


Abstract
Several homeodomains and homeodomain-containing proteins enter live cells through a receptor- and energy-independent mechanism. Translocation through biological membranes is conferred by the third α-helix of the homeodomain, also known as Penetratin. Biophysical studies demonstrate that entry of Penetratin into cells requires its binding to surface lipids but that binding and translocation are differentially affected by modifications of some physico-chemical properties of the peptide, like helical amphipathicity or net charge. This suggests that the plasma membrane lipid composition affects the internalization of Penetratin and that internalization requires both lipid binding and other specific properties. Using a phase transfer assay, it is shown that negatively charged lipids promote the transfer of Penetratin from a hydrophilic into a hydrophobic environment, probably through charge neutralization. Accordingly, transfer into a hydrophobic milieu can also be obtained in the absence of negatively charged lipids, by the addition of DNA oligonucleotides. Strikingly, phase transfer by charge neutralization was also observed with a variant peptide of same charge and hydrophobicity in which the tryptophan at position 6 was replaced by a phenylalanine. However, Penetratin, but not its mutant version, is internalized by live cells. This underscores that charge neutralization and phase transfer represent only a first step in the internalization process and that further crossing of a biological membrane necessitates the critical tryptophan residue at position 6.
INTRODUCTION
Homeoproteins define a large class of transcription factors sharing an evolutionary conserved DNA-binding domain, the homeodomain (1). Homeodomains fold into a globular structure made of two antiparallel α-helices followed by a third, orthogonal one (2–5). Upon DNA binding, the third α-helix of the homeodomain fits into the major groove of cognate DNA sites where amino acid residues make specific contacts with the base pairs. In addition, the extended N-terminal arm of the homeodomain specifically interacts with base pairs via the double helix minor groove.
In addition to DNA binding, the homeodomain achieves other functions like binding to RNA (6), making protein-to-protein contacts (4,5,7–11) and acting as a transcriptional activation or repression domain (12–15). Among the new roles discovered for some homeodomains, the most unforeseen one is their involvement in secretion and internalization, thus intercellular trafficking, of homeoproteins (16–22). Homeoprotein and homeodomain internalization property is conferred by a 16-amino acid long peptide corresponding to the third α-helix of the homeodomain (19). Homeoprotein secretion is dependent upon another sequence overlapping with helices 2 and 3 of the homeodomain and presenting a nuclear export signal activity (20,21). These data strongly suggest a paracrine function for some homeoproteins and show that the homeodomain provides the necessary signals for internalization and secretion. However, the exact cellular and molecular mechanisms underlying intercellular trafficking are not fully understood.
The third α-helix of the Antennapedia homeodomain, hereafter referred to as Penetratin, is internalized by several cell types at 37°C and at 4°C and reaches the cell nucleus (23). Retro, enantio and retro-inverso forms of Penetratin are also efficiently captured at 4°C, demonstrating that cellular entry is energy and receptor independent (23). In the proposed model of internalization through the transient formation of inverted micelles (17,24), the interaction between negatively charged phospholipids and Penetratin is critical. Indeed, Penetratin contains seven arginine or lysine residues, and an alanine scan mapping has shown that the mutation of any of these residues significantly reduces cellular entry (25).
Many experiments, however, suggest that the lipid-binding affinity of Penetratin and of its internalized variants is not sufficient to explain their capacity to enter live cells (25,26). Modulating the hydrophobicity and the hydrophobic moment of the peptide differently affects its binding to lipid bilayer models depending on their content in charged lipids. In addition, there is no correlation between the helical amphipathicity of the variant peptides and their internalization. The substitution of several hydrophobic residues causes a decrease in Penetratin internalization. Of particular interest is the almost full loss of Penetratin internalization that follows the change of tryptophan 6 residue (W6) into a phenylalanine (W6F mutant) (16,19).
A very recent study addressing the mode of interaction of Penetratin with lipid bilayers provided evidence that wild type and W6F mutant Penetratins only interact with negatively charged phospholipids, therefore showing that peptide–lipid association is primarily due to electrostatic interactions (27). Furthermore, Christiaens et al. (27) showed that the W6 residue deeply inserts into the lipid bilayer, and that the W6F Penetratin mutant shows lower affinity for lipids than wild type Penetratin.
