


Abstract
Thermotoga neapolitana (Tne) DNA polymerase belongs to the DNA polymerase I (Pol I) family. The O-helix region of these polymerases is involved in dNTP binding and also plays a role in binding primer–template during DNA synthesis. Here we report that mutations in the O-helix region of Tne DNA polymerase (Arg722 to His, Tyr or Lys) almost completely abolished the enzyme’s ability to catalyze the template-independent addition of a single base at the 3′-end of newly synthesized DNA in vitro. The mutations did not significantly affect the DNA polymerase catalytic activity and reduced base misinsertions 5- to 50-fold. The same Arg722 mutations dramatically increased the ability of the enzyme’s 3′→5′ exonuclease to remove mispaired 3′ bases in a primer extension assay. These mutant DNA polymerases can be used to accurately amplify target DNA in vitro for gene cloning and genotyping analysis.
INTRODUCTION
Based upon its primary amino acid sequence and catalytic mechanism, Thermotoga neapolitana (Tne) DNA polymerase belongs to the DNA polymerase I (Pol I) family. Similar to Escherichia coli DNA polymerase I, but unlike Taq DNA polymerase, Tne DNA polymerase contains both 3′→5′ and 5′→3′ exonulease activity. Crystal structures of E.coli DNA polymerase I (1,2), Taq DNA polymerase (3) and Bacillus stearothermophilus DNA polymerase I (4) delineate the organization of these three separate domains. Comparison of the structures of all known DNA polymerases, including distantly related reverse transcriptase, show a similar topology and organization of the polymerase active site (5,6). The structural alignment of different polymerases combined with mutagenesis studies of the Klenow fragment of Pol I of E.coli indicate that the polymerase active site residues are located primarily in the palm region (7). The dNTP-binding region is located in the fingers sub-domain encompassing the exposed face of the O-helix (residues 754–766 of Klenow fragment) and in an adjacent region of the palm domain (8). Extensive biochemical, genetic and mutagenesis studies combined with crystallographic data have helped to elucidate the overall mechanism of and the role of individual amino acids in DNA synthesis (5–9).
Maintaining the fidelity of DNA synthesis inside the living cell is of fundamental importance in biology for accurate transmission of genetic information. In this process, DNA polymerase must select and incorporate correct nucleotides and extend a correctly base paired primer terminus during DNA synthesis. In most cases, accurate polymerization by DNA polymerase is achieved by the concerted efforts of its proofreading 3′→5′ exonuclease and polymerase activities (10–12). The relative level of proofreading activity during DNA synthesis by the associated 3′→5′ exonuclease activity of DNA polymerase is a major factor contributing to the fidelity.
Thermostable DNA polymerases are routinely used in PCR to amplify DNA targets. Among these, Taq DNA polymerase is the most widely used, although it does not have a proofreading function. The error rate of Taq DNA polymerase has been estimated to be approximately 1 × 10–4 to 2 × 10–5, depending upon the assay system used (13,14). This error rate is a serious concern in applications such as gene cloning and genotyping that require enhanced accuracy of DNA synthesis. Researchers performing genotyping using Taq DNA polymerase frequently have difficulty determining the correct size of the alleles they are amplifying. This is due to the tendency of Taq DNA polymerase to add a nucleotide in a template-independent fashion to PCR products (15).
In this work, we show that substitutions for the Arg722 residue in the O-helix of Tne DNA polymerase have a dramatic effect on the fidelity of DNA synthesis and non-templated nucleotide addition. The level of proofreading activity and the level of nucleotide misincorporation during DNA synthesis are influenced in part by the amino acids present at this position. Improved fidelity and reduced non-templated nucleotide addition make these modified DNA polymerases useful for a variety of genetic applications, including PCR and genotyping.
MATERIALS AND METHODS
Cloning of Tne DNA polymerase and its mutants
The Tne DNA polymerase gene was cloned from the genomic DNA of T.neapolitana (DSM 5068) by following a method described elsewhere (16). The gene was expressed under control of either the lac or the tac promoter in E.coli. The Tne DNA polymerase mutants were generated by site-directed mutagenesis according to the procedure of Kunkel (17) or by PCR using synthetic oligonucleotides.
Mutant DNA polymerases
Tne DNA polymerase contains both 5′→3′ and 3′→5′ exonuclease activities. Mutations were introduced into Tne DNA polymerase so as to inactivate either one (D137A) or both (D137A/D323A) exonucleases and then further mutations were introduced into the O-helix domain. Tne polymerase containing mutations D137A and D323A refers to inactivated 5′→3′ and 3′→5′ exonuclease activities, respectively. The mutants derived from Taq DNA polymerase had wild-type 5′→3′ exonuclease activity and a mutation in the O-helix region.
