPCRs are thought to use the Gα N-terminus to tilt Gβγ (making extensive contacts with the Gα N-terminus) away from Gα, thereby opening the β3/α2 lip (Iiri et al., 1998). Our structure of the GEF peptide KB-752 bound to Gαi1 supports the Bourne model. By binding between the switch II and α3 helices, KB-752 pushes the α2 helix away from nucleotide, similar to the proposed levering action of Gβγ. Displacement of switch II results in the β3/α2 loop (part of the proposed occlusive lip (Iiri et al., 1998)) also being pulled away from nucleotide in a way that might allow more efficient GDP egress. Whereas switch II is displaced by the binding of the RGS14 GoLoco peptide, the β3/α2 loop remains essentially unaltered in conformation compared to the Gβγ-bound, heterotrimeric structure. These results further highlight the potential role of the β3/α2 loop as an occlusive lip preventing GDP release, as both Gβγ and GoLoco (each with GDI activity) position the β3/α2 loop to block the proposed GDP egress route, whereas KB-752 (with GEF activity) removes the loop from this position. Importantly, not only does KB-752 displace the β3/α2 loop from its occlusive orientation but also makes several contacts with this loop, which presumably serves to stabilize its reorientation. Although the precise structural determinants of GPCR-mediated GEF activity will clearly be distinct from that of our artificial phage-display peptide GEF, the structural changes in Gαi1 induced by KB-752 provide support for the Gβγ-levering model of receptor GEF function by suggesting that repositioning of switch II and the β3/α2 loop is critical for GDP release. In this model, the proposed egress route for GDP is towards the Gβγ binding face of Gα, which is more accessible following the displacement of the occlusive β3/α2 loop.
An alternative opinion on receptor-mediated heterotrimer activation (Cherfils and Chabre, 2003) suggests that GPCRs use the Gα N-terminus to maneuver Gβγ in an opposite fashion to that proposed in the Bourne model. In this “gear-shift” model, Gβγ is shifted towards Gα resulting in a closely packing Gα-Gβ interface stabilized by a proposed binding of the Gγ N-terminus to the Gα helical domain (Cherfils and Chabre, 2003). This Gβγ shift is proposed to alter the conformation of the α5 helix, previously implicated in the receptor-catalyzed nucleotide exchange reaction (Marin et al., 2002). Our structure of Gαi1·GDP/KB-752, while not invalidating the receptor GEF model of Cherfils and Chabre given lack of sequence similarity between KB-752 and known Gα regulators, certainly does not support their model of GPCR GEF activity for three reasons: (i) KB-752 causes switch II to be displaced away from the GDP pocket rather than being packed more tightly, (ii) the proposed GDP exit route induced by KB-752 binding is on the Gβ-binding face, and (iii) KB-752 does not cause significant alterations in α5 helix conformation.
In addition to affecting switch II (α2 helix) and maneuvering the β3/α2 loop in a manner consistent with the Gβγ-lever model (Iiri et al., 1998), KB-752 binding also alters switch I. In contrast to displacement of switch II away from the nucleotide binding pocket, switch I is displaced slightly towards this pocket into a similar conformation to that of GTPγS- and GDP·AlF4--bound Gα. In the Gαi1·GDP/Gβ1γ2 heterotrimer and Gαi1·GDP/R14GL complex, R178 of switch I forms a salt bridge with E43 (an interaction not observed in free Gαi1·GDP), forming a “seatbelt” over bound GDP thought to aid in the stabilization of Gα·GDP by Gβγ or GoLoco binding (Kimple et al., 2002; Lambright et al., 1996; Wall et al., 1995). Switch I in the Gαi1·GDP/KB-752 structure reveals an R178 conformation out of bonding distance to E43, similar to that seen in the structure of free Gαi1·GDP, in which R178 is thought to be quite flexible (Mixon et al., 1995). The loss of the R178/E43 interaction in both the Gαi1·GDP/KB-752 (GEF) structure as well as free Gαi1·GDP (which has higher spontaneous nucleotide exchange compared to Gβγ-bound) supports the loss of this interaction as coinciding with nucleotide exchange. Thus, breaking the R178/E43 “GDP seatbelt” is a potentially crucial step in GDP release and subsequent GTP binding. Surprisingly, the R178 side chain is in a nearly identical conformation in the Gαi1·GDP/KB-752 structure compared to the Gαi1·GDP·AlF4- structure (Figure 7B), indicating that this residue potentially adopts a conformation that is suitable for both GDP/GTP exchange and GTP hydrolysis. Having the R178/E43 interaction disrupted, along with creating a feasible exit route by modulating the switch II helix, may contribute to enhanced GDP release and, thus, an enhanced nucleotide exchange rate observed upon KB-752 binding.