Here, we compare the ability of Penetratin and its W6F mutant variant to reach a hydrophobic environment through charge neutralization, by a simple phase transfer assay. We also compare the capacity of these peptides to promote the entry of a non-conjugated anionic polymer, i.e. a double-stranded DNA (ds DNA) oligonucleotide, into live cells. While the peptides are equally competent for the phase transfer of negatively charged molecules, only the wild type Penetratin allows the cellular uptake of DNA in the absence of endocytosis, at 4°C. This strongly suggests that internalization detected in the presence of Penetratin is a two step event that requires (i) Penetratin binding to the cell surface, basically by electrostatic interactions with lipids, and (ii) translocation across the lipid bilayer possibly through a tryptophan-induced destabilization of the membrane.
MATERIALS AND METHODS
Phase transfer assay
Phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and phosphatidic acid (PA) were conditioned in chloroform. Penetratin (sequence: 6 NH2-RQIKIWFQNRRMKWKK-CONH2), the W6-to-F6 Penetratin mutant (W6F) and NBD-Penetratin were synthesized and provided by Synt:em SA (Nîmes, France). Peptides were blocked at both N- and C-terminal extremities and were >98% pure, as monitored by HPLC. Peptide aliquots were conditioned in Tris 10 mM, pH 7.4. DNA oligonucleotides (5′-CGAAATTGATTGATTGATGCTTTAATTGAC-3′ and 5′-GTCAATTAAAGCATCAATCAATCAATTTCG-3′; Eurogentec) were end labeled by T4 polynucleotide kinase (NEBiolabs) and 32P-dCTP (Amersham), as recommended by the manufacturer.
For the phase transfer assays, 1 ml of the water phase (Tris 10 mM, pH 7.4), containing the peptide, and/or dsDNA oligonucleotides when indicated, was mixed to 1 ml of organic phase, with or without lipids, by vortexing (30 s). Both phases were decanted overnight at 4°C or separated by centrifugation (800 g, 15 min). The NBD-Penetratin recovered from the water phase was quantified by fluorimetry (Perkin Elmer LS30 fluorimeter). NBD emits fluorescence at a wavelength of 537 nm after excitation at 464 nm. 32P-labeled oligonucleotides retrieved from both phases were dosed by radioactivity counting (liquid scintillation in Ultima-Gold or Pico-Fluor 40, Packard).
Cellular internalization of Penetratin and DNA oligonucleotides
COS7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal calf serum (PAA laboratories, GmbH), 100 IU/ml of penicillin and 100 µg/ml of streptomycin (Sigma), at 37°C in a humidified, 5% CO2 atmosphere. Twenty-four hours before peptide, DNA or peptide–DNA internalization, cells proliferating exponentially were trypsinized and 3 or 6 × 104 cells were plated per well in 24-well plates. Before adding the peptide (Penetratin or the W6F variant), DNA, or peptide–DNA mix, cells were rinsed twice in phosphate buffered saline (PBS). Peptides, DNA or peptide–DNA mix were conditioned in DMEM (without serum and antibiotics) and added to the cells for 2 h at 37 or 4°C.
To quantify the entry of radioactively labeled DNA into cells, the culture medium was collected and processed for liquid scintillation counting, the cells were washed twice with PBS-NaCl 0.5 M and PBS, trypsin treated and lysed in Triton ×100 2% for counting the internalized radioactivity. To visualize internalized FITC-labeled oligonucleotides (Gibco BRL Life Technologies), cells grown on glass coverslips were exposed to DNA or to peptide–DNA mix, rinsed, fixed in 4% paraformaldehyde and mounted in Vectashield mounting medium (Vector laboratories). Oligonucleotides were protected from degradation by phosphorothioate bonds. Intracellular distribution of DNA was determined by confocal microscopy (Axiovert 135 M, Zeiss).
