

Abstract
The free radical scavenger WR1065 (SH) is the active thiol form of the clinically approved cytoprotector amifostine. At doses of 40 μM and 4 mM it can activate the redox-sensitive nuclear transcription factor κB (NFκB) and elevate the expression of the antioxidant gene manganese superoxide dismutase (MnSOD) in human microvascular endothelial cells (HMEC). MnSOD contains binding motifs for a number of transcription factors other than NFκB and codes for a potent antioxidant enzyme localized in the mitochondria that is known to confer enhanced radiation resistance to cells. The purpose of this study was to determine the effect of WR1065 exposure on the various transcription factors known to affect MnSOD expression along with the subsequent kinetics of intracellular elevation of MnSOD protein levels and associated change in sensitivity to ionizing radiation in HMEC. Cells were grown to confluence and exposed to WR1065 for 30 min. Affects on the transcription factors AP1, AP2, CREB, NFκB, and Sp1 were monitored as a function of time ranging from 30 min to 4 h after drug exposure using a gel-shift assay. Only NFκB exhibited a marked activation and that occurred 30 min following the cessation of drug exposure. MnSOD protein levels, as determined by Western blot analysis, increased up to 16-fold over background control levels by 16 h following drug treatment, and remained at 10-fold or higher levels for an additional 32 h. MnSOD activity was evaluated using a gel-based assay and was found to be active throughout this time period. HMEC were irradiated with X-rays either in the presence of 40 μM or 4 mM WR1065 or 24 h after its removal when MnSOD levels were most elevated. No protection was observed for cells irradiated in the presence of 40 μM WR1065. In contrast, a 4 mM dose of WR1065 afforded an increase in cell survival by a factor of 2. A “delayed radioprotective” effect was, however, observed when cells were irradiated 24 h later, regardless of the concentration of WR1065 used. This effect is characterized as an increase in survival at the 2 Gy dose point, i.e., a 40% increase in survival, and an increase in the initial slope of the survival curve by a factor of 2. The anti-inflammatory sesquiterpene lactone, Helenalin, is an effective inhibitor of NFκB activation. HMEC were exposed to Helenalin for 2 h at a nontoxic concentration of 5 μM prior to exposure to WR1065. This treatment not only inhibited activation of NFκB by WR1065, but also inhibited the subsequent elevation of MnSOD and the delayed radioprotective effect. A persistent marked elevation of MnSOD in cells following their exposure to a thiol-containing reducing agent such as WR1065 can result in an elevated resistance to the cytotoxic effects of ionizing radiation and represents a novel radioprotection paradigm.
Keywords: WR1065, Nonprotein thiols, Nuclear transcription factor κB, Manganese superoxide dismutase, Radiation protection, Free radicals
Abbreviations: NPT, nonprotein thiols; NAC, N-acetyl-l-cysteine; MnSOD, manganese superoxide dismutase; ROS, reactive oxygen species; NFκB, nuclear transcription factor κB; AP1 and 2, activator proteins 1 and 2; Sp1, specificity protein 1; CREB, adenosine 3′,5′-cyclic monophosphate-regulator element-binding factor; HMEC, human microvascular endothelial cells; PBS, phosphate-buffered saline; DTT, dithiotrethiol; PMSF, phenylmethysulfonyl fluoride; EMSA, electrophoretic mobility-shift assays; DMSO, dimethyl sulfoxide; SDS, sodium dodecyl sulfate; NBT, nitroblue tetrazolium; TNFα, tumor necrosis factor-α; αPER, α-protection enhancement ratio
Introduction
The deleterious effects of ionizing radiation are known to be mediated both through the direct deposition of energy to critical biological molecules, the direct effect, and indirectly through the generation of highly reactive free radicals that in turn can go on to induce free radical damage to important biological targets such as DNA, the indirect effect [1]. The free radical-mediated indirect effect is known to account for approximately 75% of the subsequent damage to cells exposed to low LET ionizing radiation. For this reason the historical development of chemical compounds to protect against radiation damage has centered on antioxidative free radical scavenging agents that for the most part contain free thiol moieties that facilitate the removal of these highly reactive damage-causing molecules [1]. The most effective radioprotective drug developed to date, and the only agent approved by the FDA, for use in the protection of normal tissues in patients treated with radiation is amifostine (S-2-[3-aminopropylamino]ethylphosphorothioic acid) [2]. Other nonprotein thiols (NPT) possessing antioxidative properties that are currently approved for clinical use include: captopril ([S]-1-[3-mercapto-2-methyl-1-oxo-propyl]- l-proline), mesna (sodium-2-mercaptoethanesulfonate), and N-acetyl- l-cysteine (NAC).
The development of radioprotective strategies against oxidative damage induced by ionizing radiation is not limited to just exogenously applied antioxidants such as amifostine. The inherent radiation resistance of cells can also be augmented by affecting endogenous intracellular antioxidant enzyme levels. An example of such an enzyme is manganese superoxide dismutase (MnSOD). This enzyme is a nuclear-encoded mitochondrial enzyme that scavenges superoxide radicals in the mitochondrial matrix, and has been shown to be highly protective against radiation-induced reactive oxygen species (ROS) damage that can lead to cell lethality [3–5]. Mitochondria have been demonstrated to play an important role in the apoptotic processes induced by ionizing radiation, including the release of cytochrome c and the activation of caspase 3 that leads to PARP cleavage and subsequent DNA strand breaks [4,6,7]. Consistent with the role of mitochondrial damage leading to cell death, overexpression of MnSOD in cells induced by tranfection of an MnSOD transgene gives rise to an enhanced radioresistance and to an inhibition of radiation-induced apoptosis [8–10]. Furthermore, it has been demonstrated that localization of superoxide dismutase within the mitochondria is a requirement for subsequent radioprotection against cellular damage [4].
The generation of highly reactive free radical species by radiation is a very rapid process, with the preponderance of radiation damage fixation complete by 10−3 s. Most of the exogenous antioxidant agents such as amifostine are effective for only a limited period of time ranging from 15 min to 1 h under in vivo conditions [11]. Therefore, to be effective agents such as amifostine must be administered shortly before radiation exposure. Administration following irradiation is ineffective in affording protection against free radical damage [12,13].
Amifostine, as well as NPT in general, can participate in reductive/oxidative reactions that in turn can affect redox-sensitive cellular processes involving the activation of certain transcription factors, expression of genes, and activities of proteins [14–18]. In particular, amifostine, NAC, and oltipraz have each been observed to be effective in activating the redox sensitive nuclear transcription factor κB (NFκB) and enhancing the expression of the antioxidant gene MnSOD [14,16,18]. While the MnSOD gene contains binding motifs for a number of transcription factors including NFκB, activator proteins 1 (AP1) and 2 (AP2), specificity protein 1 (Sp1), and adenosine 3′,5′-cyclic monophosphate-regulator element-binding factor (CREB), it is the NFκB transcription factor that has been linked most closely with the process of inducible as compared to constitutive expression of MnSOD [19–22]. Sp1 is essential for transcription of MnSOD, while AP2 down-regulates transcription via its interaction with Sp1 [19]. These transcription factors are involved with the constitutive expression of MnSOD [19,21]. Inhibition of NFκB activation either through the use of inhibitors such as BAY 11-7082 ([E]3-[(4-methylphenyl)-sulfonyl]-2-propenenitrile), a propenenitrile compound that inhibits IκB phosphorylation and subsequent NFκB activation, or through the stable transfection of a mutant IκBα gene in cells prevents both an enhancement of MnSOD expression and the elevation of MnSOD protein levels following exposure to NPT [23].