Despite the GEF activity of KB-752 towards Gαi1, the structure of the complex contains bound GDP. This seemingly paradoxical observation is explained by several considerations. The nucleotide-free state of isolated Gα is very unstable, likely reflecting an instantaneous conformation as nucleotide binding is extremely rapid (Ferguson et al., 1986; Sprang, 1997); stable trapping of the nucleotide-free state has only recently been successfully described following binding to the non-receptor GEF RIC-8 (Tall and Gilman, 2004). The Gα/RIC-8 interface is likely more extensive than with the small KB-752 peptide, which would add substantial stability to the nucleotide-free conformation. Similarly, establishing the nucleotide-free state of small GTPases also necessitates a large stabilizing interface with respective GEFs (e.g., Worthylake et al., 2000) that cannot be provided by the small KB-752 peptide. Along with the fact that the Gαi1 /KB-752 complex was crystallized in the presence of 5 μM GDP, these factors likely impeded the chances of capturing Gαi1 in a nucleotide-free state.
In conclusion, our identification and structural analysis of a novel Gα·GDP binding peptide with GEF activity towards Gαi1-3 subunits provides support of the “Gβγ lever” hypothesis of GPCR GEF activity. The activity of KB-752 as a GEF for Gαi suggests a future utility of this peptide as a new molecular tool to study heterotrimeric G-protein signaling in vitro and in vivo.
Experimental Procedures
Unless otherwise noted, all reagents were from Sigma. Peptides were synthesized by Anaspec (San Jose, CA). Biotinylated peptides were synthesized by Dr. Michael Berne and colleagues of the Tufts University Core Facility: biotinylation was performed on resin-bound, Fmoc group-protected synthetic peptides that were selectively deprotected only at their N-termini, assuring biotin conjugation solely at the free amine.
Phage selection. Biotinylated Gαi1 was purified from E. coli as described (Kimple et al., 2004): presence of an N-terminal AviTag sequence (GLNDIFEAQKIEWHE) allowed for selective in vivo biotinylation on the lysine residue during expression in E. coli strain AVB 100 that also expresses biotin ligase (BirA) and fermentation in free biotin-containing medium as per manufacturer's instructions (Avidity LCC, Denver, CO). Nineteen different, random peptide bacteriophage libraries were obtained from New England Biolabs (PhD7, PhD12) or prepared by Karo*Bio USA using published methods (Sparks et al., 1996). Immulon 4 plates (96-well; Dynatech) were coated with streptavidin in 0.1 μNaHCO3, blocked with 1.0% BSA in 0.1 M NaHCO3, then incubated for 1 hr at 25 °C with 10 pmoles/well of biotin-Gαi1 in buffer A (20 mM HEPES pH 7.5, 1 mM EDTA, 16 mM MgCl2, 1 mM DTT, 0.05% Tween-20) with either 5 μM GDP or GTPγS. Iterative selection of binding phage was performed using published methods (Sparks et al., 1996). Briefly, after incubating phage libraries with immobilized biotin-Gαi1 for 3 hrs at 25 °C, non-specifically bound phage were removed by washing with TBST buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.05% Tween-20) with 0.5 mM biotin. Bound phage were eluted sequentially with a low pH glycine buffer and a high pH ethanolamine buffer; after neutralizing the pH, phage were amplified and subjected to repeat rounds of selection (Sparks et al., 1996).
