


Abstract
Fluorescence is the favored signaling technology for molecular diagnoses. Fluorescence energy transfer-based methods are powerful homogeneous assay tools. A novel oligonucleotide probe, named MagiProbe, which is simple to use, is described, and information given about the duplex formed with a target. The probe internally has a fluorophore and an intercalator. Its fluorescence is quenched by the intercalator in the absence of a target sequence. On hybridization with a target sequence, the probe emits marked fluorescence due to the interference in quenching by intercalation. Furthermore, MagiProbe hybridized with a single-base mismatched target emits less fluorescence than with a perfect matched target. It therefore can detect a single base difference in a double-stranded form with a target.
INTRODUCTION
In the post-genome era, there is an ever-increasing demand for more rapid, high-throughput and cost-effective techniques for nucleic acid detection. Fluorescence-based methods are very powerful tools in this field (1). The superiority of fluorescence-based technologies for nucleic acid detection is that they can be used in homogeneous assays.
Homogeneous fluorescence assay methods are coupled with various nucleic acid amplification methods, in particular with PCR, TaqMan (2), molecular beacon (3), sunrise primer (4), Scorpion (5), adjacent hybridization probe (6) and invader (7). All have been used successfully to detect the presence of a target sequence and single base alterations. To detect single base alterations, some of these methods exploit the high specificity intrinsic to enzymes such as DNA polymerase or endonuclease, others depend on hybridization specificity. Enzyme recognition and hybridization are the most widely used means of detecting single base alterations. The disadvantage of the former is that it is derived from the enzyme itself and therefore influenced by sample quality and the need for complicated reagents. The latter requires the laborious work of optimization of the probe design.
A novel electrocatalytic method for detecting single base alterations (8) has been developed, in which specificity depends on electronic coupling within the base pair stack rather than on the thermodynamics of base pairing. Furthermore, small molecules which bind to the base-mismatch site have been shown to be molecular recognition elements (9).
To overcome the limitations inherent in the current methods, a new type of oligonucleotide probe has been developed, named MagiProbe, which recognizes a target sequence by hybridization but depends neither on the thermodynamics of the duplex nor enzymatic assistance to detect single base alterations. MagiProbe functions as a molecular sensor that distinguishes a duplex with a perfect matched target from one with a one-base mismatched target. Although Yamana et al. (10) reported previously a similar conceptual probe, it only is applicable to the DNA–RNA duplex. Shinozuka et al. (11) developed a sophisticated probe based on fluorescence resonance energy transfer (FRET), in which the oligonucleotide carries fluorescence and acridine at its 5′ end. This probe emits fluorescence on irradiation at 360 nm, which corresponds to the excitation maximum of the acridine moiety, in the absence of a target sequence, whereas its fluorescence diminishes in the presence of a target sequence. This difference is caused by inhibition of FRET due to intercalation of acridine to the duplex formed. The extent of fluorescence change, however, is not significant, and there seems to be few combinations of fluorophore and intercalator. In addition, Tyagi et al. (12) developed a molecular beacon, a single-stranded DNA molecule composed of a hairpin-shaped oligonucleotide that bears both a fluorophore and quencher group. In that probe, direct energy transfer is considered to be the quenching mechanism because efficient quenching occurs without spectral overlap between the donor’s emission and acceptor’s absorption spectra. Taking into account this phenomenon, I designed a novel fluorescence quenching-based oligonucleotide probe carrying a fluorophore and an intercalator (13,14). Recently, Ranasinghe et al. (15) also reported similar probes to MagiProbe.
Here I describe details of MagiProbe and demonstrate its great ability for single-base change discrimination.
MATERIALS AND METHODS
Oligonucleotide synthesis
All the modified oligonucleotides were synthesized in an ABI 392 synthesizer by standard phosphoramidite reactions. To synthesize the probe for the real-time PCR, 3′-phosphate CPG (Glen Research) was used to block extension. Fluorescein was incorporated internally into the oligonucleotide by the use of fluorescein phosporamidite (Glen Research). A Uni-Link Aminomodifier (Clontech) was used to introduce a primary N-group for subsequent derivatization with an intercalator or quenching dye. After deprotection, the N-derivatized oligonucleotide was coupled with a succinimidyl derivative as reported previously (16). The species used in the reactions (all from Molecular Probes) were succinimidyl 7-diethylaminocoumarin-3-carboxylic acid (D-1412), succinimidyl 1-pyrenebutanoic acid (P-130) and succinimidyl 4-{[4-(dimethylamino)phenyl]azo}benzoic acid (Dabcyl: D-2245). TAMRA-NHS (Applied Biosystems) was used to introduce Tamra. After the reaction, excess dye was removed by size exclusion chromatography in a Sephadex G-50 column and elution with 50 mM trietyl ammonium bicarbonate buffer. Modified oligonucleotides were purified by reversed-phase HPLC in a C18 column then eluted with an acetonitrile gradient in triethyl ammonium acetate buffer. In the HPLC, the desired modified oligonucleotide bearing two dyes (e.g. pyrene and fluorescein) showed three to four peaks because of the two asymmetric carbon atoms in the linkers. This was confirmed as a single band by denaturing polyacrylamide gel electrophoresis. All the conjugates also were characterized by their specific absorption and emission spectra. Unmodified oligonucleotides used as targets were purchased from a supplier and purified by denaturing polyacrylamide gel electrophoresis.