Purification of DNA polymerases
A single colony of E.coli DH10B (Invitrogen) carrying a plasmid that contained the wild-type or mutant DNA polymerase gene was inoculated into 20 ml of LB containing 100 µg/ml ampicillin. After incubation at 30°C overnight, the culture was transferred into 330 ml of the same growth medium. At an OD590 of about 1.0, isopropyl-β-d-thiogalactoside was added to a final concentration of 1.0 mM and the cells were grown for an additional 3.5 h. Cells were harvested by centrifugation at 6000 r.p.m. (Sorvall GS3 rotor) and 2–3 g of cells were resuspended in 15 ml of Mono Q column buffer (50 mM Tris–HCl, pH 8.0, 5% glycerol, 5 mM 2-mercaptoethanol, 50 mM NaCl, 1 mM EDTA) and 0.5 mM PMSF. Suspended cells were disrupted by sonication for 2 min (550 Sonic Dismembrator; Fisher Scientific). The sonicated sample was heated at 80°C for 20 min (to inactivate E.coli polymerases and to precipitate large amounts of the E.coli proteins) and then placed in ice water for 5 min. After addition of 0.6 ml of 5 M NaCl (20 mM final concentration) and 0.6 ml of 10% polyethyleneimine, the nucleic acids and the precipitated proteins were removed by centrifugation at 13 000 r.p.m. (Sorvall SS34 rotor). The Tne DNA polymerase was precipitated by adding ammonium sulfate (305 g/l) to the supernatant. The precipitate was collected by centrifugation at 13 000 r.p.m. as above and resuspended in 4 ml of Mono Q column buffer. The sample was dialyzed against 1 l of Mono Q column buffer overnight. Following centrifugation at 13 000 r.p.m. as above to remove insoluble material, the dialyzed sample was loaded onto a 1 ml Mono Q column (HR5/5; Pharmacia). The column was washed with Mono Q column buffer until the OD280 reached baseline and then was eluted with a linear gradient of 50–300 mM NaCl in 20 ml of Mono Q column buffer. Fraction contents were characterized by Coommassie blue staining following 8% SDS–PAGE and by an assay for DNA polymerase activity (described below). The fractions containing active and pure Tne DNA polymerase were pooled and dialyzed against 500 ml of dialyzing buffer (20 mM Tris–HCl, pH 8.0, 0.1 mM EDTA, 1 mM DTT, 0.04% w/v NP-40, 0.04% w/v Tween-20, 50% glycerol) for 12 h and stored at –20°C.
DNA polymerase activity assay
Activated salmon sperm DNA (Sigma) was prepared by a protocol described elsewhere (18). Purified DNA polymerase activity was measured in a 50 µl reaction mixture containing 25 mM TAPS, pH 9.3, 2.0 mM MgCl2, 50 mM KCl, 1.0 mM DTT, 200 µM each dNTP, 25 µg activated salmon testes DNA, 1.05 µCi [32P]dCTP. After incubation at 72°C for 10 min, the reaction was terminated by addition of 10 µl of 500 mM EDTA. Incorporation of radioactivity into acid-insoluble products was determined as described elsewhere (19). One unit of DNA polymerase represents incorporation of 10 nmol dNTPs into acid-precipitable material at 72°C in 30 min.
Primer extension assay
The primer extension assay was carried out based on the method described by Goodman with minor modifications (12). A 32P-labeled primer–template (2 nM) was incubated with different amounts of Tne DNA polymerase in 10 µl of reaction mixture containing 20 mM Tris–HCl, pH 8.4, 50 mM KCl, 1.5 mM MgCl2, 1.0 mM DTT in the presence 200 µM each dNTP at 72°C for 2 min. After addition of 5 µl of sequencing stop buffer and heating at 90°C for 2 min, the sample (5 µl) was loaded onto a 12% polyacrylamide–1× TBE–7 M urea sequencing gel and subjected to electrophoresis at 1200 V for 1.5 h. For autoradiography, the gel was vacuum dried on Whatman filter paper and X-ray film was overlayed for exposures. For some experiments, the radioactivity in bands of the dry sequencing gel was analyzed with a phosphorimager.