We recently reported that activation of NFκB and subsequent enhancement of MnSOD expression following exposure of mouse SA-NH tumor cells to WR1065 (SH) (2-[aminopropylamino]ethane thiol) or other NPT gave rise to over a 15-fold increase in active intracellular MnSOD protein levels in these cells relative to control levels, peaking at about 24 h after drug exposure [18,23,24]. Since MnSOD has been shown in multiple cell and animal models to be radioprotective, it was of interest to determine whether enhanced MnSOD levels induced by NPT exposure in these cells would be sufficient to confer a concomitant enhanced radiation resistance. When SA-NH tumor cells were irradiated 24 h following a 30-min exposure to various NPT with radiation doses of 2 and 4 Gy and then assayed for clonogenic survival, survival increased 20 to 40% over comparably irradiated control cells not exposed to NPT. This phenomenon was identified as a NPT-induced “delayed radioprotective effect” mediated by elevated intra-cellular levels of MnSOD [24]. Exposure of cells to inhibitors of NFκB prevented the elevation of MnSOD levels by NPT along with the subsequent increase in radiation resistance [23]. Both gene expression and protein levels of MnSOD are routinely abnormal in malignant cells, with both generally diminishing as cells progress through the carcinogenic process [25]. For this reason we have expanded our study to include nontumor endothelial cells from human dermis, i.e., human microvascular endothelial cells (HMEC). A demonstration of a similar NPT-induced delayed radioprotective effect in these cells will have important consequences in the development of novel strategies for radioprotection. We therefore have included in this investigation not only the study of the effects of the free thiol form of amifostine, i.e., WR1065, on intracellular MnSOD protein level and activity, but also the determination of possible concomitant effects of WR1065 on both the immediate and delayed response of HMEC to ionizing radiation. Two different doses of WR1065 are used, 40 μM and 4 mM. These concentrations of WR1065 have been demonstrated to be maximally effective in protecting against radiation-induced mutagenesis and cell killing, respectively [13].
Materials and methods
Cells and culture conditions
Endothelial cells from human dermis immortalized with SV40 (HMEC) were originally obtained from Dr. T.J. Lawley, Biological Products Branch, Centers for Disease Control, Atlanta, Georgia [26]. Cells were maintained in endothelial basal medium MCDB131 (Gibco/BRL, Grand Island, NY) and supplemented with 15% fetal bovine serum (FBS) (Sigma, St. Louis, MO), 10 ng/ml epidermal growth factor (Collaborative Biochemical Products, Bedford, MA), 1 μg/ml hydrocortisone (Sigma), penicillin, and streptomycin (Gibco/BRL). HMEC were subcultured weekly with 0.25% trypsin and 1 mM EDTA (Gibco/BRL) and incubated in a humidified atmosphere of 5% CO2 and 95% air at 37°C. New cell stocks were used every 3–6 months. All cell survival experiments were performed with HMEC grown to confluence in order to reflect the experimental conditions used in the determination of WR1065 effects on NFκB activation, MnSOD gene expression, and MnSOD protein levels.
WR1065
WR1065, the active thiol metabolite of amifostine also designated as SH, was supplied by the Drug Synthesis and Chemistry Branch, Division of Cancer Treatment, National Cancer Institute, Bethesda, Maryland. Immediately prior to use, WR1065 was dissolved at a concentration of 1 M in phosphate-buffered saline (PBS; 8.1 mM Na2HPO4, 1.5 mM KH2PO4, 140 mM NaCl; Gibco/BRL). Final concentrations were chosen to reflect the optimum dose affording either the maximum level of immediate radioprotection (4 mM) or the prevention of mutation induction (40 μM) [13,27]. It is important to note that while millimolar levels of WR1065 are not achievable under in vivo conditions, concentrations of 40 μM are readily detectable in the serum of patients treated with radioprotective doses of amifostine [28].
Preparation of nuclear extracts
Nuclear extracts were prepared from confluent cultures grown in 150-mm tissue culture dishes. The experimental groups investigated included both untreated control cells and cells exposed to either 40 μM or 4 mM WR1065 for 30 min in complete medium. Immediately following treatment, cells were washed free of drug using PBS and then grown in drug-free medium. Nuclear protein was isolated either immediately or at times of 30 min or 1, 2, 3, and 4 h after WR1065 exposure. Cells were washed with 10 ml of ice-cold PBS, scraped from the dish, and pelleted by centrifugation at 1000 rpm for 5 min at 4°C. Cell pellets were resuspended in 400 μl of ice-cold buffer A (10 mM Hepes, pH 7.9; 10 mM KCl; 0.1 mM EDTA; 1 mM dithiotrethiol [DTT]; 0.5 mM phenylmethysulfonyl fluoride [PMSF]; 1 μg/ml leupeptin; 5 μg/ml aprotinin) and allowed to swell on ice for 15 min. Following the addition of 25 μl of 10% NP-40, the suspension was vortexed for 10 s to lyse the cells and centrifuged at 14,500 rpm for 1 min at 4°C. Nuclei were resuspended in 50 μl of ice-cold buffer B (20 mM Hepes, pH 7.9; 0.4 M NaCl; 1 mM EDTA; 1 mM DTT; 1 mM PMSF; 25% glycerol; 1 μg/ml leupeptin; 5 μg/ml aprotinin) and incubated on ice for 15 min. The nuclear suspension was then centrifuged at 14,500 rpm for 5 min at 4°C to pellet the nuclei, and the supernatant containing nuclear proteins was transferred to a clean tube. Protein concentrations for each sample were determined by the Bradford method [29] (Bio-Rad, Richmond, CA), and were adjusted to 2 μg/μl by the addition of buffer B.
Electrophoretic mobility-shift assays (EMSA)
Assays were performed using the Promega (Madison, WI) gel-shift assay system. NFκB (5′-AGTTGAGGGGACTTTCCCAGGC-3′), AP1 (5′-CGCTTGATGAGTCAGCCGGAA-3′), AP2 (5′- GATCGAACTGACCGCCCGCGGCCCGT-3′), Sp1 (5′-ATTCGATCGGGGCGGGGCGAGC-3′), and CREB (5′-AGAGATTGCCTGACGTCAGAGAGCTAG-3′) concensus sequence oligonucleotides were end-labeled with [γ32P]ATP using T4 polynucleotide kinase. Binding reactions contained the following: 5 μl nuclear extract (10 μg protein), 2 μl distilled deionized water, and 2 μl of 5× gel shift binding buffer (20% glycerol; 5 mM MgCl2; 2.5 mM DTT; 250 mM NaCl; 50 mM Tris-HCl, pH 7.5; 0.25 mg/ml poly(dI-dC)•poly(dI-dC). The reactions were incubated at room temperature for 10 min and then 1 μl (1.75 pmol) of 32P-labeled oligonucleotide was added and the reactions incubated at room temperature for an additional 20 min. Reactions were stopped by adding 1 μl of 10× gel loading buffer (250 mM Tris-HCl, pH 7.5; 0.2% bromophenol blue; 40% glycerol), loaded on an 8% non-denaturing polyacrylamide gel, and electrophoresed at 5 V/cm for 2 h. The gel was dried and exposed to Kodak BioMax MR film, and binding activity was quantified by laser densitometry. The same nuclear extracts were used to determine the activity of each transcription factor.
NFκB inhibitor Helenalin
The NFκB inhibitor Helenalin was used to determine its effect on WR1065’s ability to activate NFκB, elevate intracellular MnSOD protein levels, and induce a delayed radioprotective effect. Helenalin (Biomol, Plymouth Meeting, PA) was dissolved in DMSO at a concentration of 1 mM and filter-sterilized. Toxicity was examined by exposing HMEC grown to confluence to Helenalin at concentrations ranging from 1 to 20 μM for 2.5 h. To assess its effects on WR1065-induced radioprotection, HMEC were exposed to WR1065 at doses of 40 μM or 4 mM for 30 min or 1 or 5 μM Helenalin for 2 h and then exposed to WR1065 for 30 min at 37°C. Cells were washed twice with PBS warmed to 37°C and then incubated in drug-free medium.
Protein isolation and Western blot analysis
Protein isolation and Western blot analysis were performed following methods previously described [24]. Briefly, cell lysates were prepared from HMEC that were grown to confluence in 100-mm tissue culture dishes. Following isolation and determination of protein concentrations [24,29], MnSOD protein levels were assessed using the WesternBreeze chemiluminescent Western blot immunodetection system (Invitrogen, Carlsbad, CA). Twenty micrograms of total cellular protein was electrophoresed on a 12% SDS polyacrylamide gel according to the method of Laemmli [30]. Equal protein loading was confirmed by Coomassie blue R250 staining of gels (data not shown). Proteins were transferred onto polyvinylidene difluoride (PVD) membranes, blocked in Blocking Solution (Invitrogen) at room temperature for 1 h, and then incubated with primary antibody (1:1000 dilution of rabbit anti-MnSOD; Cat. No. 06-984, Upstate Biotechnology, New York) at room temperature for 1 h. The blots were washed four times with Washing Buffer (Invitrogen), and then they were incubated with goat anti-rabbit IgG conjugated with alkaline phosphatase (Invitrogen) for 30 min. The blots were washed four times with Washing Buffer, and the protein bands were visualized using Chemiluminescent Substrate (Invitrogen) as per instructions. The membrane was exposed to BioMax XAR film (Kodak, Rochester, NY) and scanned using a HP ScanJet 5300C scanner (Hewlett-Packard, Palo Alto, CA). Band intensities were analyzed using NIH ImageJ software.