RESULTS
Charged phospholipids allow the transfer of Penetratin into a hydrophobic environment
To evaluate the relationship between the nature of phospholipids and their ability to interact with Penetratin, we followed the transfer of the peptide from an aqueous into a hydrophobic phase, i.e. chloroform, in the presence of different lipids. Penetratin was N-terminally labeled with a NBD fluorophore and phase transfer was quantified by measuring the residual NBD fluorescence in the water phase. In the absence of lipid, about one-half of added NBD-Penetratin (2.2 µM) was detected in the water phase (relative fluorescence of 100; see Fig. 1). The addition of zwitterionic lipids (PC or PE) to the organic phase, with a lipid:peptide molar ratio of 17:1, slightly decreased (up to two-fold) NBD-Penetratin phase transfer (Fig. 1).
Figure 1.

Negatively charged lipids promote the water-to-hydrophobic phase transfer of Penetratin. NBD-Penetratin was dissolved in Tris 10 mM, pH 7.4, to reach a concentration of 2.2 µM. Lipids were conditioned in chloroform. After mixing and decanting the phases, the NBD fluorescence corresponding to the NBD-Penetratin remaining in the water phase was quantified. Bars indicate the standard variation of at least three independent experiments.
In strong contrast, negatively charged PS or PA enhanced the transfer of NBD-Penetratin into the hydrophobic phase. As illustrated in Figure 1, the addition of PS and PA (lipid:peptide molar ratio of 17:1) decreased water-soluble NBD-Penetratin to 15 and 50% of that measured in control conditions, respectively.
Varying the lipid:peptide molar ratio from 2 × 10–3:1 to 34:1, we observed a maximal NBD-Penetratin transfer in the presence of PS when reaching a 8:1 molar ratio (Fig. 2). Considering that NBD-Penetratin harbors seven positive charges at pH 7.2, optimal peptide transfer occurs when reaching a lipid:peptide ratio that allows charge neutralization. Also illustrated in Figure 2 is the absence of NBD-Penetratin phase transfer in presence of PC, even at very high lipid:peptide ratios (up to 34:1). Rather, addition of PC impaired phase transfer of the peptide. Taken together, these experiments suggest that peptide–lipid association is primarily due to electrostatic interactions and that charge neutralization is the first step allowing the peptide to enter into a hydrophobic environment.
Figure 2.

PS induces an optimal phase transfer of Penetratin when reaching a lipid:peptide molar ratio of 8:1. NBD-Penetratin was dissolved in Tris 10 mM, pH 7.4, to reach a concentration of 4.4 µM. PS and PC were conditioned in chloroform. After mixing and decanting the phases, the NBD fluorescence corresponding to the NBD-Penetratin remaining in the water phase was quantified. Bars indicate the standard variation of at least three independent experiments.
Penetratin promotes the transfer of non-conjugated DNA oligonucleotides into a hydrophobic environment
If phase transfer is due to Penetratin charge neutralization by anionic lipids, oligonucleotides, as anionic polymers, might bind Penetratin and find access, through peptide neutralization, to the hydrophobic phase. In the absence of peptide, ∼60% of added radioactive dsDNA (16 pM) remained in the water phase (Fig. 3). However, the addition of Penetratin (4.4 µM) allowed the passage of up to 80% of the dsDNA oligonucleotides into the hydrophobic phase (Fig. 3). Similarly, Penetratin also enhanced phase transfer of single-stranded DNA oligonucleotides, but with a somewhat lower efficiency (data not shown).
Figure 3.

Penetratin allows the transfer of DNA oligonucleotides to a hydrophobic environment. Various amounts of Penetratin (Pen) or W6F mutant peptide (PenWF) were added to 32P-labeled dsDNA at 16 pM, conditioned in Tris 10 mM pH 7.4. PS was conditioned in chloroform. After mixing and decanting the phases, the radioactivity retrieved from both phases was counted and reported to that of the input (relative c.p.m. of 100). Only the data corresponding to the water phase are shown. Bars indicate the standard variation of at least three independent experiments.
The charge neutralization hypothesis was further tested by a competition experiment where the peptides and PS were added together. Figure 3 illustrates that the addition of PS, at a concentration of 37 µM, inhibits oligonucleotide transfer when Penetratin was used at 0.44 µM, compared with a transfer of 60% of the oligonucleotide in the absence of lipid. However, a high Penetratin concentration (4.4 µM) restored oligonucleotide transfer to levels similar to those observed in the absence of PS. Taken together these results suggest that PS competes with the oligonucleotide for Penetratin binding. It is noteworthy that, even in the absence of PS, Penetratin-mediated transfer of DNA was effective only in presence of a large excess of peptide.