Coupled mismatch primer extension assay
A 5′-32P-end-labeled substrate (1 nM) was incubated in 10 µl of extension reaction at 72°C for 2 min as in the primer extension assay using various amounts of DNA polymerase. The reaction mixtures were cooled on ice for 5 min, 10 U of BglII were added to the reaction and the mixture was incubated at 37°C for 1 h. After addition of 5 µl of sequencing stop buffer and heating the mixture at 94°C for 2 min, 5 µl of the samples were electrophoresed through a 10% polyacrylamide–1× TBE–8 M urea sequencing gel.
In vivo fidelity assay
The assay was done similarly to a procedure described before (20). Purified DNA polymerase (0.1–1.0 U) was used to amplify 100 pg of pUC19 that was linearized at the unique BsrFI site. The oligos used were 5′-CGATAGCGGAGCCGGTGAGCGTGGGTCTC-3′ and 5′-CCTGAGTCCGCACCGGCTCCAGATTTATC-3′. The PCR mixture contained 400 nM oligos (each), 200 µM each dNTP, 20 mM Tris–HCl, pH 8.3, 50 mM KCl and 1.5 mM MgCl2 in a total reaction volume of 50 µl. The sample was heated at 94°C for 1 min, followed by 35 cycles of 94°C for 10 s, 55°C for 15 s and 72°C for 2.5 min, with a final 3 min extension at 72°C. A small aliquot (∼5 µl) of PCR product was run out on a 0.8% agarose gel with a low mass DNA ladder (Invitrogen) to estimate the amount of product. The rest of the PCR product was phenol extracted, ethanol precipitated, resuspended in TE (10 mM Tris–HCl, pH 8.0, 1 mM EDTA) and digested with BsrFI (New England Biolabs). The sample was phenol extracted and ethanol precipitated and ligated overnight at 16°C. The ligation reaction products were transformed into DH10B cells (Invitrogen). The appropriate dilution of cells to give ∼200 colonies/plate was plated on LB agar containing ampicillin (100 µg/ml) and X-gal (0.1 mg/ml) overnight at 37°C. The error rate of DNA synthesis resulting from the primer extension process was calculated as previously described (21). Briefly, mutation frequency (MF) was determined to be the number of light blue and white colonies (lacZα–) divided by the total number of colonies. Error rate was determined as MF/(bp × d), where bp is the number of base pairs amplified and the doublings d was determined using the equation 2d = (amount of PCR product)/(amount of initial target).
RESULTS AND DISCUSSION
DNA polymerase activity of modified polymerases
The O-helix of an E.coli DNA polymerase I-type DNA polymerase contains four highly conserved and surface-exposed amino acids, Arg, Lys, Phe and Tyr (Fig. 1). These four amino acids have been implicated mainly in dNTP binding. In addition, at least one of the amino acids, Tyr766 in the Klenow fragment, is involved in primer–template binding. To delineate the role of these conserved amino acids in DNA synthesis, we have generated mutants altered in this O-helix region of Tne DNA polymerase. Since wild-type Tne DNA polymerase contains both 5′→3′ exonuclease and 3′→5′ exonuclease activities, mutations (D137A and D323A) were introduced into Tne DNA polymerase so as to inactivate the exonucleases and then further mutations were introduced into the O-helix domain. Mutations D137A and D323A resulted in an approximately >100-fold reduction of 5′→3′ and >1000-fold reduction of 3′→5′ exonuclease activity, respectively (22).
Figure 1.

Amino acid alignment and structure of the O-helix of members of the DNA polymerase I family. (A) The amino acid sequences and the numbers are taken from Astatke et al. (8). The conserved amino acids in the O-helix are in black. (B) The structure of the O-helix with four conserved side chains was adapted from Joyce and Steitz (5).
DNA polymerase activity of the mutants carrying substitutions in the O-helix was determined in heat-treated (20 min at 80°C) crude extracts or purified preparations as described in Materials and Methods. Mutants carrying the substitutions K726H and K726Q produced undetectable levels of polymerase activity (data not shown). This result is in agreement with previous work with Taq DNA polymerase (23). In addition, the mutation at the homologous amino acid position in E.coli Pol I, K758A, is also reported to have a 330-fold reduction in catalytic activity (8) compared with the wild-type protein. These results suggest that this Lys residue is essential for catalytic activity. However, for Arg722, the effect on DNA polymerase activity varied with amino acid substitution. The mutant DNA polymerases R722K, R722H, R722Q, R722Y and R722L retained ∼50–90% of the parent exonuclease-deficient (D137A/D323A) DNA polymerase activity. The DNA polymerase activity of mutant R722A was undetectable. A similar mutation, R754A, in E.coli Pol I also reduced the polymerase activity by 33-fold (8).