MnSOD activity gel assay
MnSOD activity was measured in the presence of 0.75 mM NaCN by a method based on the inhibition of reduction of nitroblue tetrazolium (NBT) (Sigma, St. Louis, MO) by SOD as described elsewhere [31]. HMEC were harvested as described above and sonicated in 50 mM potassium phosphate buffer (pH 7.8) and a total of 200 μg protein/lane was electrophoresed into a native polyacrylamide gel consisting of a 5% stacking gel (pH 6.8) and a 12% running gel (pH 8.8) at 4°C. To visualize MnSOD activity, gels were incubated in 2.43 mM NBT, 0.028 mM riboflavin, and 280 mM N,N,N′,N′-tetramethylethylenediamine (TEMED) (Sigma) in 50 mM potassium phosphate buffer (pH 7.8) for 20 min in the dark. Gels were then washed in deionized water and illuminated under fluorescent light until clear zones of MnSOD activity were evident on a blue background. The bands were visualized and quantified with a computerized digital imaging system using AlphaImager 2000 software (Alpha Innotech, San Leandro, CA).
Irradiation conditions and survival assay
All survival assays were performed using HMEC grown to confluence. Cells were irradiated at room temperature using a Philips X-ray generator operating at 250 kVp and 15 mA at a dose rate of 1.65 Gy per minute. Radiation doses ranged from 0 to 6 Gy. To assess the “immediate radioprotective” effectiveness of WR1065, HMEC were exposed to 40 μM or 4 mM doses for 30 min just prior to irradiation. Immediately after exposure to radiation, the cells were washed free of drug, trypsinized, counted, and plated into fresh growth medium at appropriate numbers to give rise to about 100 colonies per dish. Five dishes were used per experimental point and experiments were repeated two to three times. Cells were incubated under standard growth conditions for 21 days and resultant colonies were stained with 20% crystal violet and colonies containing 50 or more cells were counted. The same procedure was followed to assess the delayed radioprotective effects of WR1065 with one difference. Cells were exposed to WR1065 for 30 min, washed free of the drug, and then incubated in growth medium for 24 h before being exposed to ionizing radiation. The resulting survival curves were analyzed using an α/β model and compared using a Z test to determine levels of significant difference. Because MnSOD has been demonstrated to primarily affect the initial slope of the survival curve [4,9,23,24], an analysis and comparison of the α parameters of the survival curves were made. To facilitate a comparison between the various treatment conditions an α-protection enhancement ratio (αPER), defined as αcontrol/αWR1065, was used. In addition, survival curves were also analyzed using the multitarget model to determine D0 values representing the reciprocals of the slopes of the terminal regions of each survival curve using SigmaPlot 8.0 (SPSS, Chicago, IL) and Microsoft Excel. Comparison of the resulting D0 values was performed using a Z test to determine levels of significance. Pairwise comparisons of surviving fractions at 2 Gy between each of the experimental conditions were performed using a two-tailed t test. To assess the effects of the NFκB inhibitor Helenalin at a concentration of 5 μM on the radiation response of HMEC irradiated in the presence or absence of WR1065, i.e., 40 μM and 4 mM doses, pairwise comparisons of surviving fractions at 2 Gy were also performed and levels of significance determined using a two-tailed t test.
Results
The MnSOD gene contains a number of transcription factor-binding motifs that include those for AP1, AP2, Sp1, CREB, and NFκB. In our previous reports we focused our attention on characterizing the ability of NPT to induce NFκB activation [18,23,24]. Presented in Fig. 1 are representative gel shifts describing the effects of a 30-min exposure of WR1065 at a concentration of 40 μM on the binding of various transcription factors in HMEC that were grown to confluence. The relative changes in band density representing the magnitudes of the various transcription factors binding to their respective motifs as a function of time following exposure to WR1065 were measured and contrasted to that of untreated control cells. Consistent with our earlier reports, NFκB activation increased by a robust 20-fold in cells exposed to WR1065. In contrast, no change was measured for either AP2 or Sp1 and only a minimal 1.1-fold increase was detected for both AP1 and CREB in these cells.
Fig. 1.
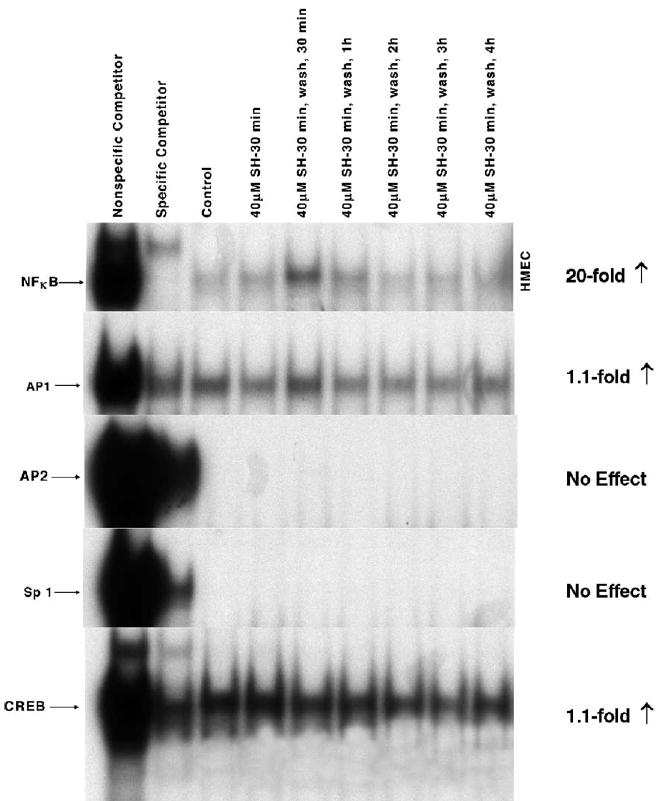
Comparative effects of 40 μM WR1065 (SH) on NFκB, AP1, AP2, Sp1, and CREB activation in HMEC exposed to drug for 30 min. Nuclear protein extracts were prepared immediately following drug exposure and at 30 min, 1, 2, 3, and 4 h after drug removal. The same nuclear extract was used to determine the activity of each transcription factor. Presented is a representative gel from three separate experiments.
Tumor necrosis factor-α (TNFα) is a potent inducer of MnSOD expression in cells [18,24]. For this reason it was included as a positive control with which to contrast the ability of WR1065 to affect this endpoint in HMEC. MnSOD protein levels were measured in HMEC as a function of WR1065 and TNFα exposure using Western blot analysis. A representative blot is presented in Fig. 2. HMEC were exposed to either 40 μM WR1065 or 10 ng/ml TNFα for 18 h, at which time cell cultures were washed with PBS, covered with fresh medium, and isolated to determine intracellular MnSOD levels 4, 8, 24, 48, and 72 h later. The corresponding activity gel is presented in Fig. 3. These data demonstrate that both protein level and enzyme activity are increased following exposure of HMEC to WR1065 and TNFα, with the TNFα-induced effect being much more robust. The effects of a 30-min exposure of WR1065 at a concentration of 40 μM on the elevation of intracellular MnSOD protein levels in HMEC as a function of time were also determined as shown in Fig. 4. Data describing the kinetics of the intracellular changes in MnSOD protein under each of these conditions are presented for comparison in Fig. 5. Maximal levels of MnSOD (>10-fold) were detected between 16 and 24 h following WR1065 exposure and appeared to persist for at least 48 h.
Fig. 2.
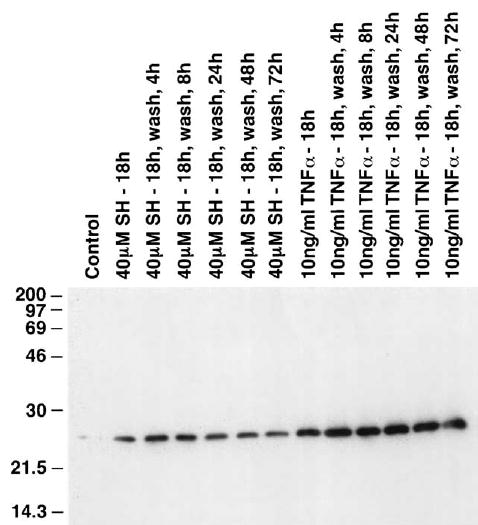
Representative Western blot showing the MnSOD protein levels in HMEC exposed to 40 μM WR1065 (SH) or, for comparative purposes, 10 ng/ml TNFα for 18 h. MnSOD levels were measured immediately following exposure and at 4, 8, 24, 48, and 72 h after drug removal.