Penetratin enhances the cellular entry of non-conjugated DNA both at 37°C and 4°C
Since Penetratin promoted the passage of anionic polymers, like DNA, to a hydrophobic phase, we postulated that it may also trigger the entry of non-conjugated DNA into live cells. COS7 cells were exposed to DNA pre-incubated, or not, with Penetratin. Radioactive dsDNA (6.5 nM) was maintained constant and diluted with non-labeled dsDNA (up to 65 nM final). Penetratin and DNA were mixed in a molar ratio varying from 5 × 102 to 5 × 103 and added to the cells. After 2 h, the culture medium was recovered for counting (data not shown) and the intracellular amount of radioactive material was evaluated after washings and trypsin treatment.
Although Penetratin stimulated the entry of DNA at all DNA concentrations, optimal internalization was obtained with 6.5 nM of dsDNA and a 5 × 103 molar excess of Penetratin (32 µM; Fig. 4). At 4°C, Penetratin induced a 60-fold increase in DNA internalization with respect to a control situation (no peptide added). At 37°C, the amount of internalized DNA was higher but the Penetratin effect was 30-fold, suggesting that, at this temperature, part of the oligonucleotide is captured through classical endocytosis. Internalization experiments were also performed with FITC-labeled DNA (0.2–2 µM), in the presence or in the absence of Penetratin (100 µM). Interestingly, in the presence of Penetratin, a strong signal was detected within the nucleus of all the cells following exposure at 4°C (confocal microscopy, Fig. 5). Thus, the Penetratin peptide is effective to carry along non-conjugated DNA into cells.
Figure 4.

Penetratin promotes the cellular entry of non-conjugated DNA oligonucleotides both at 37 and at 4°C. 32P-labeled dsDNA oligonucleotide (6.5 nM) was preincubated in DMEM medium in the absence of peptide (control), in the presence of Penetratin (Pen) (32 µM) or in the presence of the W6F mutant peptide (PenWF) (32 µM), and added to COS7 cells for 2 h. Cells were then lysed and processed to quantify the internalized radioactivity. Bars indicate the standard variation of at least three independent experiments.
Figure 5.
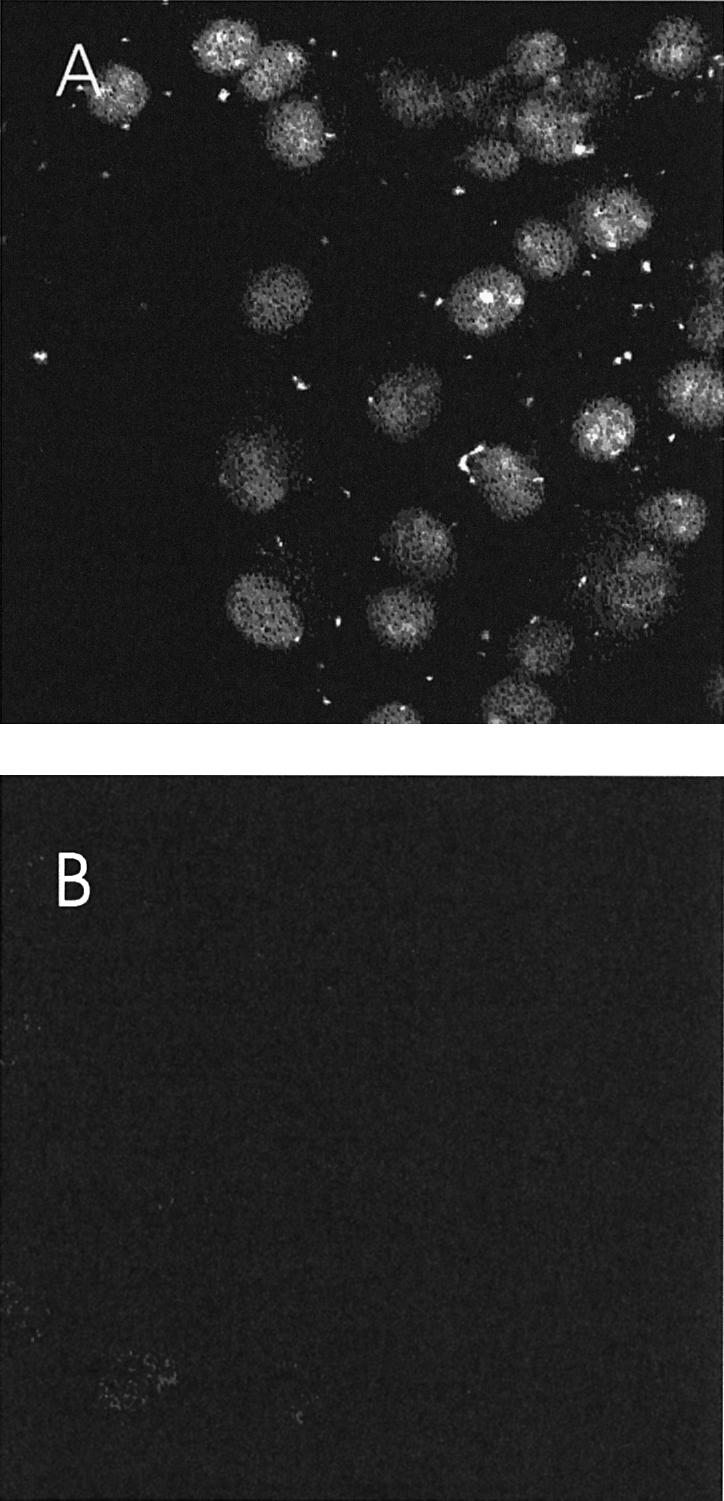
Internalized DNA oligonucleotides reach the nucleus of COS7 cells at 4°C. Penetratin (100 µM) and/or FITC-labeled dsDNA oligonucleotides (0.2 µM) were preincubated in DMEM medium and added to COS7 cells for 2 h at 4°C. Cells were then processed for visualization under confocal microscopy. (A) Intracellular fluorescence detected after incubation with Penetratin and FITC dsDNA, at 4°C. (B) Intracellular fluorescence detected after incubation at 4°C with FITC dsDNA oligonucleotides alone.