The results with the other substitutions at Arg722 of Tne polymerase were surprising and intriguing due to the fact that the corresponding position of Taq DNA polymerase, Arg659, appeared to be immutable in a selection assay (23). This was based upon the fact that mutants of Taq DNA polymerase at this position did not complement a polAts mutant of E.coli in vivo.
In order to compare the effect of mutation(s) at this position of the O-helix on the DNA polymerase activity of two members of the Pol I family, Taq and Tne, three purified Taq DNA polymerase mutants, R659K, R659H and R659Y, were analyzed. These mutant Taq DNA polymerases retained only 5–10% of the activity of wild-type Taq DNA polymerase. It is possible that the low level of residual polymerase activity of these mutants may not have been sufficient to complement the DNA synthesis rate required by the E.coli mutant (polAts) to grow and form colonies (23,24).
Several studies on Pol I family members have demonstrated the importance of the amino acid side chain at the position corresponding to Tne DNA polymerase Phe730 for discrimination against dideoxynucleotides and for fidelity of DNA synthesis (25,26). In this study, therefore, the attention was focused on analysis of mutations at Arg722 of Tne DNA polymerase.
Primer extension by mutant DNA polymerases
The method described by Goodman (12) was used to estimate the intrinsic fidelity of Tne DNA polymerase and its mutants. Overall misinsertion of three non-complementary nucleotides in the absence of the first complementary nucleotide in the template–primer substrate is shown in Figure 2A. The primer extension was carried out using homogeneous preparations of exonuclease-deficient (D137A/D323A) and modified enzymes (D137A/D323A in addition to a substitution at Arg722) under identical experimental conditions. A typical autoradiogram (Fig. 2B) shows the primer extension experiment in the presence of only three deoxynucleotides. The template–primer was designed in such a way that we could also quantitatively measure the efficiency of mismatched primer extension, following the insertion of a non-complementary nucleotide. In the presence of all four nucleotides, exonuclease-deficient and all three of the mutant Tne DNA polymerases at position Arg722 elongated almost all of the primer to full-length product (Fig. 2B, lanes 1, 6, 11 and 16). Each of the enzymes tested extended the primer to full length at 72°C in only 2 s (data not shown).
Figure 2.

Misinsertion of non-complementary nucleotides by Tne DNA polymerases. (A) Substrate used in the assay of misinsertion of a non- complementary nucleotide by DNA polymerase. N indicates A, T, G or C in this position in the template. (B) An autoradiograph of the reaction products. The assay was carried out as described in Materials and Methods with 0.25 U of Tne DNA polymerase. All DNA polymerases used in this experiment are deficient in both 5′→3′ exonuclease (D137A) and 3′→5′ exonuclease (D323A) activities. The results with D137A/D323A, R722K, R722H and R722Y are shown in lanes a–d, respectively. The dNTP complementary to the N position of the template shown in (A) was omitted in the reaction mixture and indicated as -dNTP. The dash (-) indicates that no dNTP was omitted. Unextended primer is shown in lane 0. The arrow indicates extended 48 nt product without template-independent base addition to the 3′-end.
The analysis of the insertion of non-complementary nucleotides by Tne DNA polymerases has been summarized in Table 1. Exonuclease-deficient Tne DNA polymerase extended 66, 72, 68 and 74% of the primer molecules when dATP, dTTP, dGTP or dCTP was omitted from the reaction mixture, respectively. In contrast, each of the three mutant derivatives at position Arg722 only extended about 1–16% of the primer molecules depending on which of the dNTPs was omitted from the reaction. Thus, mutations at this position reduced base misinsertion by 5- to 50-fold. This result suggests that Arg722 of Tne polymerase plays a pivotal role in the selection between correct and incorrect nucleotides prior to incorporation into the growing chain.
Table 1. Misincorporation opposite N of the template.
| Enzyme | Fraction of primer extended | Relative misincorporation (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| –A | –T | –G | –C | –A | –T | –C | –G | |
| D137A/D323A | 0.66 | 0.72 | 0.68 | 0.74 | 100.0 | 100.0 | 100.0 | 100.0 |
| R722K | 0.03 | 0.10 | 0.02 | 0.16 | 4.8 | 14.2 | 2.8 | 21.9 |
| R722Y | 0.02 | 0.04 | 0.01 | 0.01 | 3.6 | 5.3 | 1.9 | 2.1 |
| R722H | 0.01 | 0.08 | 0.03 | 0.10 | 1.9 | 11.6 | 4.7 | 14.7 |
The radioactivity in the bands shown in Figure 2 was analyzed with a phosphorimager. All DNA polymerases used in this experiment are deficient in both 5′→3′ exonuclease (D137A) and 3′→5′ exonuclease (D323A) activities. The fraction of primers extended is defined as the radioactivity present in the extended product divided by the total input. Total input is the sum of radioactivity in the extended products and in the primers. The values are averages of four experiments. –A, –T, –G and –C indicate the reaction in the absence of dATP, dTTP, dGTP and dCTP, respectively.