Fluorescence measurement of MagiProbe with a complementary oligonucleotide
180 nmol of MagiProbe was mixed with an equimolar amount of unmodified oligonucleotide in 50 µl of 10 mM Tris–HCl pH 7.5, 1 mM EDTA, 50 mM NaCl and 10 ng/µl of carrier DNA. The mixture was denatured in a thermal cycler at 94°C for 10 min then cooled at 2°C/min to 40°C. The solution obtained was diluted 1/45 with the same buffer, and its fluorescence measured at 538 nm on excitation at 485 nm by POLAstar (bMG). EC1 was used as an unrelated oligonucleotide for the sequences of EFN3 and EFN4, and EC2, as well as EC1, for those of EFN1 and EFN2.
Melting profiles
UV absorbance versus temperature profiles was measured at 260 nm in a Shimazu UV-2100 spectrophotometer equipped with a thermostated cell holder. The buffer contained 10 mM phosphate buffer pH 7.0, 0.1 mM EDTA and 150 mM NaCl. All initial and final absorbance values were between 0.5 and 2.0 OD. Melting curves were obtained by increasing the temperature from 25 to 85°C at the ramp rate of 3°C/min. Fluorescence melting curves were obtained by the use of LightCycler™. Each reaction mixture was made from the buffer used for UV melting measurements plus 210 nM of MagiProbe and the complementary oligonucleotide. Data for melting curves generated by heating the mixture from 35 to 85°C at the ramp rate of 6°C/min were collected continuously during that period.
One-base discrimination experiments for the human β-globin gene
The unmodified oligonucleotide sequences used as targets were 5′-ACAGGGCAGTAACCGCAGACTTCTCCTCAGG AGTCAGGTGCACCATGGTGTC: BNF52A (complementary to the A-allele) and 5′-ACAGGGCAGTAACCGCAGA CTTCTCCACAGGAGTCAGGTGCACCATGGTGTC: BN F52AM (complementary to the T-allele), in which underlining shows the polymorphic position. Fluorescence profiles of MagiProbes for the human β-globin gene were obtained in the presence of the perfect matched oligonucleotide (BNF52A), the one-base mismatched oligonucleotide (BNF52AM) and in the absence of the complementary oligonucleotide. Each mixture consisted of 20 µl of 100 nM MagiProbe, 400 nM complementary oligonucleotide, 10 mM phosphate buffer pH 7.0, 0.1 mM EDTA, 150 mM NaCl and 10 ng/µl carrier DNA. The mixture was denatured and cooled to 35°C at 6°C/min. Fluorescence data were collected continuously by increasing the temperature to 85°C at 6°C/min.
Real-time PCR assays using MagiProbe
Human genomic DNA was purchased from Roche Diag nostics. Primer sequences that amplify the 110 bp fragment of the human β-globin gene were identical to those reported previously (17). Each PCR reaction mixture was made up of 2 µl of DNA solution (15 ng/µl, 1.5 ng/µl, 150 pg/µl, 15 pg/µl), 2 µl of 10× Stoffel Buffer (Perkin Elmer), 1 µl of a 5 µM solution of each primer, 2 µl of a 2.5 mM solution of dNTPs, 3.6 µl of a 25 mM solution of MgCl2, 1 µl of a 10 µg/µl solution of BSA, 2 µl of a 0.2 µM solution of Py/F-BNF5, 0.125 µl (1.25 U) of Stoffel Fragment (Perkin Elmer) and water, final volume 20 µl. For the negative control, DNA was replaced with sterile water. After denaturation at 95°C for 1 min, 45 cycles of amplification (95°C for 5 s, 55°C for 5 s, 72°C for 5 s) were done in a LightCycler™. Fluorescence was recorded at the end of the annealing step.