The translocation-defective W6F Penetratin mutant allows the phase transfer but not the cellular entry of non-conjugated DNA
The translocation-defective W6F Penetratin mutant (19) harbors the same net charge and hydrophobicity as the wild type peptide. We tested whether it can promote the transfer of DNA to a hydrophobic phase through charge neutralization. As observed with the wild type peptide, the W6F mutant allowed the passage of 80% of the dsDNA oligonucleotides into the hydrophobic phase (Fig. 3). Moreover, addition of PS in the assay competed with the oligonucleotide transfer. This inhibitory effect was quantatively similar to that observed with the wild type Penetratin, and was also released at higher peptide concentration (4.4 µM; Fig. 3). This shows that Penetratin and its W6F mutant derivative equally set up electrostatic interactions with negatively charged molecules.
The W6F mutant peptide was therefore assayed for its capacity to stimulate the cellular uptake of oligonucleotides. While Penetratin enhanced the entry of DNA into cells at both 4 and 37°C, its W6F variant only promoted DNA internalization at 37°C (15-fold stimulation as compared with control, Fig. 4). Indeed, at 4°C, the stimulatory effect observed with Penetratin was almost lost when the W6 residue was changed into F (from 60- to 6-fold, Fig. 4). Thus, the W6F amino acid substitution, which did not affect the capacity of the peptide to enter a hydrophobic milieu through charge neutralization, impaired its ability to cross biological membranes. This therefore strongly suggests that internalization at 37°C has a strong endocytotic component stimulated by the peptide–DNA complex but that the uptake of DNA molecules in the absence of endocytosis is dependent on the presence of the wild type peptide.
DISCUSSION
Penetratin is an amphipathic peptide that can cross biological membranes and gain access to the cytoplasm as well as to the nucleus of live cells. This cellular uptake occurs efficiently despite the net positive charge of the peptide. By the use of a phase transfer assay, we provided evidence that Penetratin may enter a hydrophobic environment upon interaction with negatively charged molecules, like PS or PA. Accordingly, Penetratin may bring hydrophilic, negatively charged polymers like DNA oligonucleotides into a hydrophobic phase, presumably by virtue of charge neutralization. In fact, the negatively charged lipid PS, which permits phase transfer of Penetratin, inhibits to some extent that of Penetratin-DNA, as shown by PS versus DNA competition experiments.