Elimination of template-independent one base addition
It has been shown that Taq DNA polymerase and other thermostable DNA polymerases (that lack a 3′→5′ exonuclease activity) used in PCR accumulate amplified products containing non-templated 3′-terminal nucleotides (15,27–29). A careful examination of the autoradiogram shown in Figure 2B (lanes 1, 6, 11 and 16) demonstrates a template-independent extra nucleotide (n + 1) addition (the band above the arrow) in the case of exonuclease-deficient but not in the case of Arg722 mutants in the same exonuclease-deficient background.
Template-independent nucleotide addition is a slow process compared with template-directed DNA synthesis (27,28). Since the level of (n + 1) formation is also affected by the sequence several nucleotides distant from the 5′-end of the template (29), the sequence of the last 5 nt of the template was randomized as shown in Figure 3A in order to minimize sequence bias. About 65% of the DNA product synthesized by the exonuclease-deficient (D137A/D323A) DNA polymerase contained a one base addition at the 3′-terminus (Fig. 3B, lane 1). However, replacement of Arg722 in the same exonuclease-deficient Tne DNA polymerase by any of five amino acids (Fig. 3B, lanes 2–6) almost completely eliminated the template-independent nucleotide addition. However, a different O-helix mutant, F730Y (lane 7), had no significant effect on this property. The degree of non-templated one nucleotide addition can be reduced by changing the PCR conditions. However, to the best of our knowledge, this is the first demonstration of an association between a single amino acid and this activity.
Figure 3.

Elimination of template-independent base addition by mutant Tne DNA polymerases. (A) The substrate with five randomized nucleotides at the 5′-end of a 48 nt template used in the assay. (B) An autoradiograph of the reaction products. The assay was carried out as described in Materials and Methods with 1 U of Tne DNA polymerase and 100 µM dNTPs. All DNA polymerases used in this experiment are deficient in both 5′→3′ exonuclease (D137A) and 3′→5′ exonuclease (D323A) activities. The results with D137A/D323A, R722K, R722Y, R722L, R722H, R722Q and F730Y are shown in lanes 1–7, respectively. The unextended primer in the reaction mixture without DNA polymerase is shown in lane 0. The arrow indicates extended 48 nt product without template-independent base addition to the 3′-end. The band above the arrow represents the extra nucleotide addition (n + 1) product.
Fidelity of the mutant DNA polymerases in vivo
Although the misincorporation assays (Fig. 2B and Table 1) provided some information about the overall fidelity of the Tne DNA polymerase mutants, quantitative fidelity was determined in a forward mutation assay (20). This assay has several advantages over the in vitro misincorporation assay. Quantitation of the absolute frequency of mutation by the misinsertion procedure is difficult, while this assay allows one to quantitate the mutation frequency. In addition, the mutation frequency is determined in more natural conditions in the presence of all four dNTPs, whereas the misincorporation assay was done in the absence of a particular dNTP. The improper balance of the concentrations of correct and incorrect nucleotides forces DNA polymerase to select a non-complementary nucleotide.
To quantitate the fidelity of DNA synthesis, the α-peptide coding sequence of the lacZ gene was amplified and transformed into E.coli. The mutation frequencies of various mutant DNA polymerases are summarized in Table 2. Replacement of Arg722 by a Lys or Tyr in exonuclease-deficient (D137A/D323A) Tne DNA polymerase reduced error rate (increased fidelity) by 10- to 18-fold, while the fidelity of these mutants was increased 25- to 30-fold when the associated 3′→5′ exonuclease activity was present. The 3′→5′ exonuclease activity contributed about a 3- to 4-fold improvement in fidelity, while a more significant contribution was derived from mutations in the Arg722 residue, probably through correct base selection. This conclusion is consistent with the misinsertion experiment mentioned above.