After 4 iterations, clonal phage isolates were purified, amplified, and sequenced as described (Sparks et al., 1996). To detect bound phage by ELISA, biotin-Gαi1 was incubated overnight in buffer A with either 100 μM GDP or GTPγS and then 1 pmol Gαi1/well (or buffer A alone) was immobilized onto plates as previously described. 5 μL phage was added to each well in buffer A with either 100 μM GDP or GTPγS and incubated for 30 min. at 25 °C. Unbound phage was removed by TBST washes and bound phage detected with an anti-M13 antibody/horseradish peroxidase conjuguate. Assays were developed for 10 min at room temperature by adding 2,2-azinobis(3-ethylbenzothiazoline)-6 sulfonic acid and H2O2. Signal development was stopped by adding SDS to a final concentration of 1%.
Protein purification. His6-tagged human Gαi1 (full length, R144A mutant, and N-end 25 aa truncated) and human GαoA (full length) were purified from BL21(DE3) E. coli as previously described (Kimple et al., 2004). Gαi1 and Gαo were induced at an OD600 = 0.8 with 1 mM IPTG for 4 hr at 37 °C. Gαi2β1γ2 was purified from Sf9 insect cells co-infected with baculoviruses encoding Gαi2,Gβ1, and His6-Gγ2 as previously described (Hooks et al., 2003). Proteins were purified by Ni2+-NTA, anion exchange, and size exclusion chromatographies as described (Hooks et al., 2003; Kimple et al., 2004). All proteins were concentrated using YM-10 centrifugal filters (Millipore).
Surface plasmon resonance (SPR) biosensor measurements. SPR binding assays were performed at 25 °C on a BIAcore 3000. To analyze nucleotide-dependent Gα binding, N-terminally biotinylated KB-752 (diluted to 0.1 μg/ml in BIA running buffer [10 mM HEPES, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.005 % NP40]) was coupled to separate flow cells of streptavidin biosensors (Biacore) using MANUAL INJECT to a surface density of ∼250, ∼500, or ∼1000 resonance units. Prior to injection, Gα subunits were diluted in BIA running buffer with 100 μM GDP, 100 μM GDP plus 30 μM AlCl3 and 10 mM NaF, or 100 μM GTPγS and incubated at 25 °C for 2-3 hr. 30 μl Gα subunit was then simultaneously injected (using KINJECT) over flow cells at 10 μl/min followed by 300 s dissociation. Binding to a non-Gα interacting, biotinylated peptide (mNOTCH1; ref. Snow et al., 2002) was subtracted from all binding curves to correct for nonspecific binding and buffer shifts. Surfaces were regenerated with two 10 μl pulses of 500 mM NaCl / 25 mM NaOH at 20 μl/min. Binding curves and kinetic analyses were conducted using BIAevaluation ver. 3.0 and plotted using GraphPad Prism ver. 4.0b. Binding affinities were calculated using the simultaneous association (ka) and dissociation (kd) analysis parameter using generated sensorgram curves.
Gα nucleotide cycle assays. GTPγS exchange assays were conducted using a nitrocellulose filter binding method (Afshar et al., 2004), with GTPγS binding reactions performed at either 20 °C (Gαo and Gαi1-R144A) or 30 °C (Gαi1, i2, i3 and Gαi2β1γ2). Steady-state GTPase assays were carried out using a charcoal precipitation-based method (Afshar et al., 2004), with reactions incubated at 20 °C (Gαo) or 30 °C (Gαi) for 30 min.