The W6F Penetratin variant has been shown to be impaired for translocation through biological membranes (16,19). In addition, it has been reported recently that Penetratin and the W6F mutant, which still harbors a tryptophan residue at position 14, displayed similar fluorescence emission spectra in the presence of lipid vesicles indicative of a low affinity of the peptides for zwitterionic lipids and of an equal interaction with negatively charged phospholipids (27). This suggests a significant role for electrostatic interactions in lipid–peptide binding. Christiaens et al. (27) further provided evidence that the W6 residue of Penetratin was deeply inserted into the lipid bilayer in vesicles. Finally, comparing the W6F mutant with another variant in which the tryptophan residue at position 14 was also replaced by phenylalanine, these authors showed that the W6 residue displayed a tighter association with the lipids than the W14 residue. Since the W6F Penetratin mutant is defective for the entry into cells (16,19), these recent data together point to a possible involvement of the W6 residue in the interaction with the lipid acyl chains to destabilize the bilayer.
Although the phase transfer assay is only a simplified model of the interaction between the peptides and biological membrane, our results demonstrate that the W6F mutant displays the same capacity as Penetratin to interact with negatively charged molecules and to promote their phase transfer. However, Penetratin also strongly enhances the entry of dsDNA into cells in the absence of endocytosis, whereas the W6F mutant is devoid of such activity. Since the cellular uptake of DNA occurs at 4°C in the presence of Penetratin, but not of the W6F mutant peptide, it is highly probable that DNA enters the cell through the Penetratin translocation route.
Some reports on the intracellular delivery of cationic cell-penetrating peptides and of oligonucleotides suggest that cell washing and fixation cause artifactual uptake or intracellular distribution of these molecules (28–30). This is not the case here for the following reasons. First, fluorescent Antennapedia-like peptides can be localized within unfixed live cells by confocal microscopy (31). Second, the wild type peptide, but not the W6F variant, is internalized and internalizes dsDNA (this study), and is retrieved from live cells by cell fractionation (19). Third, the activity of cargoes linked to the peptide occurs in live cells (reviewed in 32). Fourth, the quantitation of Penetratin-DNA entry into cells (this study) did not involve cell fixation and the washing steps included a trypsin treatment, thus precluding that peptides bound to the extracellular membrane are internalized (30). Fifth, the mild fixative protocol we used to detect FITC-labeled oligonucleotides was reported not to influence the uptake and intracellular localization of oligonucleotides (28). In conclusion, although some redistribution during fixation cannot be entirely precluded, we are confident that peptide and dsDNA entry and nuclear accumulation is not an artifact.
Altogether our data fit with a model suggesting that the entry of Penetratin into cells requires at least two consecutive events (16,19,33, and references therein). Firstly, the peptide should interact with and enter the membrane bilayer. This first step would depend on the hydrophobicity and the hydrophobic moment of the peptide, as well as on charge neutralization, for which both Penetratin and the W6F variant are similar. The second step should involve membrane destabilization and further intracellular delivery, for which the W6 residue seems to play a critical role. As proposed by others, the Penetratin peptide may enter live cells by the transient formation of inverted micelles (16,17,24) provoked by the tryptophan in position 6 (W6). Accordingly, it has been suggested that the substitution of the W6 residue into F6 would impair this transient membrane destabilization.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a BIOTECH grant from the European Commission (BIO4 CT98-0227). C.M. holds a PhD fellowship from the FRIA (National Fund for Scientific Research, FNRS, Belgium). O.B. is supported by the ‘Ministère de la Région Wallonne’ contract no. 14540 (PROTMEM). M.-P.M.-L. is supported by the ‘Ministère de la Région Wallonne’ (contract NANOMEMB) and the ‘Université Catholique de Louvain’ (FSR2002). M.-P.M.-L. is Senior Research Associate at the FNRS. R.B. is Research Director at the FNRS. R.R. is Postdoctoral Researcher of the FNRS.