Fig. 3.
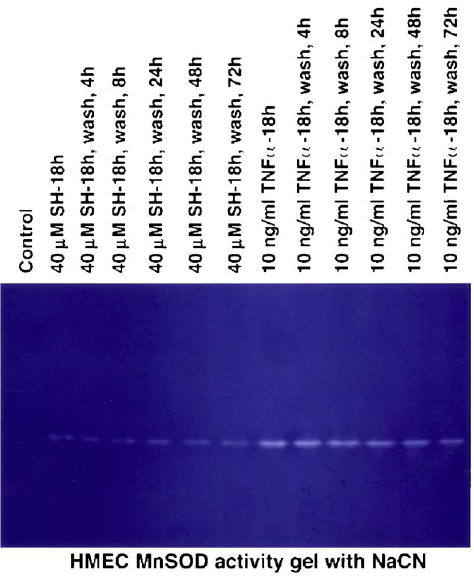
Representative activity gel showing the inhibition of the reduction of nitroblue tetrazolium by MnSOD. Gels were illuminated under fluorescent light in the presence of NaCN, which inhibits Cu/ZnSOD activity, until clear zones of active MnSOD were evident on a blue background. MnSOD activity was measured immediately following exposure to 40 μM WR1065 (SH) or, for comparative purposes, 10 ng/ml TNFα for 18 h, and at 4, 8, 24, 48, and 72 h after drug removal.
Fig. 4.

Representative Western blot showing MnSOD protein levels measured in HMEC following a 30-min exposure to 40 μM WR1065 (SH). MnSOD levels were measured at 4, 8, 16, 20, 24, 28, 32, and 48 h after drug removal.
Fig. 5.
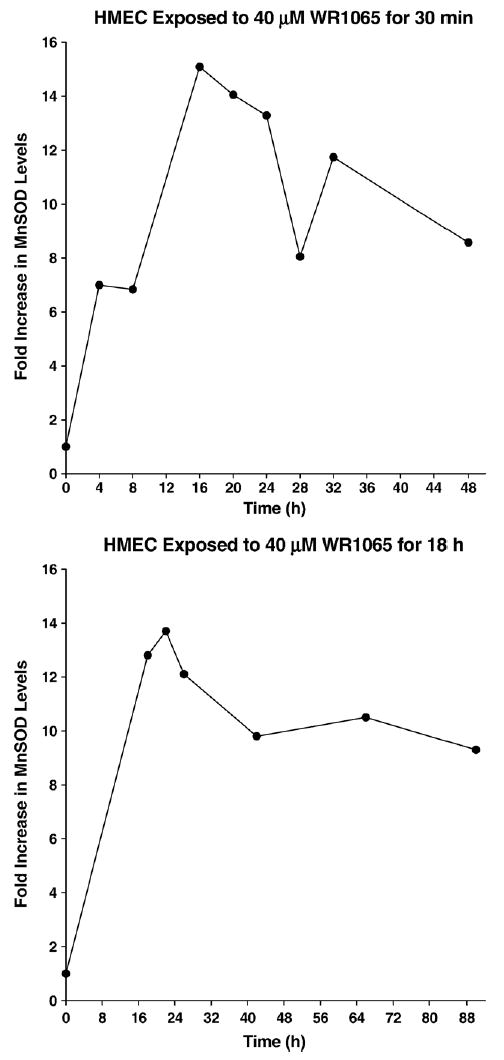
Kinetics of the changes in MnSOD protein levels after exposure of HMEC to 40 μM WR1065 for 30 min or 18 h. Maximum levels of MnSOD were detected between 16 and 24 h following drug exposure.
Helenalin is an anti-inflammatory sesquiterpene lactone that can effectively inhibit NFκB through the selective alkylation of the p65 subunit [32]. We determined the effect of this agent on WR1065-induced NFκB activation and the subsequent elevation of MnSOD. Presented in Fig. 6 is a dose response of HMEC survival as a function of Helenalin concentration following a 2.5-h exposure. Concentrations of 10 and 20 μM were found to be significantly toxic. For this reason all Helenalin studies related to NFκB and MnSOD in HMEC were limited to concentrations of 1 or 5 μM.
Fig. 6.
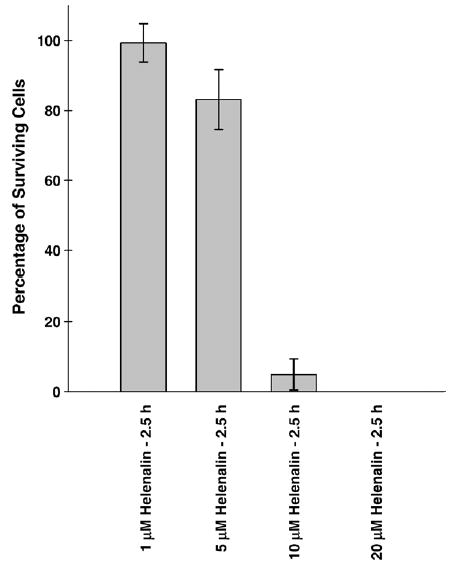
Bar graph depicting the percentage survival of HMEC exposed to Helenalin concentrations ranging from 1 to 20 μM for 2.5 h. Each bar represents the mean ± SE of three separate experiments.
To assess the inhibitory effects of Helenalin on WR1065-induced activation of NFκB in HMEC grown to confluence, cells were exposed to either 1 or 5 μM Helenalin for 2 h. In the presence of Helenalin, 40 μM or 4 mM WR1065 was added and cells were incubated for an additional 30 min and then washed free of drugs and harvested 30 min later to coincide with the optimum time for NFκB activation by WR1065 as demonstrated in Fig. 1. Presented in Fig. 7 is a representative gel shift that describes the inhibitory effect of Helenalin on NFκB activation by WR1065. While both concentrations of Helenalin were found to be effective, the 5 μM concentration was the more effective. To determine whether the inhibitory effects of Helenalin on NFκB activation by WR1065 would also inhibit the subsequent elevation of MnSOD, HMEC exposed to 5 μM Helenalin for 2 h followed by a 30 min exposure to 40 μM WR1065 were harvested 24 h later and extracts were made to assess intracellular protein levels. A representative Western blot describing relative MnSOD levels following exposure of HMEC to Helenalin and WR1065 is presented in Fig. 8. Helenalin not only inhibited the activation of NFκB by WR1065, but also inhibited the subsequent elevation of MnSOD protein.
Fig. 7.

Representative gel shift showing the inhibitory effects of Helenalin on WR1065 (SH)-mediated NFκB activation. HMEC were exposed to 1 or 5 μM Helenalin for 2 h followed by a 30-min exposure to either 40 μM or 4 mM WR1065 for 30 min. Also included for comparison purposes are nuclear protein extracts prepared from HMEC 30 min after exposure to 40 μM or 4 mM WR1065, which represents the maximal time of NFκB activation following drug exposure.
Fig. 8.
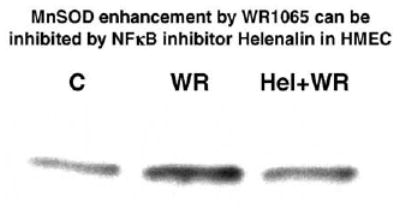
Representative Western blot showing MnSOD protein levels measured in untreated control HMEC (C), and 24 h after exposure of HMEC to 40 μM WR1065 (WR) for 30 min or a 2 h exposure to 5 μM Helenalin followed by a 30 min exposure to 40 μM WR1065 (Hel+WR).