Allelic discrimination of CYP2C19 by real-time PCR
The allele-specific MagiProbes for CYP2C19 are 5′-GAT TGTAAGCACCCCCTGG(Py)(F)ATCCA: Py/F-CYP636G, 5′-GATTGTAAGCACCCCCTGA(Py)(F)ATCCA: Py/F-CYP636A, 5′-CCCG(Py)(F)GGAACCCATAACAAATTA: Py/F-CYP681G and 5′-TCCC(F)(Py)AGGAACCCATAAC AAATTA: Py/F-CYP681A, in which the underlining denotes polymorphic sites. PCR primers that amplify exons 4 and 5 of CYP2C19 were identical to those reported previously (18,19). Plasmid DNA was prepared from human DNA by the standard cloning method with PCR. Individual sequences were confirmed by restriction fragment length polymorphism. PCR conditions were the same as those described for the β-globin gene amplification, except for the cycling temperature. The cycling condition for both exons 4 and 5 was 95°C for 5 s, 55°C for 15 s and 72°C for 15 s. Fluorescence was recorded at the extra step of 65°C for 5 s.
RESULTS
Design and preparation of MagiProbe
Figure 1 shows the design and working principles of MagiProbe. The fluorophore and intercalator are introduced internally to the oligonucleotide probe. In this structure, the distance between the fluorophore and intercalator must be close enough to permit direct energy transfer, and the fluorescence of MagiProbe is quenched by the intercalator in the absence of a target sequence. Upon hybridization, however, the probe emits greater fluorescence due to the interference in quenching by intercalation. In addition to the homogeneous assay format for the detection of specific sequences, MagiProbe discriminates single base alterations because its intercalation is less stable with a one-base mismatched duplex than with a perfect matched one, in which the mispair is located close to the intercalator. As a result, the fluorescence intensity in the one-base mismatched duplex is less than that in the perfect matched duplex.
Figure 1.

MagiProbe working principle. MagiProbe fluorescence is quenched by pyrene in the absence of a target. On hybridization with a perfect match sequence, MagiProbe emits fluorescence due to dequenching by intercalation. In the mismatched duplex form, its fluorescence remains quenched because misparing bases disturb the intercalation of pyrene.
First, respectively, fluorescein and pyrene were chosen for the fluorophore and intercalator. Fluorescein, the most common fluorophore, is relatively stable and less prone to interact with nucleic acid (20). As the intercalator, pyrene conjugated to oligonucleotide binds to DNA, perhaps by intercalation (21,22). It is desirable that MagiProbe be prepared by automated synthesizer protocols without significant change relative to the synthesis of common unmodified oligonucleotides. Fluorescein-linked phosphoramidite and primary amine group-linked phosporamidite therefore are used. Both are extendable for chain elongation and have the same backbone structures and linker arms (Fig. 2). The intercalator, e.g. pyrene, is attached to the primary N-group post-synthetically by means of amine-reactive derivatives. This post-synthetic method for the introduction of the intercalator is preferable for the systematic study of various intercalators.
Figure 2.

MagiProbe structure.
Effects of the intercalator in MagiProbe
Four oligonucleotide models with artificial sequences were prepared to examine the effects of the intercalator in MagiProbe (Table 1). Two characteristic sequences surround pyrene, a GC- and an AT-rich sequence when the duplex forms. From another perspective, pyrene and fluorescein are inserted consecutively into the sequences or one base is inserted between them. Probes carrying pyrene or without pyrenyl modification were compared to determine the intercalator effect. Figure 3 shows the fluorescence intensities of MagiProbe in the double- and single-stranded forms. The intensities of the probes not carrying pyrene tended to increase on hybridization (Fig. 3A). The extent of fluorescein interaction with the nucleo-base in the single-stranded form is decreased by formation of the duplex, and quenching is weakened. In contrast, the increase in fluorescence intensity is striking in the probes carrying pyrene. In the comparison of the pyrenyl-modified and -non-modified probes, fluorescence intensities in the double-stranded form are nearly equal, whereas those in the single-stranded one differ markedly. This confirms the working hypothesis of MagiProbe.
Table 1. MagiProbes with artificial sequences and the complementary oligonucleotides.

EC1 is complementary to Py/F-EFN1 and Py/F-EFN2, EC2 to Py/F-EFN3 and Py/F-EFN4. For convenience, the respective structural forms of the inserted pyrene and fluorescein are referred to as (Py) and (F).
Figure 3.

Comparison of fluorescence properties of probes without (A) and with (B) pyrenyl modification (MagiProbe). Each probe was mixed with an equimolar amount of an unrelated oligonucleotide (hatched bar) or a complementary oligonucleotide (black bar) or the probe only (white bar) in 10 mM Tris–HCl, 1 mM EDTA, 50 mM NaCl, 10 ng/µl carrier DNA. The mixture was denatured and annealed. After appropriate dilution, fluorescence was measured at 538 nm after excitation at 485 nm.