REFERENCES
- 1.Banerjee-Basu S., Ryan,J.F. and Baxevanis,A.D. (2000) The homeodomain resource: a prototype database for a large protein family. Nucleic Acids Res., 28, 329–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gehring W.J., Qian,Y.Q., Billeter,M., Furukubo-Tokunaga,K., Schier,A.F., Resendez-Perez,D., Affolter,M., Otting,G. and Wuthrich,K. (1994) Homeodomain-DNA recognition. Cell, 78, 211–223. [DOI] [PubMed] [Google Scholar]
- 3.Wolberger C. (1996). Homeodomain interactions. Curr. Opin. Struct. Biol., 6, 62–68. [DOI] [PubMed] [Google Scholar]
- 4.Piper D.E., Batchelor,A.H., Chang,C.P., Cleary,M.L. and Wolberger,C. (1999) Structure of a HoxB1-Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell, 96, 587–597. [DOI] [PubMed] [Google Scholar]
- 5.Passner J.M., Ryoo,H.D., Shen,L., Mann,R.S. and Aggarwal,A.K. (1999) Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex. Nature, 397, 714–719. [DOI] [PubMed] [Google Scholar]
- 6.Niessing D., Driever,W., Sprenger,F., Taubert,H., Jackle,H. and Rivera-Pomar,R. (2000) Homeodomain position 54 specifies transcriptional versus translational control by Bicoid. Mol. Cell., 5, 395–401. [DOI] [PubMed] [Google Scholar]
- 7.Kim Y.H., Choi,C.Y., Lee,S.J., Conti,M.A. and Kim,Y. (1998) Homeodomain-interacting protein kinases, a novel family of co- repressors for homeodomain transcription factors. J. Biol. Chem., 273, 25875–25879. [DOI] [PubMed] [Google Scholar]
- 8.Ohneda K., Mirmira,R.G., Wang,J., Johnson,J.D. and German,M.S. (2000) The homeodomain of PDX-1 mediates multiple protein–protein interactions in the formation of a transcriptional activation complex on the insulin promoter. Mol. Cell. Biol., 20, 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zappavigna V., Falciola,L., Helmer-Citterich,M., Mavilio,F. and Bianchi,M.E. (1996) HMG1 interacts with HOX proteins and enhances their DNA binding and transcriptional activation. EMBO J., 15, 4981–4991. [PMC free article] [PubMed] [Google Scholar]
- 10.Zappavigna V., Sartori,D. and Mavilio,F. (1994) Specificity of HOX protein function depends on DNA–protein and protein–protein interactions, both mediated by the homeo domain. Genes Dev., 8, 732–744. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H., Hu,G., Wang,H., Sciavolino,P., Iler,N., Shen,M.M. and Abate-Shen,C. (1997) Heterodimerization of Msx and Dlx homeoproteins results in functional antagonism. Mol. Cell. Biol., 17, 2920–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H., Catron,K.M. and Abate-Shen,C. (1996). A role for the Msx-1 homeodomain in transcriptional regulation: residues in the N-terminal arm mediate TATA binding protein interaction and transcriptional repression. Proc. Natl Acad. Sci. USA, 93, 1764–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnabel C.A. and Abate-Shen,C. (1996) Repression by HoxA7 is mediated by the homeodomain and the modulatory action of its N-terminal-arm residues. Mol. Cell. Biol., 16, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang N., Shen,W., Ho,A.D. and Lu,M. (1996) Three distinct domains in the HOX-11 homeobox oncoprotein are required for optimal transactivation. Oncogene, 13, 1781–1787. [PubMed] [Google Scholar]
- 15.Li X., Murre,C. and McGinnis,W. (1999) Activity regulation of a Hox protein and a role for the homeodomain in inhibiting transcriptional activation. EMBO J., 18, 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prochiantz A. (1996) Getting hydrophilic compounds into cells: lessons from homeopeptides. Curr. Opin. Neurobiol., 6, 629–634. [DOI] [PubMed] [Google Scholar]
- 17.Prochiantz A. (2000) Messenger proteins: homeoproteins, TAT and others. Curr. Opin. Cell Biol., 12, 400–406. [DOI] [PubMed] [Google Scholar]