MnSOD has been described by many investigators as being an effective endogenous antioxidant enzyme capable of conferring enhanced radiation resistance to cells [4,5,8,10]. Since maximal elevation of MnSOD occurs between 16 and 24 h following exposure of cells to WR1065, it was of interest to determine whether irradiation of cells at this time would result in an enhanced cellular resistance to the cytotoxic effects of ionizing radiation. Two concentrations of WR1065 were chosen for investigation. WR1065 when present during irradiation at a concentration of 4 mM can significantly protect cells from radiation toxicity [12,13,27]. This effect is best described as immediate radioprotection and is routinely demonstrated by administering WR1065 30 min prior to irradiation. The radioprotective effect, however, diminishes rapidly as the concentration of WR1065 is reduced [12,13,27]. At a concentration of 40 μM, WR1065 is ineffective for protection against cell killing but is sufficient for preventing radiation-induced mutagenesis through a distinctly separate mechanism of action [13]. Presented in Fig. 9 are radiation dose response curves obtained following the irradiation of HMEC alone or 30 min following the administration of WR1065 to the culture medium. While the radiation survival curves presented in Fig. 9 were fitted using a linear quadratic model, the survival data were also analyzed using a multitarget model to determine D0 values (data not shown) [23]. Consistent with previous reports, increased cell survival indicating radioprotection was only evident when WR1065 was present at a concentration of 4 mM [13,27].
Fig. 9.
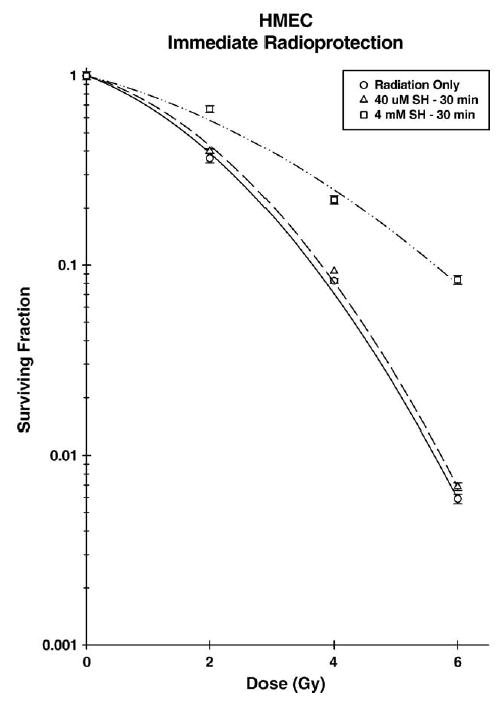
Survival of HMEC exposed to 250 kVp X-rays in the absence (open circles) or presence of 40 μM (open triangles) or 4 mM (open squares) WR1065 (SH). WR1065 was added to cells 30 min prior to irradiation and was removed immediately after. Points represent the means ± SE of three separate experiments.
To evaluate a potential radioprotective effect induced by WR1065 exposure but mediated by elevated MnSOD, HMEC were first exposed to WR1065 at either a 40 μM or 4 mM concentration for 30 min, washed free of drug, and then irradiated 24 h later when MnSOD levels were elevated over 10-fold. In contrast to the immediate radioprotective effect that was concentration dependent, the induced delayed radioprotective effect was not dependent upon the concentration of WR1065 used but rather correlated with the subsequent elevation of MnSOD (see Fig. 10). Elevation of cellular radiation resistance by MnSOD has historically been observed as an enhancement of the initial slope or α component of the survival curve [4,8–10,23]. To facilitate a more quantitative analysis of the protective effects of MnSOD, protection enhancement ratios (PER), defined by the ratio of the α components of the radiation survival curves describing the survival responses of control and WR1065 treated HMEC, i.e., αC/αWR1065, were determined and are listed in Table 1. Protection factors were also determined using a multitarget survival curve model, comparing the ratio of terminal slopes of the resultant survival curves, i.e., ratio of D0’s (see Table 1). At a concentration of 40 μM, WR1065 exhibited no effect on immediate radioprotection, but had a PER of about 2 for the induced delayed radioprotective effect. In contrast, WR1065 elicited both an immediate and an induced delayed protective effect when administered at a concentration of 4 mM. However, if radiation protection factors were determined not by comparing changes in survival in the lower dose range as described by the initial slope α, but rather in the more traditional manner by comparing the D0’s that describe the terminal regions of the survival curves using the multitarget model of analysis, no change in the D0 and therefore no significant protection factor could be demonstrated for a delayed effect regardless of the dose of WR1065 used ( P = 0.16). Analysis of these survival curves using the α/β model, however, showed that both 40 μM WR1065 (α/β = 2.19 ± 0.87) and 4 mM WR1065 (α/β = 3.24 ± 0.90) induced significant delayed radioprotection ( P = 0.04 and 0.06, respectively). This is consistent with the hypothesis that the immediate radioprotective effect is mediated directly by WR1065, while the delayed radioprotective effect is due to elevated levels of MnSOD. These data were further analyzed by comparing survival levels at 2 Gy as a function of treatment protocol (see Table 2). Again, WR1065 induced comparable levels of elevated delayed radiation resistance regardless of the concentration used, but afforded immediate radioprotection only at a concentration of 4 mM.
Fig. 10.
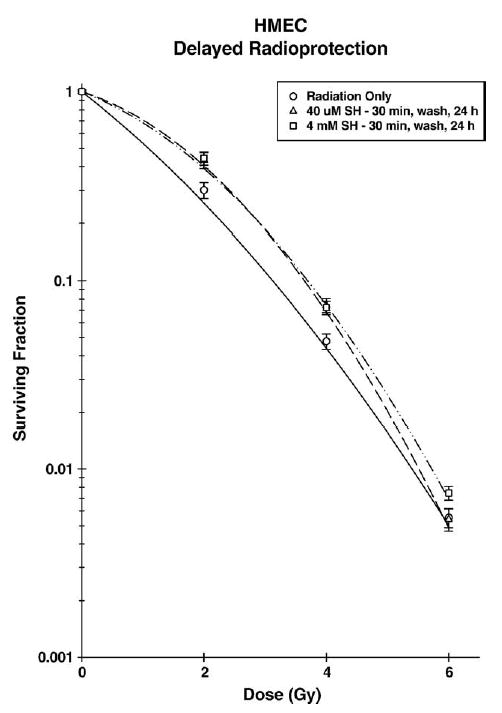
Survival of HMEC exposed to 250 kVp X-rays 24 h after a 30-min treatment with either 40 μM (open triangles) or 4 mM (open squares) WR1065 (SH) compared with untreated control cells (open circles). Points represent the means ± SE of three separate experiments.
Table 1.
| ± WR1065 | PER ± SE | P valuee | Do ± SE | Protection factor ± SE | P valuee | |
|---|---|---|---|---|---|---|
| Immediatec | — | 1.25 ± 0.03 | ||||
| 40 μM SH | 1.25 ± 0.26 | P = 0.17 | 1.28 ± 0.03 | 1.02 ± 0.03 | P = 0.24 | |
| 4 mM SH | 1.42 ± 0.32 | P = 0.09 | 2.56 ± 0.05 | 2.05 ± 0.05 | P < 0.01 | |
| Delayedd | — | 1.22 ± 0.02 | ||||
| 40 μM SH | 2.45 ± 0.74 | P = 0.03 | 1.32 ± 0.03 | 1.08 ± 0.02 | P = 0.02 | |
| 4 mM SH | 2.00 ± 0.42 | P = 0.01 | 1.37 ± 0.03 | 1.12 ± 0.03 | P < 0.01 |
Ratio of αc/αWR1065.
Ratio of Do’s ± WR1065.
HMEC were treated with WR1065 (SH) for 30 min, irradiated, and plated for survival.
HMEC were treated with WR1065 (SH) for 30 min, washed to remove the drug, irradiated 24 h after drug treatment and plated for survival.
Comparison between irradiated cells and cells treated with WR1065 (SH) and then irradiated. P values were calculated using the Z test.
Table 2.
Protection ratios at 2 Gy
| Treatment | Mean surviving fractionc ± SE | Protection ratio ± SE | P valued | |
|---|---|---|---|---|
| Immediatea | 2 Gy | 0.37 ± 0.02 | ||
| 40 μM SH-2 Gy | 0.40 ± 0.01 | 1.05 ± 0.22 | P > 0.05 | |
| 4 mM SH-2 Gy | 0.67 ± 0.03 | 1.81 ± 0.40 | P < 0.0001 | |
| Delayedb | 2 Gy | 0.30 ± 0.03 | ||
| 40 μM SH-2 Gy | 0.43 ± 0.04 | 1.43 ± 0.77 | P = 0.0151 | |
| 4 mM SH-2 Gy | 0.44 ± 0.04 | 1.47 ± 0.72 | P = 0.0044 |
HMEC were treated with WR1065 (SH) for 30 min, irradiated, and plated for survival.
