


Abstract
A general problem that exists in the assembly of large and organized DNA structures from smaller fragments is secondary structure that blocks or prevents it. For example, it is common to assemble longer synthetic DNA and RNA fragments by ligation of smaller synthesized units, but blocking secondary structure can prevent the formation of the intended complex before enzymatic ligation can occur. In addition, there is a general need for protecting groups that would block reactivity of some DNA bases in a sequence, leaving others free to react or hybridize. Here we describe such a strategy. The approach involves the protecting group dimethylacetamidine (Dma), which we show to remain intact on exocyclic amines of adenine bases while other bases carrying commercially available ‘ultra mild deprotection’ protecting groups are removed by potassium carbonate in methanol. The intact Dma groups prevent unwanted hybridization at undesired sites, thus encouraging it to occur where intended, and allowing for successful ligations. The Dma group is then deprotected by treatment with ammonia in methanol. Other common amine protecting groups such as benzoyl and allyloxycarbonyl were not successful in such a strategy, at least in part because they did not prevent hybridization. We demonstrate the method in the synthesis of a circular 54mer oligonucleotide composed of nine human telomere repeats, which was not possible to assemble by conventional methods.
INTRODUCTION
The joining of DNA fragments is a ubiquitous chemical reaction that makes possible many of the current technological breakthroughs in modern biology and medicine. For example, enzyme-catalyzed ligation of DNA has formed the basis for modern cloning. Furthermore, enzymatic and chemical ligations of DNA have recently become useful in genetic screening methods (1–9) and in the preparation of longer DNA and RNA fragments from smaller synthetic segments (10–14).
Although many methods have been developed for joining of DNAs, there remain broad classes of DNA sequence that are extremely difficult, if not impossible, to ligate: namely, repetitive and/or highly structured sequences, particularly when they are single stranded. Repetitive DNA sequences are quite common throughout the human genome, and repeating sequences are also associated with a number of diseases (15–20). The unusual secondary structures that arise as a result of this sequence repetition are thought to be important contributors to these diseases. Furthermore, the telomeric sequences at the ends of eukaryotic chromosomes are also highly repetitive and highly structured (21–23). In order to understand the structure, biochemistry and biology of such repetitive and highly structured sequences it is necessary to find convenient ways to construct them. While small segments (∼100 nt and less) can be readily produced on an automated synthesizer, longer segments cannot, and most of these biologically important repeats occur over much longer lengths than 100 nt. Unfortunately, making larger segments of these repeats is virtually impossible by performing ligations (enzymatic or chemical) of smaller segments.
The reason for this is simple. Nearly all enzymatic and chemical methods of joining DNAs rely on the template effect, whereby the complementary binding of one unbroken template strand hybridizing across the two broken ends brings the reactive ends into close proximity. If two single-stranded ends to be ligated are part of a longer repeating sequence, such a template, or splint, has several (or many) alternative sites in which to bind, and ligation fails. In addition, these repetitive sequences usually form ordered and stable secondary structures that also can prevent splints from binding productively. Here we report on a solution to this general problem, in which we use an orthogonal protecting group strategy to selectively encourage a template DNA to hybridize at its ends by preventing unwanted secondary structure, allowing for successful ligation.
There have been some recent reports of orthogonal protecting groups for oligonucleotide synthesis, and some of these have enabled chemical modifications and conjugations that would otherwise have been difficult. For example, the allyloxycarbonyl (alloc) protecting group has been useful in the modification of DNA attached to a solid support since the alloc group can be removed without simultaneously cleaving the DNA from the support on which it is prepared (24,25). Alternatively, the alloc protecting group can allow for phosphate group deprotection and removal of an oligonucleotide from the solid support without deprotection of the DNA bases (U.M.Lindström and E.T.Kool, unpublished results).
Here we describe a different orthogonal protecting group strategy, which became necessary in the synthesis and ligation of linear and circular DNA oligonucleotides of repeating telomere sequence. Human telomeric DNA sequences consist of a long hexanucleotide repeat, and these are under widespread investigation for their roles in aging and cancer. The present strategy was developed to enable the synthesis of circular oligonucleotides containing telomeric repeats; these molecules have recently been shown to co-catalyze the synthesis of artificial telomeres with DNA polymerases (U.M.Lindström, R.A.Chandrasekaran, L.Orbai, S.A.Helquist, G.P.Miller, E.Oroudjev, H.G.Hansma and E.T.Kool, manuscript submitted).
MATERIALS AND METHODS
Dimethylacetamidine (Dma)-protected nucleoside phosphoramidites
The N6-dimethylacetamidine-dA (Dma-dA) phosphoramidite was prepared according to the method described by McBride et al. (26). Spectroscopic data were in accordance with published data.