To ensure that intercalation is essential for MagiProbe rather than just quenching, the typical fluorescein quenchers Tamra and Dabcyl and the intercalator coumarin were introduced to the probe. Figure 4 shows marked changes in fluorescein intensity in the probes with coumarin but not in those with Tamra and Dabcyl. In this construction, fluorescein quenching by Dabcyl is slight or absent, whereas quenching by Tamra is effective and is not dequenched on hybridization. Although the mechanisms of quenching and dequenching are not yet known, clearly the combination of fluorescein and an intercalator is essential to MagiProbe.
Figure 4.

Comparison of fluorescence properties of combinations of fluorescein and various quenchers. Probes in (A) had pyrene and fluorescein, in (B) coumarin and fluorescein, in (C) DABCYL and fluorescein, in (D) TAMRA and fluorescein. Each probe was mixed with an equimolar amount of complementary oligonucleotide (black bar) or probe only (white bar) in 10 mM Tris–HCl, 1 mM EDTA, 50 mM NaCl, 10 ng/µl carrier DNA and the mixture denatured and annealed. After appropriate dilution, fluorescence was measured at 538 nm after excitation at 485 nm.
Thermal stability of the duplex with MagiProbe
Solutions of duplexes containing MagiProbe were subjected to UV melting analysis to see how the introduction of fluorescein and pyrene to this construction affects the thermal stability of the duplex. In all cases, modification decreased the melting point from 6 to 11°C as compared with the melting points of the unmodified control probes (Fig. 5A and B). Melting profiles of MagiProbes also were obtained by consecutive fluorescence measurement in a LightCycler™ instrument (Fig. 5C). The plotted curves derived from fluorescence monitoring are roughly similar to those from UV analysis even though the experimental conditions differed as to the oligonucleotide concentration and temperature gradient rate. This indicates that the curve plotted as a function of the temperature of the fluorescence intensity of MagiProbe in the presence of a complementary sequence may represent the melting curve.
Figure 5.

Melting profiles of MagiProbe. (A) Comparison of UV melting profiles of MagiProbes (Py/F-EFN1, green line; Py/F-EFN2, red line) and unmodified oligonucleotide (dark pink line) in the presence of an equimolar amount of complementary oligonucleotide (EC1). (B) Comparison of the UV melting profiles of MagiProbes (Py/F-EFN3, blue line; Py/F-EFN4, brown line) and unmodified oligonucleotide (pink line) in the presence of an equimolar amount of complementary oligonucleotide (EC2). (C) Melting profiles obtained by fluorescence measurement. Solid lines correspond to the fluorescence signals of the MagiProbes (green, Py/F-EFN1; red, Py/F-EFN2; blue, Py/F-EFN3; brown, Py/F-EFN4) in the presence of an equimolar amount of complementary oligonucleotide. Dashed lines correspond to the fluorescence signal of the MagiProbes (green, Py/F-EFN1; red, Py/F-EFN2; blue, Py/F-EFN3, brown, Py/F-EFN4) in the absence of a complementary oligonucleotide.
MagiProbe for human β-globin gene detection
The human β-globin gene was chosen as the target sequence to evaluate MagiProbe for the sequence dependency and position of modification for detection of a single base alteration. A series of MagiProbes covering codon 6 of β-globin, who’s GAG to GTG mutation is responsible for sickle-cell disorders (23), was prepared (Fig. 6). Although all the probes showed similar fluorescence change properties on hybridization, the degree of change and background fluorescence in the absence of a complementary sequence differed for each one. Figure 7 shows the fluorescence intensity curves of representative probes as a function of temperature. In the absence of the complementary sequence, the fluorescence signals of Py/F-BNF2 and Py/F-BNF3 increased markedly at temperatures below 60°C, whereas the fluorescence signal of Py/F-BNF5 remained constant throughout. This difference is attributable to intra- or intermolecular base pairing. The self-complementary sequences thought to be responsible for the high-background also are shown in Figure 7. Unlike Py/F-BNF2 and Py/F-BNF3, Py/F-BNF1 shows less background fluorescence even though it has a self-complementary sequence of 10 bp (not shown in Fig. 7). This is because pyrenyl modifications of Py/F-BNF2 and Py/F-BNF3 are located within the duplex that they form, whereas the modification of Py/F-BNF1 is located outside of the duplex with the dangling single-stranded form. Whether the background fluorescence is caused by intramolecular or intermolecular base pairing is not clear. Whichever structure is responsible for the high- background, careful design of the probe preparations is necessary.
Figure 6.