HMEC were treated with WR1065 (SH) for 30 min, washed to remove the drug, irradiated 24 h after drug treatment, and plated for survival.
Average from three separate experiments.
Comparison between cells irradiated with 2 Gy and cells treated with WR1065 (SH) and then irradiated. P values were calculated using a two-tailed t test.
Helenalin pretreatment of HMEC prevented WR1065 induction of both NFκB activation and subsequent elevation of MnSOD protein. To test its potential inhibitory effect of WR1065-mediated induced delayed radioprotection, HMEC were exposed to 2 Gy of ionizing radiation 24 h following no drug treatment, exposure to 5 μM Helenalin for 2.5 h, exposure to Helenalin in combination with a 30 min exposure to either 40 μM or 4 mM WR1065 for 30 min, or exposure only to 40 μM or 4 mM WR1065. Helenalin treatment alone did not alter the radiation sensitivity of HMEC (P = 0.91), but it completely inhibited the WR1065-induced delayed radioprotective effect (P = 0.01 and 0.001 for 40 μM or 4 mM WR1065-treated cells, respectively; P = 0.65 and 0.73 for Helenalin + 40 μM or 4 mM WR1065 treatment, respectively) (see Fig. 11).
Fig. 11.
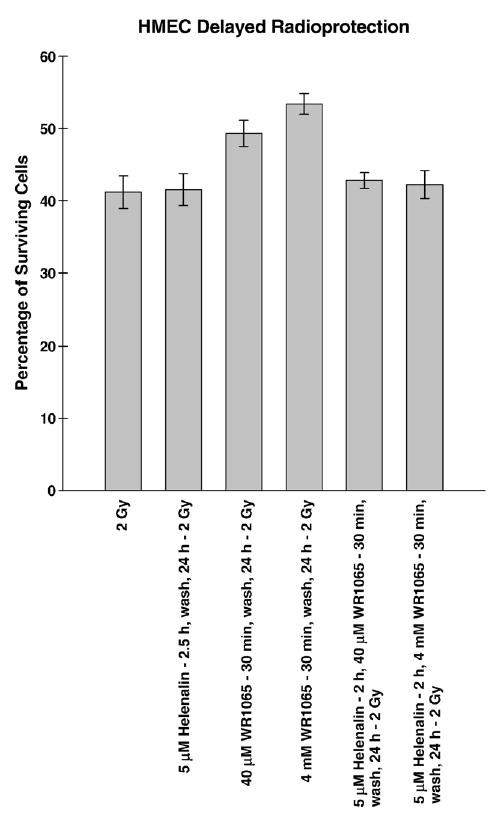
Bar graph depicting the percentage survival of HMEC exposed to 2 Gy of 250 kVp X-rays 24 h after exposure to 5 μM Helenalin for 2 h followed by a 30-min treatment with either 40 μM or 4 mM WR1065 contrasted with that of cells irradiated 24 h after WR1065 treatment alone. HMEC exposed to 2 Gy or irradiated 24 h after exposure to 5 μM Helenalin for 2.5 h are included for comparison purposes. Each bar represents the mean ± SE of two separate experiments.
Discussion
In earlier studies we reported that exposure of cells to WR1065 along with a number of other thiol-containing reducing agents resulted in the activation of NFκB and enhanced expression of MnSOD [18,23,24,33,34]. Using a supershift assay we demonstrated that WR1065 affected the p50–p65 heterodimer, but not homodimers or heterodimers containing p52 or c-Rel subunits of NFκB, and that neither catalase nor pyruvate when added to the culture medium to minimize hydrogen peroxide production could inhibit NFκB activation [18]. Furthermore, while both WR1065 and TNFα exposure led to an enhanced expression of MnSOD, WR1065, in contrast to TNFα, did not induce the expression of inflammatory genes known to have NFκB-binding motifs such as intercellular adhesion molecule-1 (ICAM-1) [18,34].
Manganese superoxide dismutase is an antioxidant enzyme that is localized in the mitochondria. When elevated in cells, it is known to confer among other things an enhanced resistance against the cytotoxic effects of ionizing radiation [4,8–10,23,24]. The MnSOD gene contains multiple transcription factor-binding motifs that include sites for AP1, AP2, Sp1, CREB, and NFκB [19–22]. As shown in Fig. 1, only NFκB appears to be susceptible to activation following exposure to WR1065, a thiol-containing drug having strong reductive capabilities. This finding is in agreement with the observations of others who demonstrated a similar specificity in NFκB activation in both normal and malignant cells following exposure to thiol-containing drugs such as N-acetyl- l-cysteine, dithiothreitol, 2-mercaptoethanol, WR1065, and oltipraz [14,16,18]. This is not surprising since NFκB is a well-characterized redox-sensitive transcription factor whose activation is known to be induced by both oxidative stress [14–18] and reducing agents that are capable of altering the redox state of the cysteine 62 residue of its p50 subunit [35]. Correlating with NFκB activation is the robust expression of the MnSOD gene. This has been demonstrated by others as well as our laboratory following exposure of cells to TNFα (see Figs. 2 and 3). While this gene contains several NFκB-binding motifs [19–21], it is an intronic NFκB site that has been identified as being most associated with a stress and/or redox-mediated inducible effect on MnSOD transcription [22].
We previously reported on the time course of changes in MnSOD gene expression in HMEC exposed to either 40 μM or 4 mM WR1065. Gene expression increased steadily as a function of time following WR1065 exposure and peaked between 18 and 22 h later [18]. Changes in MnSOD protein levels following WR1065 exposure reflected a similar pattern regardless of the mode of exposure used (see Figs. 2–4). If HMEC were exposed to 40 μM WR1065 continuously for 18 h or longer, MnSOD protein levels peaked at 22 h following the initiation of exposure but remained significantly elevated out to 90 h following the onset of exposure (Figs. 2 and 5). The activity gel presented in Fig. 3 showing the inhibition of the reduction of nitroblue tetrazolium by MnSOD demonstrates that this protein was active. A similar kinetic profile of MnSOD elevation, however, was also observed under conditions where HMEC were exposed to 40 μM WR1065 for only 30 min, after which time the drug was washed off and the cells were exposed to fresh medium (see Figs. 4 and 5). MnSOD increased to peak levels between 16 and 24 h after WR1065 exposure and remained elevated out to 48 h, the last time point evaluated.
Previous experiments to measure intracellular levels of WR1065 following exposure of cells in complete medium to micromolar amounts of the protector showed that WR1065 is rapidly depleted from the medium and concentrated within the cells [27]. When intracellular levels of drug were measured during incubation for 30 min in medium containing 40 μM WR1065, the level of WR1065 in the medium fell to less than 3 μM while the intracellular levels rose to 150 μM, indicating that cells can rapidly take up and concentrate WR1065 by about a factor of 50-fold within a 30-min period. Therefore, while two different modes of exposure were used, the rapid cellular uptake of WR1065 resulted in essentially similar effective exposures to the cells and similar kinetics of MnSOD elevation and persistence.
The stimulatory effect of WR1065 on elevation of MnSOD levels in HMEC, while somewhat similar in magnitude to that observed in mouse SA-NH tumor cells exposed under similar conditions, differed in that it appeared to be more persistent. Elevated MnSOD levels in SA-NH tumor cells returned to background levels by 48 h, while the effect remained at robust levels in HMEC even at time periods of 48 h or longer [23,24]. The prolonged duration of elevated MnSOD in the nontumor HMEC as compared to murine SA-NH tumor cells may be a reflection of the differential status of MnSOD in tumor as compared to normal cells. Cancer cells are generally lower in MnSOD activity than their normal tissue counterparts [36–38], and increasing MnSOD activity in transformed cells has been reported to lead to a reversal of the malignant phenotype [36–39]. This inherent deficiency of MnSOD in cancer as opposed to noncancer cells might account for the differences observed regarding the persistence of the effect following exposure of these two types of cells to WR1065.
The relationship(s) among WR1065 exposure, NFκB activation, and elevated MnSOD protein levels was further investigated using the NFκB inhibitor Helenalin. Preincubation of cells with 5 μM Helenalin was effective in not only inhibiting NFκB activation (see Fig. 7), but also in inhibiting elevation of MnSOD induced by WR1065 exposure (see Fig. 8).