Oligonucleotide synthesis
DNA oligonucleotides were synthesized on an Applied Biosystems 392 synthesizer using β-cyanoethylphosphoramidite chemistry. Ultra-mild deprotection phosphoramidites were purchased from Glen Research. No changes to the standard protocol were needed for the couplings of the Dma-dA phosphoramidite. 5′-Phosphorylation was carried out with a phosphoramidite reagent (Glen Research). Deprotection was done with 0.05 M K2CO3/MeOH for 4–12 h at room temperature for the ultra-mild deprotection bases (a minimum of 8 h is needed when the 5′-phosphorylation reagent is used) following the protocol provided by Glen Research. Neutralization of the carbonate solution was accomplished by adding an equal volume of 2 M tetraethylammonium acetate. For the Dma-dA base, cleavage was achieved with either NH4OH for 8 h at 55°C or with ammonia/methylamine (AMA) reagent [NH4OH/MeNH2 (40% in water) 1:1] for at least 8 h at room temperature. Room temperature is preferred for circular DNA. Cleavage of the oligomer from the support was simultaneously accomplished under all of these conditions. Oligomers were purified by preparative 20% denaturing polyacrylamide gel electrophoresis (PAGE) and quantified by absorbance at 260 nm. Molar extinction coefficients for the oligonucleotides were calculated using the nearest neighbor method.
Spectroscopic data for oligodeoxynucleotides
d(CAcAPacGiPr-PacADmaT)-CPG → d(CAGAT) (AMA, 12 h, room temperature): MALDI-TOF-MS calculated for C49H63N20O27P4 (M+H): 1488.03. Found: 1490.76.
d(CAcAPacGiPr-PacADmaT)-CPG → d(CAGADmaT) [K2CO3/MeOH, 12 h, room temperature]: MALDI-TOF-MS calculated for C53H70N21O27P4 (M+H): 1557.14. Found: 1559.52.
d(CAGADmaT) → d(CAGAT) [NH4OH, 8 h, 55°C]: MALDI-TOF-MS calculated for C49H63N20O27P4 (M+H): 1488.03. Found: 1488.57.
d(CAGADmaT) → d(CAGAT) [NH4OH/MeNH2 (40% in water) 1:1, 8 h, room temperature]: MALDI-TOF-MS calculated for C49H63N20O27P4 (M+H): 1488.03. Found: 1489.51.
Ligation reactions using orthogonal protecting groups
Ligation of the Dma-modified, 5′-phosphorylated DNA, d(5′- pAACCCTAACCCTAACCCTAACCCTAACCCTAACCC TAACCCTAACCCTAACCCT-3′), where underlines represent Dma-protected bases, was carried out using an 18mer DNA template, d(5′-GTTAGGGTTAGGGTTAGG-3′), to align the reactive ends and T4 DNA ligase (New England Biolabs) to achieve the ligation. The reactions were typically carried out in 50 mM Tris-buffer (pH 7.5) that contained 1 µM linear precircle, 1.2 µM template strand, 10 mM MgCl2, 5 µM ATP, 10 mM DTT and 0.34 U/µl ligase. One doubling of DNA concentrations can be done without affecting the yield. Reactions were incubated at room temperature for 18 h. The mixtures were then dialyzed against distilled water and lyophilized. Preparative purification of circular products was carried out using denaturing 20% polyacrylamide gels. Circular DNA was detected as a significantly slower moving band by UV-shadowing. Analytical gels were visualized with Stains-All dye (Sigma). Isolation of DNA was accomplished by crushing the gel and extracting with 0.2 M NaCl for 12 h. Conversion of precircle to circle often appeared high (∼50–70%) by UV-shadowing, but isolated yields based on the linear precursor were usually <30%. Oligonucleotides were obtained as the sodium salt after dialysis and yields of the purified circular products were determined by UV absorbance at 260 nm. Final deprotection of circle was done by overnight treatment with AMA at room temperature. Then, lyophilization afforded the final product. Confirmation of circularity was provided by nicking with S1 endonuclease; initial cleavage of circle produces a single band with the mobility of the full-length linear precursor. The S1 experiment was carried out as follows. The circular product and the linear precursor DNAs (0.2 nmol) were dried down in two separate tubes, and then water (0.5 µl) and 2× S1 buffer (1.5 µl) were added to each tube. The samples were briefly vortexed and centrifuged before S1 endonuclease (1 µl; 1 U/µl) was added to each reaction. After incubating for 5 min at 37°C, the reaction was stopped by adding gel-loading buffer (3 µl) and heating briefly at 65°C. The products of cleavage were analyzed by 20% denaturing PAGE.