Crystallization and structure determination. Crystals of KB-752 bound to Gαi1 were obtained by vapour diffusion from hanging drops (3 μL) containing a 1:1 (v/v) ratio of protein solution (6 mg/ml Gαi1ΔN25 and 1.3-fold molar excess KB-752 in 20 mM Tris, pH 7.5, 20 mM NaCl, 1 mM MgCl2, 10 μM GDP, 1 mM DTT, 5 % glycerol) to well solution (50 mM sodium citrate, pH 5.0, 10 % (w/w) PEG-8000, 10 % (w/w) sucrose). Crystals formed in 3-5 days at 4 °C in the space group P21 (a = 72.9 Å, b = 112.8 Å, c = 109.5 Å, α = 90°, β = 93.7°,γ= 90°), with four Gαi1·GDP/KB-752 heterodimers in the asymmetric unit. To collect data at 100K, crystals were cryoprotected in 30% glycerol for 1 min then submerged in liquid N2. A native data set was collected at the SER-CAT 22-ID beamline at APS, Argonne National Laboratory. Data was scaled and indexed using the program HKL2000. The structure of Gαi1·GDP·Mg2+ (PDB accession code 1BOF), excluding the 25 aa N-terminus, aa 177-184 (switch I), aa 200-218 (switch II) and aa 233-239 (switch III), and waters and sulphates, was used for molecular replacement with AMoRe (Navaza, 1994). Model building was done ino (Jones et al., 1991) with successive rounds of simulated annealing, minimization, B group, and torsion angle refinements being completed using CNS (Brunger et al., 1998). All refinement was completed with non-crystallographic symmetry restraints and each of the 4 Gαi1·GDP/KB-752 dimers are essentially identical. Electron density maps for model building as well as the simulated annealing composite omit map were generated using CNS. Gαi1 residues 26-33 (extreme N-terminus), 113-116 (αB-αC loop, dubbed ‘switch IV’, within the all-helical domain; Mixon et al., 1995), and 345-354 (extreme C-terminus) were not included in the final model given incomplete electron density; prior to removal, each region had refined B-factors of >150 indicative of low statistical certainty and relative disorder. Additionally, in 3 of the 4 Gαi1 subunits (molecules ‘A’, ‘C’, and ‘D’) in the asymmetric unit, residues 234-239 of switch III were removed from the final model. All structural images were made using PyMol (DeLano Scientific, San Carlos, CA, USA).
Acknowledgements
We thank Dr. Richard Neubig for Gαioi and Drs. Dana Fowlkes and Elliott Ross for early guidance on this project. F.S.W. and M.B.J. are postdoctoral fellowthe American Heart Association and the PhRMA Foundation, respectively. This work was supported in part by Karo*Bio USA and by NIH grants R01 GM062338 (to D.P.S.), and P01 GM065533 (to T.K.H., J.S., and D.P.S.). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
References
- Afshar K, Willard FS, Colombo K, Johnston CA, McCudden CR, Siderovski DP, Gonczy P. RIC-8 is required for GPR-1/2-dependent Galpha function during asymmetric division of C. elegans embryos. Cell. 2004;119:219–230. doi: 10.1016/j.cell.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Ashraf SS, Anderson E, Duke K, Hamilton PT, Fredericks Z. Identification and characterization of peptide probes directed against PKCalpha conformations. J Pept Res. 2003;61:263–273. doi: 10.1034/j.1399-3011.2003.00056.x. [DOI] [PubMed] [Google Scholar]
- Bourne HR. How receptors talk to trimeric G proteins. Curr Opin Cell Biol. 1997;9:134–142. doi: 10.1016/s0955-0674(97)80054-3. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Pt 5. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24:765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- Chen Z, Singer WD, Sternweis PC, Sprang SR. Structure of the p115RhoGEF rgRGS domain-Galpha13/i1 chimera complex suggests convergent evolution of a GTPase activator. Nat Struct Mol Biol. 2005;12:191–197. doi: 10.1038/nsmb888. [DOI] [PubMed] [Google Scholar]
- Cherfils J, Chabre M. Activation of G-protein Galpha subunits by receptors through Galpha-Gbeta and Galpha-Ggamma interactions. Trends Biochem Sci. 2003;28:13–17. doi: 10.1016/s0968-0004(02)00006-3. [DOI] [PubMed] [Google Scholar]