MnSOD is known to confer radiation resistance to cells when elevated over control levels [4,5,8,9,38]. Therefore, it is reasonable to expect that cells experiencing increased MnSOD levels will also exhibit an enhanced radiation-resistant phenotype corresponding to the kinetics and relative magnitude of MnSOD elevation following exposure to an inducing agent such as the nonprotein thiol WR1065. Treatment of cells with WR1065 30 min prior to irradiation will significantly protect against radiation-induced cell killing in a dose-dependent manner, i.e., an immediate protective effect [1,2,13]. The data presented in Fig. 9 are consistent with these findings. To assess this immediate or classical radioprotective effect, resultant survival curves are routinely analyzed using a multitarget model to determine D0 values representing the reciprocals of the slopes of their terminal regions. A protection factor defined as the ratio of the D0’s of the radioprotector treated to the radiation only survival curves was determined for both treatment conditions. Cell survival in HMEC exposed to 4 mM WR1065 was elevated at all radiation dose points in contrast to the lack of a protective effect observed when the protector concentration was reduced to 40 μM, resulting in protection factors of 2.05 and 1.02, respectively.
In contrast, if HMEC were exposed to either dose of WR1065 for 30 min, then washed free of the protector and irradiated 24 h later corresponding to the time at which thiol-induced MnSOD levels were maximally elevated (see Figs. 2 and 4), cell survival was significantly enhanced regardless of the dose of WR1065 used (see Fig. 10). This cytoprotective effect is not due to WR1065′s ability to directly scavenge free radicals, but rather is a reflection of an induced delayed protective effect. Since MnSOD has been demonstrated by others to only affect the initial slope of the resulting survival curve, survival curves were analyzed using a α/β model in order to quantify changes in the initial slope. α protection factors were determined in this manner to facilitate comparisons between treatment groups (see Table 1). Alternatively, radiation protection ratios were also determined for all treatment groups using a multitarget model to calculate respective D0 values and survival values at a dose of 2 Gy, a radiation dose routinely used in radiation therapy (see Tables 1 and 2). Both the αPER and 2 Gy survival endpoint approaches demonstrate a novel WR1065-induced delayed radioprotective effect that is correlated in time with measurements of elevated MnSOD levels. This relationship is further strengthened by the demonstration that exposure of HMEC to Helenalin inhibited the ability of WR1065 exposure to activate NFκB (see Fig. 7), elevate MnSOD (see Fig. 8), and to induce the delayed radioprotective effect (see Fig. 11).
Qualitative differences in the survival responses of HMEC describing the immediate and delayed radioprotective effects of WR1065 can be attributed to differences in the intracellular localization of the responsible protective moieties. Immediate radioprotection of HMEC is the result of the localization of the free thiol WR1065 within two sensitive subcellular targets for radiation damage, the mitochondria and the nuclear DNA compartment [40,41]. If exposure to ionizing radiation occurs at this time, both of these sensitive targets can be protected. Since the relative effect on cell survival increases as the dose of radiation increases, the immediate radioprotective effect exhibited by WR1065 is clearly a dose-modifying effect. In contrast, the delayed radioprotective effect is the result of the induction of elevated levels of MnSOD by WR1065 only within the mitochondria of HMEC and it does not increase with increasing dose. However, the magnitude of the delayed effect is comparable to that of the immediate effect at low radiation doses in the range of 2 to 4 Gy. It is important to note that 2 Gy is the dose per fraction used in conventional radiotherapy regimens.
The delayed radioprotective effect induced by WR1065 was first reported in a murine sarcoma tumor model designated SA-NH [23,24]. The ability of nonprotein thiols such as WR1065 to induce activation of NFκB and enhance expression of MnSOD was not limited, however, to just mouse tumors and HMEC but has also been reported for pulmonary adenocarcinoma cells, human glioma tumor cells, and rat hepatocytes, suggesting that this may be a general phenomenon [14,16, 33,34]. Furthermore, the ability to induce the concomitant delayed radioprotective effect has been demonstrated for other MnSOD-inducing nonprotein thiols including N-acetyl- l-cysteine, captopril, and mesna [23], also suggesting that this is a general property for the NPT class of compounds.
These findings have implications both for patients undergoing radiation therapy and for radioprotection in general. Amifostine is currently the only radioprotective drug approved by the FDA for use in the clinical treatment of cancer by radiation therapy [2]. Its indication is limited for the use of reducing xerostomia in patients undergoing postoperative radiation treatment for head and neck cancer where the radiation port includes a substantial portion of the parotid glands. However, patients undergoing radiation therapy may be exposed to other nonprotein thiols such as captopril, mesna, or NAC. Captopril, for example, is an angiotensin-converting enzyme inhibitor that is widely used by millions of people in the treatment of hypertension [42]. Each of these nonprotein thiols can elicit a similar delayed radioprotective effect 24 h following their use. Since this effect is manifested primarily as an elevated radiation resistance to radiation doses routinely given in a radiation therapy setting, i.e., 2 Gy per fraction, the potential exists that patients undergoing a conventional course of radiation therapy may be compromised. Conversely, the persistent elevation of MnSOD in the normal tissues of individuals following their ingestion of low nontoxic doses of NPT such as cysteine might offer a novel approach to elevating their inherent radiation resistance. This would be especially important for workers in occupations that make them at risk for exposure to radiation sources such as radiation workers and first responders. Several caveats remain regarding the translational application of the delayed radioprotection phenomenon to such occupational populations. It is not at present clear whether a prolonged elevation of MnSOD can be maintained in normal cells following a chronic exposure to nonprotein thiols, or if such an elevated state were maintained whether concomitant catalase and/or glutathione peroxidase activity would be sufficient to prevent the buildup of toxic levels of hydrogen peroxide, a product of the dismutation of the superoxide radical. Regardless, the ability of nonprotein thiols represented by WR1065 to activate NFκB and subsequently elevate MnSOD levels in cells 24 h later that leads to a concomitant elevation of radiation resistance, i.e., the delayed radioprotective effect, is a novel discovery having important implications in the fields of radiation oncology and radioprotection in general.
Supplementary Material
Acknowledgments
The authors acknowledge Dr. Dingcai Cao, Biostatistical Consulting Laboratory, The University of Chicago, for his help with all of the statistical analyses. This work was supported by NIH/NCI R01 Grants CA37435 and CA99005, and DOE Grant DE-FG02-05ER64086 to D.J. Grdina.
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.freeradbiomed.2005.10.060.
References
- 1.Giambarresi, L.; Jacobs, A. J. In: Conklin, J. J., Walker, R. I., eds., Radioprotectants in military radiobiology. Academic Press, Orlando, FL, pp. 265–301; 1987.