Schematic representation of the MagiProbe sequence for the human β-globin gene. The underline shows the polymorphic position (A→T), triangles the positions at which pyrene and fluorescein are inserted. All the MagiProbes have a perfect match to the A-allele and a one-base mismatch to the T-allele.
Figure 7.

Temperature-dependent fluorescence properties for one-base discrimination of the human β-globin gene. Each probe was mixed with the target oligonucleotide (red line, perfect match; green line, one-base mismatch; blue line, without the target oligonucleotide) then heated at 95°C. Melting curves were obtained during a 0.1°C/s decrease to 35°C in a LightCycler™. Self-complementary sequences shown under each graph were found with GENETYX software. Triangles in the palindromic sequences mark the positions of pyrene and fluorescein.
As stated, MagiProbe discriminates a completely matched duplex from a one-base mismatched duplex by the conformational change between them. In Figure 7, the fluorescence intensity curves of MagiProbe in the presence of a complementary oligonucleotide carrying a perfect matched sequence or a one-base mismatched sequence are compared. The mismatch position in all the probes was deliberately placed next to the pyrenyl modification. As seen in the figure, they all discriminated a perfect match from a one-base mismatch. Py/F-BNF1 is the most prominent probe used for single nucleotide change discrimination. In it, two bases (GG) at the proximal end after fluorescein and pyrene and an unpaired base (A) may not be able to form base pairs with the complementary DNA that carries a mismatched base; therefore, there is no base-stacking region for the intercalation of pyrene. Instead, in a perfect match, the three bases between the pyrenyl modification and its proximal end form stable base pairs with the perfect match DNA, providing a base stacking region for the intercalation of pyrene. This seems to be why Py/F-BNF1 has the clear ability to discriminate a single-base change.
To verify the above consideration for the ability for single-base change discrimination and establish a general rule for the design of a superior MagiProbe, a series of MagiProbes were prepared, in which the number of bases between the pyrenyl modification and the probe’s proximal end was changed systematically. Figure 8 shows the fluorescence intensity curves of six of these probes as a function of temperature. All are 25 nt long probe, and the base discrimination site is on the side of the proximal end adjacent to the pyrenyl modification. The fluorescence intensity of the probes with a perfectly matched sequence tends to decrease gradually on hybridization together with a reduction in the number of bases. Eventually, the fluorescence intensity curve of the probe (in which the polymorphic position is located at the 5′ end) with the perfect matched duplex becomes identical to that with the single-base mismatched duplex (Fig. 8F). The fluorescence intensity curves do not, however, entirely vary in the probes with the single-base mismatched duplex. Presumably, pyrene may interact with both sides of the base stacking regions in the perfectly matched configuration, producing relatively high fluorescence. The base pairing between pyrene and its proximal end, however, tends to become disrupted with the decrease in the number of bases, causing a gradual decrease in fluorescence. In the case of a single-base mismatch, pyrene interacts only with the base-stacking region on the distal end side irrespective of the number of bases between pyrene and its proximal end. On the basis of these findings, the MagiProbe mechanism may involve dynamic quenching as a result of collision between fluorescein and pyrene whose motion is constrained by its dynamic interaction with the base-stacking region.
Figure 8.

Effect of the position of the pyrenyl modification on one-base discrimination. The number of bases between the pyrenyl modification and proximal end was decreased systematically. Fluorescence measurement was done as described in Figure 7 (red line, perfect match; green line, one-base mismatch; blue line, without target oligonucleotide).
Application of MagiProbe to real-time PCR
Because MagiProbe is a kind of self-reporting probe like a molecular beacon, direct monitoring of nucleic acid synthesis in vitro or in living cells is possible. I therefore have shown how MagiProbe detects human β-globin gene in a real-time PCR. A MagiProbe with the same sequence as that in the previous experiments but with an additional 3′ phosphate is used directly in a conventional PCR for amplification of the human β-globin gene. By measuring the fluorescence intensity at the end of the annealing step, the PCR can be conducted in a standard real-time format in a LightCycler™. Optimization is required only for the probe concentration that gives the highest intensity signal. As expected, typical real-time detection curves that correspond to the template concentrations were obtained (Fig. 9).
Figure 9.