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- Coleman DE, Sprang SR. Crystal structures of the G protein Gi alpha 1 complexed with GDP and Mg2+: a crystallographic titration experiment. Biochemistry. 1998;37:14376–14385. doi: 10.1021/bi9810306. [DOI] [PubMed] [Google Scholar]
- Ferguson KM, Higashijima T, Smigel MD, Gilman AG. The influence of bound GDP on the kinetics of guanine nucleotide binding to G proteins. J Biol Chem. 1986;261:7393–7399. [PubMed] [Google Scholar]
- Fields TA, Casey PJ. Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. Pt 3. Biochem J. 1997;321:561–571. doi: 10.1042/bj3210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist A, Mazzoni MR, Dineen B, Dice A, Linden J, Proctor WR, Lupica CR, Dunwiddie TV, Hamm HE. Antagonists of the receptor-G protein interface block Gi-coupled signal transduction. J Biol Chem. 1998;273:14912–14919. doi: 10.1074/jbc.273.24.14912. [DOI] [PubMed] [Google Scholar]
- Hamm HE. How activated receptors couple to G proteins. Proc Natl Acad Sci U S A. 2001;98:4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessling J, Lohse MJ, Klotz KN. Peptide G protein agonists from a phage display library. Biochem Pharmacol. 2003;65:961–967. doi: 10.1016/s0006-2952(02)01653-2. [DOI] [PubMed] [Google Scholar]
- Higashijima T, Burnier J, Ross EM. Regulation of Gi and Go by mastoparan, related amphiphilic peptides, and hydrophobic amines. Mechanism and structural determinants of activity. J Biol Chem. 1990;265:14176–14186. [PubMed] [Google Scholar]
- Higashijima T, Ferguson KM, Sternweis PC, Smigel MD, Gilman AG. Effects of Mg2+ and the beta gamma-subunit complex on the interactions of guanine nucleotides with G proteins. J Biol Chem. 1987;262:762–766. [PubMed] [Google Scholar]
- Hooks SB, Waldo GL, Corbitt J, Bodor ET, Krumins AM, Harden TK. RGS6, RGS7, RGS9, and RGS11 stimulate GTPase activity of Gi family G-proteins with differential selectivity and maximal activity. J Biol Chem. 2003;278:10087–10093. doi: 10.1074/jbc.M211382200. [DOI] [PubMed] [Google Scholar]
- Hyde-DeRuyscher R, Paige LA, Christensen DJ, Hyde-DeRuyscher N, Lim A, Fredericks ZL, Kranz J, Gallant P, Zhang J, Rocklage SM, et al. Detection of small-molecule enzyme inhibitors with peptides isolated from phage-displayed combinatorial peptide libraries. Chem Biol. 2000;7:17–25. doi: 10.1016/s1074-5521(00)00062-4. [DOI] [PubMed] [Google Scholar]
- Iiri T, Farfel Z, Bourne HR. G-protein diseases furnish a model for the turn-on switch. Nature. 1998;394:35–38. doi: 10.1038/27831. [DOI] [PubMed] [Google Scholar]
- Ja WW, Roberts RW. In vitro selection of state-specific peptide modulators of G protein signaling using mRNA display. Biochemistry. 2004;43:9265–9275. doi: 10.1021/bi0498398. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Pt 2. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kimple RJ, Kimple ME, Betts L, Sondek J, Siderovski DP. Structural determinants for GoLoco-induced inhibition of nucleotide release by Galpha subunits. Nature. 2002;416:878–881. doi: 10.1038/416878a. [DOI] [PubMed] [Google Scholar]
- Kimple RJ, Willard FS, Hains MD, Jones MB, Nweke GK, Siderovski DP. Guanine nucleotide dissociation inhibitor activity of the triple GoLoco motif protein G18: alanine-to-aspartate mutation restores function to an inactive second GoLoco motif. Biochem J. 2004;378:801–808. doi: 10.1042/BJ20031686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- Marin EP, Krishna AG, Sakmar TP. Disruption of the alpha5 helix of transducin impairs rhodopsin-catalyzed nucleotide exchange. Biochemistry. 2002;41:6988–6994. doi: 10.1021/bi025514k. [DOI] [PubMed] [Google Scholar]
- Martin EL, Rens-Domiano S, Schatz PJ, Hamm HE. Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J Biol Chem. 1996;271:361–366. doi: 10.1074/jbc.271.1.361. [DOI] [PubMed] [Google Scholar]
- McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G-protein signaling: back to the future. Cell Mol Life Sci. 2005;62:551–577. doi: 10.1007/s00018-004-4462-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- Neubig RR, Siderovski DP. Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov. 2002;1:187–197. doi: 10.1038/nrd747. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Remmers AE, Engel C, Liu M, Neubig RR. Interdomain interactions regulate GDP release from heterotrimeric G proteins. Biochemistry. 1999;38:13795–13800. doi: 10.1021/bi990887f. [DOI] [PubMed] [Google Scholar]
- Rodi DJ, Makowski L, Kay BK. One from column A and two from column B: the benefits of phage display in molecular-recognition studies. Curr Opin Chem Biol. 2002;6:92–96. doi: 10.1016/s1367-5931(01)00287-3. [DOI] [PubMed] [Google Scholar]
- Rondard P, Iiri T, Srinivasan S, Meng E, Fujita T, Bourne HR. Mutant G protein alpha subunit activated by Gbeta gamma: a model for receptor activation? Proc Natl Acad Sci U S A. 2001;98:6150–6155. doi: 10.1073/pnas.101136198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM. Quantitative assays for GTPase-activating proteins. Methods Enzymol. 2002;344:601–617. doi: 10.1016/s0076-6879(02)44743-x. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Scott JK, Huang SF, Gangadhar BP, Samoriski GM, Clapp P, Gross RA, Taussig R, Smrcka AV. Evidence that a protein-protein interaction ′hot spot′ on heterotrimeric G protein betagamma subunits is used for recognition of a subclass of effectors. Embo J. 2001;20:767–776. doi: 10.1093/emboj/20.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slep KC, Kercher MA, He W, Cowan CW, Wensel TG, Sigler PB. Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature. 2001;409:1071–1077. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- Snow BE, Brothers GM, Siderovski DP. Molecular cloning of regulators of G-protein signaling family members and characterization of binding specificity of RGS12 PDZ domain. Methods Enzymol. 2002;344:740–761. doi: 10.1016/s0076-6879(02)44752-0. [DOI] [PubMed] [Google Scholar]
- Sparks AB, Adey NB, Cwirla S, Kay BK. In Phage Display of Peptides and Proteins, A Laboratory Manual. In: Kay BK, Winter J, McCafferty J, editors. Screening phage-displayed random peptide libraries. Academic Press; San Diego: 1996. pp. 227–253. [Google Scholar]
- Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem. 1997;66:639–678. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- Sukumar M, Higashijima T. G protein-bound conformation of mastoparan-X, a receptor-mimetic peptide. J Biol Chem. 1992;267:21421–21424. [PubMed] [Google Scholar]
- Sunahara RK, Tesmer JJ, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gsalpha. Science. 1997;278:1943–1947. doi: 10.1126/science.278.5345.1943. [DOI] [PubMed] [Google Scholar]
- Tall GG, Gilman AG. Purification and Functional Analysis of Ric-8A: A Guanine Nucleotide Exchange Factor for G-protein a Subunits. Methods Enzymol. 2004;390:377–388. doi: 10.1016/S0076-6879(04)90023-7. [DOI] [PubMed] [Google Scholar]
- Tall GG, Krumins AM, Gilman AG. Mammalian Ric-8A (synembryn) is a heterotrimeric Galpha protein guanine nucleotide exchange factor. J Biol Chem. 2003;278:8356–8362. doi: 10.1074/jbc.M211862200. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997a;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science. 1997b;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- Ulfers AL, McMurry JL, Miller A, Wang L, Kendall DA, Mierke DF. Cannabinoid receptor-G protein interactions: G(alphai1)-bound structures of IC3 and a mutant with altered G protein specificity. Protein Sci. 2002;11:2526–2531. doi: 10.1110/ps.0218402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- Wall MA, Posner BA, Sprang SR. Structural basis of activity and subunit recognition in G protein heterotrimers. Structure. 1998;6:1169–1183. doi: 10.1016/s0969-2126(98)00117-8. [DOI] [PubMed] [Google Scholar]
- Willard FS, Kimple RJ, Kimple AJ, Johnston CA, Siderovski DP. Fluorescence-based assays for RGS box function. Methods Enzymol. 2004;389:56–71. doi: 10.1016/S0076-6879(04)89004-9. [DOI] [PubMed] [Google Scholar]
- Worthylake DK, Rossman KL, Sondek J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 2000;408:682–688. doi: 10.1038/35047014. [DOI] [PubMed] [Google Scholar]