Table 2. Fidelity of Tne DNA polymerases.
| Enzyme substitutions | 5′→3′ exo | 3′→5′ exo | Error rate (10–6) | Relative fidelity |
|---|---|---|---|---|
| Tne D137A/D323A | – | – | 33.7 | 1.0 |
| Tne D137A/D323A R722K | – | – | 3.5 | 9.5 |
| Tne D137A/D323A R722Y | – | – | 1.9 | 17.7 |
| Tne D137A | – | + | 9.1 | 3.7 |
| Tne D137A R722K | – | + | 1.1 | 30.6 |
| Tne D137A R722H | – | + | 1.1 | 30.6 |
| Tne D137A R722Y | – | + | 1.4 | 24.0 |
| Taq (WT) | + | – | 7.3 | 4.6 |
Fidelity was determined by the lacZα forward assay as described in Materials and Methods. 5′→3′ exo indicates whether the exonuclease has been rendered deficient by the mutation D137A. 3′→5′ exo indicates whether the exonuclease has been rendered deficient by the mutation D323A. Wild-type Taq DNA polymerase contains only 5′→3′ exonuclease activity.
Improved proofreading activity of Tne DNA polymerase mutants
To test if alteration of Arg722 allowed the 3′→5 exonuclease to proofread more efficiently, the 3′→5′ exonulease activity of the Tne DNA polymerase mutants, D137A and D137A/R722K, was tested under the same reaction conditions. In the absence of dNTPs, both mutants cleaved nucleotides at the 3′-ends of both single-stranded and double-stranded DNA with similar efficiencies (data not shown). A method described by Lundberg et al. (14) was used to qualitatively measure the associated 3′→5′ exonulease activity in the presence of the four dNTPs.
To slow down the initiation of DNA synthesis by the DNA polymerase, a primer–template duplex was designed with two mispaired nucleotides at the 3′-terminus of the primer strand (Fig. 4A). The mismatches were present in the primer overlapping the sequences recognized by BglII restriction enzyme in the template. For BglII to cleave, both (but not one) mispaired nucleotides in the primer must be removed by the associated 3′→5′ exonuclease before the primer can be extended by the DNA polymerase. As shown in Figure 4B, only a small fraction of the primer molecules extended by the D137A mutant of the Tne DNA polymerase could be cleaved by BglII restriction enzyme. In contrast, the majority of the primer molecules elongated by the mutant Tne DNA polymerase (D137A/R722K) were cleaved by BglII. This result suggests that during the initiation of the reaction, a significant fraction of the mispaired primer–template duplex bound to D137A mutant of the Tne DNA polymerase was elongated without correction. However, in the case of D137A/R722K mutant Tne DNA polymerase, the mispaired primer termini were cleaved by the associated 3′→5′ exonulease prior to extension. This indicates that after the proofreading process at the 3′-terminus, the corrected primer–template substrate switches to the polymerase active site for DNA synthesis.
Figure 4.
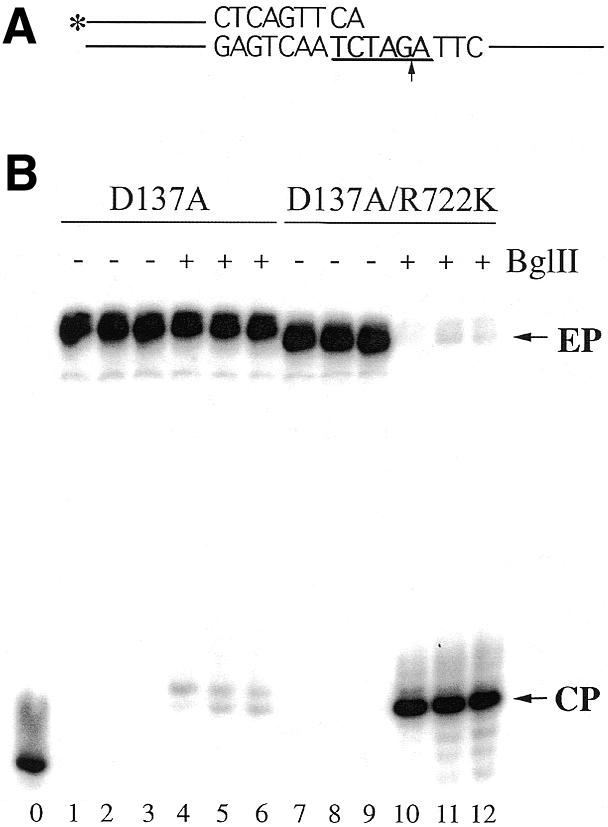
Increase in proofreading activity of Tne DNA polymerase Arg722Lys mutant. (A) The substrate used in the assay. The sequence recognized by BglII is underlined and the symbol * indicates the 32P-label at the 5′-end of the primer. The arrow indicates the cleavage position of BglII in the template. (B) An autoradiograph of the reaction products. In this experiment, mutants D137A and D137A/R722K were used. The amounts of DNA polymerase used in the reactions were 2.0 U for lanes 1, 4, 7 and 10, 1.0 U for lanes 2, 5, 8 and 11 and 0.5 U for lanes 3, 6, 9 and 12, respectively. Ten units of BglII were added to the reaction (lanes 4–6 and 10–12). EP and CP indicate the extended primer and cleavage products, respectively. The unextended primer is shown in lane 0. Because the Tne DNA polymerase was not removed from the extension reactions before incubating with BglII, the cleaved products were extended by 4 nt (indicated by CP) by the DNA polymerases at 37°C.