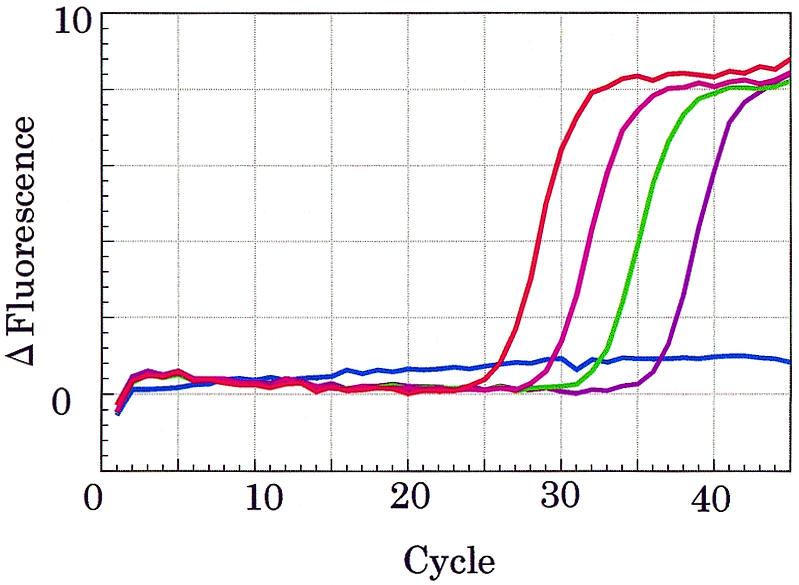
Real-time PCR for the human β-globin gene done with MagiProbe. A 110-bp fragment of human β-globin gene was amplified with specific primers from human genomic DNA (30 ng, red line; 3 ng, pink line; 300 pg, green line; 30 pg, dark pink line; negative control, blue line). All samples were amplified simultaneously and monitored at the end of the annealing phase (55°C).
To examine the distinct property by which MagiProbe recognizes a single-base alteration in a real-time PCR, the two G–A polymorphisms in exons 4 and 5 of the human CYP2C19 gene, known to be concerned with drug metabolism, were chosen as targets (18,19). Four MagiProbes, for the 636G-allele and the 636A-allele in exon 4 and for the 681G-allele and the 681A-allele in exon 5, were prepared and their fluorescence properties determined as shown in Figure 7. All had similar abilities to discriminate a single-base alteration (data not shown). Four real-time PCRs, targeting exons 4 and 5 of CYP2C19, were performed with these MagiProbes. After optimization of the measuring temperature of fluorescence, perfect discriminated results were obtained (Fig. 10).
Figure 10.

Real-time PCR for the discrimination of CYP2C19 polymorphisms. A fragment of exon 4 of CYP2C19 was amplified in the presence of MagiProbe [(A) perfect match for the CYP2C19 636G-allele; (B) perfect match for the CYP2C19 636A-allele]. A fragment of exon 5 of CYP2C19 also was amplified in the presence of MagiProbe [(C) perfect match for the CYP2C19 681G-allele; (D) perfect match for the CYP2C19 681A-allele]. Plasmid DNA bearing a fragment of each allele was used as the template (red line, G-allele of 636 and 681; green line, A-allele of 636 and 681). Fluorescence was monitored at 65°C in the extra step after the annealing phase. In all the reactions, except the negative controls, amplified products were confirmed by electrophoresis whether or not an increase in fluorescence was found.
DISCUSSION
MagiProbe is a kind of molecular fluorescence switch between the ‘on’ and ‘off’ states that is driven by the conformational change between its single- and double-stranded forms. In the single-stranded form, direct energy transfer occurs from fluorescein to pyrene because the distance between them is close. Duplex formation significantly lessens the chance of collision because of the strong interaction of pyrene with nucleic acid base pairs, called intercalation, and restores spectral appearance of free fluorescein. Although a molecular beacon acts as a molecular fluorescent switch for nucleic acid detection, there are several alternatives to the molecular beacon. Kurata et al. (24) and Crockett and Wittwer (25) reported independently that probe fluorescence is increased or decreased by hybridization because of quenching by guanine bases. They made successful applications to real-time PCR and mutation detection, but the base sequence for the probe design was restricted. Knemeyer et al. (26) reported a modified molecular beacon, in which consecutive guanine bases that form the stem structure with opposite cytosine bases act as a quencher. Another approach is a light-up probe (27,28), in which the fluorescence of the dye conjugated to the oligonucleotide probe is enhanced on hybridization. MagiProbe differs from those methods, in which hybridization behavior follows regular hybridization kinetics, and it is not like a molecular beacon, in which the extra stem–loop structure affects hybridization properties. Accordingly, concern about restriction of the target sequence for MagiProbe is unnecessary. MagiProbe therefore has different properties, its most prominent property being its action as a fluorescence chemosensor for single-base discrimination. Although some of the methods cited are applicable to single-base discrimination, all rely on the thermal stability difference between a perfect matched and a one-base mismatched duplex, which difference may be very small and may vary significantly depending on the sequence context. MagiProbe does not have this problem because its discrimination is independent of the difference in thermal stability.