- 18.LeRoux I., Joliot,A.H., Bloch-Gallego,E., Prochiantz,A. and Volovitch,M. (1993) Neurotrophic activity of the Antennapedia homeodomain depends on its specific DNA-binding properties. Proc. Natl Acad. Sci. USA, 90, 9120–9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derossi D., Joliot,A.H., Chassaing,G. and Prochiantz,A. (1994) The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem., 269, 10444–10450. [PubMed] [Google Scholar]
- 20.Joliot A., Maizel,A., Rosenberg,D., Trembleau,A., Dupas,S., Volovitch,M. and Prochiantz,A. (1998) Identification of a signal sequence necessary for the unconventional secretion of Engrailed homeoprotein. Curr. Biol., 8, 856–863. [DOI] [PubMed] [Google Scholar]
- 21.Maizel A., Bensaude,O., Prochiantz,A. and Joliot,A. (1999) A short region of its homeodomain is necessary for engrailed nuclear export and secretion. Development, 126, 3183–3190. [DOI] [PubMed] [Google Scholar]
- 22.Chatelin L., Volovitch,M., Joliot,A.H., Perez,F. and Prochiantz,A. (1996) Transcription factor hoxa-5 is taken up by cells in culture and conveyed to their nuclei. Mech. Dev., 55, 111–117. [DOI] [PubMed] [Google Scholar]
- 23.Derossi D., Calvet,S., Trembleau,A., Brunissen,A., Chassaing,G. and Prochiantz,A. (1996) Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J. Biol. Chem., 271, 18188–18193. [DOI] [PubMed] [Google Scholar]
- 24.Berlose J.P., Convert,O., Derossi,D., Brunissen,A. and Chassaing,G. (1996) Conformational and associative behaviours of the third helix of antennapedia homeodomain in membrane-mimetic environments. Eur. J. Biochem., 242, 372–386. [DOI] [PubMed] [Google Scholar]
- 25.Drin G., Mazel,M., Clair,P., Mathieu,D., Kaczorek,M. and Temsamani,J. (2001) Physico-chemical requirements for cellular uptake of pAntp peptide. Role of lipid-binding affinity. Eur. J. Biochem., 268, 1304–1314. [DOI] [PubMed] [Google Scholar]
- 26.Scheller A., Wiesner,B., Melzig,M., Bienert,M. and Oehlke,J. (2000) Evidence for an amphipathicity independent cellular uptake of amphipathic cell-penetrating peptides. Eur. J. Biochem., 267, 6043–6050. [DOI] [PubMed] [Google Scholar]
- 27.Christiaens B., Symoens,S., Vanderheyden,S., Engelborghs,Y., Joliot,A., Prochiantz,A., Vandekerckhove,J., Rosseneu,M. and Vanloo,B. (2002) Tryptophan fluorescence study of the interaction of penetratin peptides with model membranes. Eur. J. Biochem., 269, 1–9. [DOI] [PubMed] [Google Scholar]
- 28.Pichon C., Monsigny,M. and Roche,A.C. (1999) Intracellular localization of oligonucleotides: influence of fixative protocols. Antisense Nucleic Acid Drug Dev., 9, 89–93. [DOI] [PubMed] [Google Scholar]
- 29.Lundberg M. and Johansson,M. (2002) Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem. Biophys. Res. Commun., 291, 367–371. [DOI] [PubMed] [Google Scholar]
- 30.Richard J.P., Melikov,K., Vives,E., Ramos,C., Verbeure,B., Gait,M.J., Chernomordik,L.V. and Lebleu,B. (2002) Cell-penetrating peptides: a re-evaluation of the mechanism of cellular uptake. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 31.Joliot A., Pernelle,C., Deagostini-Bazin,H. and Prochiantz,A. (1991) Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl Acad. Sci. USA, 88, 1864–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derossi D., Chassaing,G. and Prochiantz,A. (1998) Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol., 8, 84–87. [PubMed] [Google Scholar]
- 33.Bouffioux O., Basyn,F., Rezsohazy,R. and Brasseur,R. (2002) Structure prediction of CPPs and iterative development of novel CPPs. In Langel,U. (ed.), Cell Penetrating Peptides. CRC Press LLC, Boca Raton, FL, USA, pp. 187–222.