The inability of 3′→5′ exonuclease-proficient Tne DNA polymerase to cleave mismatched nucleotides in the primer before polymerization could be attributed to less effective proofreading activity because of a partitioning problem. It is consistent with the results of the fidelity assay showing that 3′→5′ exonuclease activity makes very little contribution to the overall fidelity (Table 2) of the Tne DNA polymerase. It is possible that mutation of the Arg722 residue enhanced the partitioning of mispaired primer to the associated 3′→5′ exonuclease domain and, hence, improved fidelity. The detailed mechanism of the partitioning/switching between proofreading and polymerization by this mutant is being studied.
CONCLUSIONS
The mutations at Arg722 in the Tne DNA polymerase resulted in (i) improved base selection or reduced incorporation of non-complementary nucleotides, (ii) elimination of template-independent nucleotide addition to the 3′-end of newly synthesized DNA and (iii) increased proofreading by the associated 3′→5′ exonuclease activity. To the best of our knowledge, this is the first demonstration that a single amino acid mutation in the O-helix region improves all three activities of DNA polymerase resulting in a high fidelity DNA polymerase. These mutant polymerases can fulfill the need for rapid, automated methods for identifying, analyzing and typing polymorphic DNA fragments, particularly minisatellite, microsatellite or STR DNA fragments, and could be used as biochemical reagents to amplify DNA with higher fidelity.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Howard Nash (National Institutes of Health) for critical reading, helpful suggestions and editing this manuscript. The authors also thank Dr Gary Gerard for encouragement, suggestions and valuable comments and Harini Shandilya for technical support.
REFERENCES
- 1.Ollis D.L., Brick,P., Hamlin,R., Xuongc,N.G. and Steitz,T.A. (1985) Structure of a large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature, 313, 818–819. [DOI] [PubMed] [Google Scholar]
- 2.Beese L.S., Derbyshire,V. and Steitz,T.A. (1993) Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science, 260, 352–355. [DOI] [PubMed] [Google Scholar]
- 3.Eom S.H., Wang,J. and Steitz,T.A. (1996) Structure of Taq polymerase with DNA at the polymerase active site. Nature, 382, 278–281. [DOI] [PubMed] [Google Scholar]
- 4.Kiefer J.R., Mao,C., Hansen,C.J., Basehore,S.L., Hogrefe,H.H., Braman,J.C. and Beese,L. (1997) Crystal structure of a thermostable Bacillus DNA polymerase I large fragment at 2.1 Å resolution. Structure, 5, 95–108. [DOI] [PubMed] [Google Scholar]
- 5.Joyce C.M. and Steitz,T.A. (1994) Function and structure relationships in DNA polymerases. Annu. Rev. Biochem., 63, 777–822. [DOI] [PubMed] [Google Scholar]
- 6.Steitz T.A. (1999) DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem., 274, 17395–17398. [DOI] [PubMed] [Google Scholar]
- 7.Polesky A.H., Dahlberg,M.E., Benkovic,S.J., Grindley,N.D.F. and Joyce,C.M. (1992) Side chains involved in catalysis of the polymerase reaction of DNA polymerase I of Escherichia coli. J. Biol. Chem., 267, 8417–8428. [PubMed] [Google Scholar]
- 8.Astatke M., Grindley,N.D.F. and Joyce,C.M. (1995) Deoxynucleoside triphosphate and pyrophosphate binding sites in the catalytically competent ternary complex for polymerase reaction catalyzed by DNA polymerase I. J. Biol. Chem., 270, 1945–1954. [DOI] [PubMed] [Google Scholar]
- 9.Doublié S., Tabor,S., Long,A.M., Richardson,C.C. and Ellenberger,T. (1998) Crystal structure of a bacteriophage T7 replication complex at 2.2 Å resolution. Nature, 391, 251–258. [DOI] [PubMed] [Google Scholar]
- 10.Minnick D.T., Bebenek,K., Osheroff,W.P., Turner,R.M., Astatke,M., Liu,L., Kunkel,T.A. and Joyce,C.M. (1999) Side chains that influence fidelity at the polymerase active site of Escherichia coli DNA polymerase I (Klenow fragment). J. Biol. Chem., 274, 3067–3075. [DOI] [PubMed] [Google Scholar]