The current single-base discrimination of MagiProbe needs to be improved. To raise its single-base discrimination ability, the intercalator can be controlled so as to locate between fixed base pairs, of which one is involved in the mispairing in the mismatched duplex. Molecular designs for the backbone and the spacers of labels are currently being investigated. Despite its present imperfect single-base discrimination, MagiProbe does detect perfectly single-nucleotide change in conjunction with the thermal stability difference (Fig. 10).
No significant effects of the base sequences surrounding the fluorophore and intercalator have been observed, but systematic examination of such a possibility is important for many types of single nucleotide polymorphism detection. The only drawback of MagiProbe is the target-independent fluorescence caused by inter- or intramolecular base-pairing in the probe itself. This phenomenon can be controlled by reducing the self-complementary and palindromic character of the probe sequence. Even with an undesired sequence, background fluorescence can be controlled by careful positioning of the fluorophore and intercalator. Lastly, for MagiProbe color multiplexing, fluorescein can be replaced by another fluorescence dye, or an additional fluorescence dye, which acts as a fluorescence acceptor, can be introduced to MagiProbe as a third label. This approach was used successfully with the molecular beacon (29) and duplex scorpion primers (30).
Acknowledgments
ACKNOWLEDGEMENTS
I thank Dr Isao Saito and Dr Kazuhiko Nakatani of Kyoto University for their helpful discussions of this work.
REFERENCES
- 1.Didenko V.V. (2001) DNA probes using fluorescence resonance energy transfer (FRET): designs and applications. Biotechniques, 31, 1106–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Livak K.J., Flood,S.J.A., Marmaro,J., Giusti,W. and Deetz,K. (1995) Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl., 4, 357–362. [DOI] [PubMed] [Google Scholar]
- 3.Tyagi S. and Kramer,F.R. (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol., 14, 303–308. [DOI] [PubMed] [Google Scholar]
- 4.Nazarenko I.A., Bhatnagar,S.K. and Hohman,R.J. (1997) A closed tube format for amplification and detection of DNA based on energy transfer. Nucleic Acids Res., 25, 2516–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitcombe D., Theaker,J., Guy,S.P., Brown,T. and Little,S. (1999) Detection of PCR products using self-probing amplicons and fluorescence. Nat. Biotechnol., 17, 804–807. [DOI] [PubMed] [Google Scholar]
- 6.Bernard P.S., Pritham,G.H. and Wittwer,C.T. (1999) Color multiplexing hybridization probes using the apolipoprotein E locus as a model system for genotyping. Anal. Biochem., 273, 221–228. [DOI] [PubMed] [Google Scholar]
- 7.Kwiatkowski R.W., Lyamichev,V., de Arruda,M. and Neri,B. (1999) Clinical, genetic, and pharmacogenetic applications of the Invader assay. Mol. Diagn., 4, 353–364. [DOI] [PubMed] [Google Scholar]
- 8.Boon E.M., Ceres,D.M., Drummond,T.G., Hill,M.G. and Barton,J.K. (2000) Mutation detection by electorocatalysis at DNA-modified electrodes. Nat. Biotechnol., 18, 1096–1100. [DOI] [PubMed] [Google Scholar]
- 9.Nakatani K., Sando,S. and Saito,I. (2001) Scanning of guanine-guanine mismatches in DNA by synthetic ligands using surface plasmon resonance. Nat. Biotechnol., 19, 51–55. [DOI] [PubMed] [Google Scholar]
- 10.Yamana K., Zako,H., Asazuma,K., Iwase,R., Nakano,H. and Murakami,A. (2001) Fluorescence detection of specific RNA sequences using 2′-pyrene-modified oligoribonucleotides. Angew. Chem. Int. Ed., 40, 1104–1106. [DOI] [PubMed] [Google Scholar]
- 11.Shinozuka K., Seto,Y. and Sawai,H. (1994) A novel multifunctionally labeled DNA probe bearing an intercalator and a fluorophore. J. Chem. Soc. Chem. Commun., 1377–1378. [Google Scholar]
- 12.Tyagi S., Bratu,D.P. and Kramer,F.R. (1998) Multicolor molecular beacons for allele discrimination. Nat. Biotechnol., 16, 49–53. [DOI] [PubMed] [Google Scholar]
- 13.Yamane A. (2000) Smart probe: a novel fluorescence quenching-based oligonucleotide probe carrying a fluorophore and an intercalator. Nucleic Acids Symp. Ser., 44, 297–298. [DOI] [PubMed] [Google Scholar]
- 14.Yamane A. (2001) MagiProbe: a novel fluorescence quenching-based oligonucleotide probe carrying a fluorophore and an intercalator (II). Nucleic Acids Symp. Ser., 181–182. [DOI] [PubMed] [Google Scholar]
- 15.Ranasinghe R.T., Brown,L.J. and Brown,T. (2001) Linear fluorescent oligonucleotide probes with an acridine quencher generate a signal upon hybridization. Chem. Commun., 1480–1481. [Google Scholar]
- 16.Sinha N.D. and Striepeke,S. (1991) Oligonucleotides with reporter groups attached to the 5′-terminus. In Eckstein,F. (ed.), Oligonucleotides and Analogues—A Practical Approach. IRL Press, Oxford, pp. 185–210.