- 2.Grdina DJ, Murley JS, Kataoka Y. Radioprotectants: Current status and new directions. Oncology. 2002;63(Suppl 2):2–10. doi: 10.1159/000067146. [DOI] [PubMed] [Google Scholar]
- 3.Sun J, Chen Y, Li M, Ge Z. Role of antioxidant enzymes on ionizing radiation resistance. Free Radic Biol Med. 1998;24(4):586–593. doi: 10.1016/s0891-5849(97)00291-8. [DOI] [PubMed] [Google Scholar]
- 4.Epperly MW, Gretton JE, Sikora CA, Jefferson M, Bernarding M, Nie S, Greenberger JS. Mitochondrial localization of superoxide dismutase is required for decreasing radiation-induced cellular damage. Radiat Res. 2003;160:568–578. doi: 10.1667/rr3081. [DOI] [PubMed] [Google Scholar]
- 5.Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, Ogi J, Khaletskiy A, Li Z, Weydert C, Longmate JA, Huang TT, Spitz DR, Oberley LW, Li JJ. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol Cell Biol. 2003;23(7):2362–2378. doi: 10.1128/MCB.23.7.2362-2378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zamzami N, Susin A, Marchetti P, Hirsh T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 8.Li JJ, Oberley LW. Overexpression of manganese superoxide dismutase confers resistance to the cytotoxicity of TNF-α and/or hyperthermia. Cancer Res. 1997;57:1991–1998. [PubMed] [Google Scholar]
- 9.Hirose K, Longo DL, Oppenheim JJ, Matsushima K. Over expression of mitochondrial manganese superoxide dismutase promotes the survival of tumor cells exposed to interleukin-1, tumor necrosis factor, selected anti-cancer drugs, and ionizing radiation. FASEB J. 1993;7:361–368. doi: 10.1096/fasebj.7.2.8440412. [DOI] [PubMed] [Google Scholar]
- 10.Epperly MW, Sikora CA, DeFilippi SJ, Gretton J, Zhan Q, Kufe DW, Greenberger JS. Manganese superoxide dismutase (SOD2) inhibits radiation-induced apoptosis by stabilization of the mitochondrial membrane. Radiat Res. 2002;157:568–577. doi: 10.1667/0033-7587(2002)157[0568:msdsir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.Brizel DM, Wasserman TH, Henke M, Strnad V, Rudat V, Monnier A, Eschwege F, Zhang J, Russell L, Oster W, Sauer R. Phase III randomized trial of amifostine as a radioprotector in head and neck cancer. J Clin Oncol. 2000;18(19):3339–3345. doi: 10.1200/JCO.2000.18.19.3339. [DOI] [PubMed] [Google Scholar]
- 12.Grdina DJ, Nagy B, Hill CK, Wells RL, Peraino C. The radioprotector WR1065 reduces radiation-induced mutations at the HGPRT locus in V79 cells. Carcinogenesis. 1985;6:929–931. doi: 10.1093/carcin/6.6.929. [DOI] [PubMed] [Google Scholar]
- 13.Grdina DJ, Kataoka Y, Murley JS. Amifostine: mechanisms of action underlying cytoprotection and chemoprevention. Drug Metab Drug Interact. 2002;16(4):1–43. doi: 10.1515/dmdi.2000.16.4.237. [DOI] [PubMed] [Google Scholar]
- 14.Das KC, Lewis-Molock Y, White CW. Activation of NFκB and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am J Physiol. 1995;269:L588–L602. doi: 10.1152/ajplung.1995.269.5.L588. [DOI] [PubMed] [Google Scholar]
- 15.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 16.Antras-Ferry J, Maheo K, Chevanne M, Dubos MP, Morel F, Guillouzo A, Cillard P, Cillard J. Oltipraz stimulates the transcription of the manganese superoxide dismutase gene in rat hepatocytes. Carcinogenesis. 1997;18:2113–2117. doi: 10.1093/carcin/18.11.2113. [DOI] [PubMed] [Google Scholar]
- 17.Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NFκB activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 18.Murley JS, Kataoka Y, Hallahan DE, Roberts JC, Grdina DJ. Activation of NFκB and MnSOD gene expression by free radical scavengers in human microvascular endothelial cells. Free Radic Biol Med. 2001;30(12):1426–1439. doi: 10.1016/s0891-5849(01)00554-8. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Porntadavity S, St Clair DK. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (SP1) and activating protein-2 (AP2) Biochem J. 2002;362(Pt 2):401–412. doi: 10.1042/0264-6021:3620401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borrello S, Demple B. NfκB independent transcriptional induction of the human manganese superoxide dismutase gene. Arch Biochem Biophys. 1997;348(2):289–294. doi: 10.1006/abbi.1997.0355. [DOI] [PubMed] [Google Scholar]
- 21.Porntadavity S, Xu Y, Kiningham K, Rangnekar VM, Prachayasitikul V, St Clair DK. TPA-activated transcription of the human MnSOD gene: role of transcription factors SP1 and Egr1. DNA Cell Biol. 2001;20(8):473–481. doi: 10.1089/104454901316976109. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Kiningham KK, Devalaraja MN, Yeh CC, Majima H, Kasarskis EJ, St Clair DK. An intronic NFκB element is essential for induction of human manganese superoxide dismutase gene by tumor necrosis factor-α and interleukin 1β. DNA Cell Biol. 1999;18(9):709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]
- 23.Murley JS, Kataoka Y, Cao D, Li JJ, Oberley LW, Grdina DJ. Delayed radioprotection by NFκB mediated induction of Sod2 (MnSOD) in SA-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res. 2004;162:536–546. doi: 10.1667/rr3256. [DOI] [PubMed] [Google Scholar]
- 24.Murley JS, Kataoka Y, Weydert CJ, Oberley LW, Grdina DJ. Delayed cytoprotection after enhancement of Sod2 (MnSOD) gene expression in SA-NH mouse sarcoma cells exposed to WR1065, the active metabolite of amifostine. Radiat Res. 2002;158:101–109. doi: 10.1667/0033-7587(2002)158[0101:dcaeos]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 25.MacMillan-Crow LA, Cruthirds DL. Manganese superoxide dismutase in disease. Free Radic Res. 1997;34:325–336. doi: 10.1080/10715760100300281. [DOI] [PubMed] [Google Scholar]
- 26.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 27.Grdina DJ, Shigematsu N, Dale P, Newton GL, Aguilera JA, Fahey RC. Thiol and disulfide metabolites of the radiation protector and potential chemopreventive agent WR2721 are linked to both its anti-cytotoxic and anti-mutagenic mechanisms of action. Carcinogenesis. 1995;16:767–774. doi: 10.1093/carcin/16.4.767. [DOI] [PubMed] [Google Scholar]
- 28.Shaw LM, Bonner HS, Schuchter L, Schiller J, Lieberman R. Pharmacokinetics of amifostine: effects of dose and method of administration. Semin Oncol. 1999;26(Suppl 7):34–36. [PubMed] [Google Scholar]
- 29.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 30.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Oberley LW, Spitz DR. Assay of superoxide dismutase activity in tumor tissue. Methods Enzymol. 1984;105:457–469. doi: 10.1016/s0076-6879(84)05064-3. [DOI] [PubMed] [Google Scholar]
- 32.Ly G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. The anti-inflammatory sesquiterpene lactone Helenalin inhibits the transcription factor NFκB by directly targeting p65. J Biol Chem. 1998;273(50):33508–33516. doi: 10.1074/jbc.273.50.33508. [DOI] [PubMed] [Google Scholar]
- 33.Grdina DJ, Murley JS, Kataoka Y, Calvin DP. Differential activation of nuclear transcription factor κB, gene expression, and proteins by amifostine’s free thiol in human microvascular endothelial and glioma cells. Semin Radiat Oncol. 2002;12(Suppl 1):103–111. doi: 10.1053/srao.2002.31383. [DOI] [PubMed] [Google Scholar]
- 34.Kataoka Y, Murley JS, Khodarev NN, Weichselbaum RR, Grdina DJ. Activation of the nuclear transcription factor (B (NFκB) and differential gene expression in U87 glioma cells after exposure to the cytoprotector amifostine. Int J Radiat Oncol Biol Phys. 2002;53(1):180–189. doi: 10.1016/s0360-3016(01)02820-6. [DOI] [PubMed] [Google Scholar]
- 35.Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT. Thioredoxin regulates the DNA binding activity of NFκB by reduction of the disulphide bond involving cysteine 62. Nucleic Acids Res. 1992;20:3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oberley LW, Oberley TD. Role of antioxidant enzymes in cell immortalization and transformation. Mol Cell Biochem. 1988;84:147–153. doi: 10.1007/BF00421049. [DOI] [PubMed] [Google Scholar]
- 37.Oberley, T. D.; Oberley, L. W. Oxygen radicals and cancer. In: Yu, B. P., ed., Free radicals in aging. CRC Press, Boca Raton, FL, pp. 248–267; 1993.
- 38.Urano M, Kuroda M, Reynolds R, Oberley TD, St Clair DK. Expression of manganese superoxide dismutase reduces tumor control radiation dose: gene therapy. Cancer Res. 1995;55:2490–2493. [PubMed] [Google Scholar]
- 39.St Clair DK, Wan XS, Oberley TD, Muse KE, St Clair WH. Suppression of radiation-induced neoplastic transformation by over-expression of mitochondrial superoxide dismutase. Mol Carcinog. 1992;6:238–242. doi: 10.1002/mc.2940060404. [DOI] [PubMed] [Google Scholar]
- 40.Smoluk GD, Fahey RC, Ward JF. Equilibrium dialysis studies of the binding of radioprotector compounds to DNA. Radiat Res. 1986;107(2):194–204. [PubMed] [Google Scholar]
- 41.Tretter L, Ronai E, Szabados G, Hermann R, Ando A, Horvath I. The effect of the radioprotector WR2721 and WR1065 on mitochondrial lipid peroxidation. Int J Radiat Biol. 1990;57(3):467–478. doi: 10.1080/09553009014552611. [DOI] [PubMed] [Google Scholar]
- 42.Bicket DP. Using ACE inhibitors appropriately. Am Fam Physician. 2002;66:461–468. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.