- 11.Bell J.B., Eckert,K.A., Joyce,C.M. and Kunkel,T.A. (1997) Base miscoding and strand misalignment errors by mutator Klenow polymerase with amino acid substitutions at Tyrosine 766 in the O-helix of the finger subdomain. J. Biol. Chem., 272, 7345–7351. [DOI] [PubMed] [Google Scholar]
- 12.Goodman M.F. (1988) DNA replication fidelity: kinetics and thermodynamics. Mutat. Res., 200, 11–20. [DOI] [PubMed] [Google Scholar]
- 13.Kunkel T.A. (1988) Exonucleolytic proofreading. Cell, 53, 837–840. [DOI] [PubMed] [Google Scholar]
- 14.Lundberg K.S., Shoemaker,D.D., Adams,M.W., Short,J.M., Sorge,J.A. and Mathur,E.J. (1991) High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene, 108, 1–6. [DOI] [PubMed] [Google Scholar]
- 15.Smith J.M., Carpten,J.D., Brownstein,M., Ghosh,S., Magnuson,V., Gilbert,D.A., Trent,J.M. and Collins,F.S. (1995) An approach to genotyping errors caused by non-templated nucleotide addition by Taq polymerase. Genome Res., 5, 312–317. [DOI] [PubMed] [Google Scholar]
- 16.Sagner G., Ruber,R. and Kessler,C. (1991) Rapid filter assay for the detection for DNA polymerase activity: direct identification of the gene for the DNA polymerase gene from Thermus aquaticus. Gene, 97, 119–123. [DOI] [PubMed] [Google Scholar]
- 17.Kunkel T.A. (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl Acad. Sci. USA, 82, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livingstone D.M., Hinkel,D.C. and Richardson,C.C. (1975) Deoxyribonucleic acid polymerase III of E. coli: purification and properties. J. Biol. Chem., 250, 461–469. [PubMed] [Google Scholar]
- 19.Chien A., Edger,D.B. and Trela,J.M. (1976) Deoxyribonucleic acid polymerase from extreme thermophile Thermus aquaticus. J. Bacteriol., 127, 1550–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bebenek K. and Kunkel,T.A. (1995) Analyzing fidelity of DNA polymerases. Methods Enzymol., 262, 217–232. [DOI] [PubMed] [Google Scholar]
- 21.Cline J., Braman,J.C. and Hogfree,H.H. (1996) PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res., 24, 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solus J., Yang,S. and Chatterjee,D.K. (2001) Polymerases for analyzing or typing polymorphic nucleic acid fragments and uses thereof. US patent 6,306,588.
- 23.Suzuki M., Baskin,D., Hood,L. and Loeb,L.A. (1996) Random mutagenesis of Thermus aquaticus DNA polymerase I: concordance of immutable sites in vivo with the crystal structure. Proc. Natl Acad. Sci. USA, 93, 9670–9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki M., Yoshida,S., Adman,E.T., Blank,A. and Loeb,L.A. (2000) Thermus aquaticus DNA polymerase I mutants with altered fidelity. J. Biol. Chem., 275, 32728–32735. [DOI] [PubMed] [Google Scholar]
- 25.Tabor S. and Richardson,C.C. (1995) A single residue in DNA polymerases of E. coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxynucleotides. Proc. Natl Acad. Sci. USA, 92, 6339–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Astatke M., Grindley,N.D.F. and Joyce,C.M. (1998) How E. coli DNA polymerase (Klenow fragment) distinguishes between deoxy- and dideoxynucleotides. J. Mol. Biol., 278, 147–165. [DOI] [PubMed] [Google Scholar]
- 27.Clark J.M., Joyce,C.M. and Beardsley,G.P. (1987) Novel blunt-end addition reactions catalyzed by DNA polymerase I of Escherichia coli. J. Mol. Biol., 198, 123–137. [DOI] [PubMed] [Google Scholar]
- 28.Hu G. (1993) DNA polymerase-catalyzed addition of non-templated extra nucleotides to the 3′-end of a DNA fragment. DNA Cell Biol., 12, 763–770. [DOI] [PubMed] [Google Scholar]
- 29.Brownstein M.J., Carpten,J.D. and Smith,J.R. (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques, 20, 1004–1010. [DOI] [PubMed] [Google Scholar]