- 17.Saiki R.K., Scharf,S., Faloona,F., Mullis,K.B., Horn,G.T., Erlich,H.A. and Arnheim,N. (1985) Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science, 230, 1350–1354. [DOI] [PubMed] [Google Scholar]
- 18.De Morais S.M.F., Wilkinson,G.R., Blaisdell,J., Nakamura,K., Meyer,U.A. and Goldstein,J.A. (1994) The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J. Biol. Chem., 269, 15419–15422. [PubMed] [Google Scholar]
- 19.De Morais S.M., Wilkinson,G.R., Blaisdell,J., Meyer,U.A., Nakamura,K. and Goldstein,J.A. (1994) Identification of a new genetic defect responsible for the polymorphism of (S)-mephnytoin metabolism in Japanese. Mol. Pharmacol., 46, 594–598. [PubMed] [Google Scholar]
- 20.Horn T., Chang,C.A. and Urdea,M.S. (1997) Chemical synthesis and characterization of branched oligodeoxyribonucleotides (bDNA) for use as signal amplifiers in nucleic acid quantification assays. Nucleic Acids Res., 25, 4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Telser J., Cruickshank,K.A., Morrison,L.E. and Netzel,T.L. (1989) Synthesis and characterization of DNA oligomers and duplexes containing covalently attached molecular labels: comparison of biotin, fluorescein, and pyrene labels by thermodynamic and optical spectroscopic measurements. J. Am. Chem. Soc., 111, 6966–6976. [Google Scholar]
- 22.Telser J., Cruickshank,K.A., Morrison,L.E., Netzel,T.L. and Chan,C.K. (1989) DNA duplexes covalently labeled at two sites: synthesis and characterization by steady-state and time-resolved optical spectroscopies. J. Am. Chem. Soc., 111, 7226–7232. [Google Scholar]
- 23.Conner B.J., Reyes,A.A., Morin,C., Itakura,K., Teplitz,R.L. and Wallace,R.B. (1983) Detection of sickle cell βS-globin allele by hybridization with synthetic oligonucleotides. Proc. Natl Acad. Sci. USA, 80, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurata S., Kanagawa,T., Yamada,K., Tomimura,M., Yokomaku,T., Kamagata,Y. and Kurane,R. (2001) Fluorescent quenching-based quantitative detection of specific DNA/RNA using a BODIPY® FL-labeled probe or primer. Nucleic Acids Res., 29, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crockett A.O. and Wittwer,C.T. (2001) Fluorescein-labeled oligonucleotides for real-time PCR: using the inherent quenching of deoxyguanosine nucleotides. Anal. Biochem., 290, 89–97. [DOI] [PubMed] [Google Scholar]
- 26.Knemeyer J.P., Marmé,N. and Sauer,M. (2000) Probes for detection of specific DNA sequences at the single-molecule level. Anal. Chem., 72, 3717–3724. [DOI] [PubMed] [Google Scholar]
- 27.Ishiguro T., Saito,J., Yawata,H., Otsuka,M., Inoue,T. and Sugiura,Y. (1996) Fluorescence detection of specific sequence of nucleic acids by oxazole yellow-linked oligonucleotides. Homogeneous quantitative monitoring of in vitro transcription. Nucleic Acids Res., 24, 4992–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svanvik N., Westman,G., Wang,D. and Kubista,M. (2000) Light-up probes: thiazole orange-conjugated peptide nucleic acid for detection of target nucleic acid in homogeneous solution. Anal. Biochem., 281, 26–35. [DOI] [PubMed] [Google Scholar]
- 29.Tyagi S., Marras,S.A.E. and Kramer,F.R. (2000) Wavelength-shifting molecular beacons. Nat. Biotechnol., 18, 1191–1196. [DOI] [PubMed] [Google Scholar]
- 30.Solinas A., Brown,L.J., McKeen,C., Mellor,J.M., Nicol,J.T.G., Thelwell,N. and Brown,T. (2001) Duplex Scorpion primers in SNP analysis and FRET applications. Nucleic Acids Res., 29, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]