RESULTS AND DISCUSSION
Tests of several known DNA protecting groups led us to choose the Dma protecting group on adenine as a group that is stable in mildly basic deprotection conditions. Our ligation plans required that the protecting group not only withstand alkaline conditions, but that it also prevent hybridization of a complementary strand. We initially considered the benzoyl group as a suitable candidate, since it is removed slowly under very mild deprotection conditions. Surprisingly, in experiments with this strategy we found that the N6-benzoyl group on adenine does not efficiently block hybridization of a complementary DNA strand. One test sequence (a 20mer containing six N-benzoyl adenines and nine N-benzoyl cytosines) showed a Tm depression of only 1.0°C relative to the sequence lacking benzoyl groups (U.M.Lindström and E.T.Kool, unpublished results).
The above-mentioned alloc protecting group was also tested, and was observed to only weakly destabilize duplexes in which it was substituted. The use of the more sterically demanding N,N-dibenzoyl-protected dA (27) indeed afforded circular product after splint-assisted ligation, but unfortunately this strategy made the DNA more liable to depurinate under the acidic conditions of DNA synthesis, causing rapid decomposition of the circle.
The Dma protecting group, in contrast, is compatible with automated solid-phase synthesis and offers both ease of addition and removal (26), as well as successful inhibition of hybridization (see below). Although the Dma group had not been previously investigated as an orthogonal protecting group, we find that it serves admirably in this role.
We prepared an N6-Dma-protected deoxyadenosine phosphoramidite derivative following the reported methods (26). In order to monitor its efficiency in oligonucleotide synthesis, and its stability to the ultra-mild post-synthetic cleavage and deprotection conditions, we prepared a short oligomer containing the ultra-mild deprotection versions of dC, dA and dG (Glen Research), as well as the Dma-dA, and the unprotected T. No modifications to the standard solid-phase DNA-synthesis protocol were needed for the Dma-dA and the coupling yield was >98%. When the mixed-protecting-group pentamer was subjected to 0.05 M K2CO3 in MeOH for 12 h, the Dma-dA remained intact while the other bases were deprotected and the DNA was simultaneously cleaved from the support. This was confirmed by gel electrophoresis (Fig. 1, lane 2; the presence of the Dma group slightly retards the DNA on a 20% denaturing polyacrylamide gel) and by MALDI-TOF spectrometry. The resulting mono-protected pentamer could then be fully deprotected using the standard post-synthetic procedure of heating the DNA at 55°C in concentrated NH4OH for at least 8 h (Fig. 1, lanes 4 and 5). Cleavage of the Dma group could also be accomplished at room temperature (8 h) by the more powerful AMA reagent (Fig. 1, lane 3). In our hands this was the preferred method for use of the orthogonal protecting groups.
Figure 1.
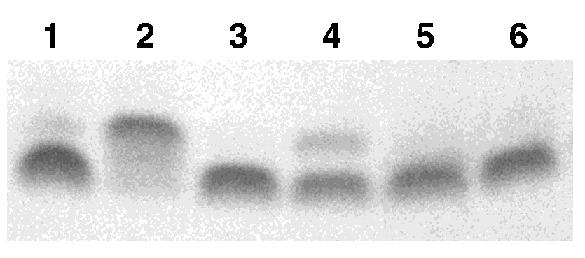
Denaturing PAGE analysis of pentamer DNA with mixed protecting groups illustrating the difference in reactivity between the Dma protecting group of dA and the ultra-mild deprotection groups for the conditions described (see also text). Slower moving bands are Dma-protected DNA. Conditions: lane 1, AMA, 8 h; lane 2, K2CO3/MeOH, 12 h; lane 3, K2CO3/MeOH, 12 h, then AMA, 8 h; lane 4, NH4OH, 55°C, 4 h; lane 5, NH4OH, 55°C, 8 h; lane 6, native ssDNA marker.
We initially considered the use of the Dma group on both dC and dA bases in order to disrupt hybridization most strongly. However, this was found not to be necessary, and in addition, the deprotection of dC was found to pose problems. When Dma-dC was included, the mass obtained after K2CO3 treatment for 12 h corresponded to a product with only the dA carrying a Dma group. Shorter deprotection times did not improve on this. These results suggest that Dma is removed significantly more rapidly from dC than from dA.
Once the orthogonality of the Dma group to the other protecting groups for the ultra-mild deprotection conditions had been established, we set out to investigate the usefulness of this strategy for the ligation of DNAs of repetitive sequences. This was done by attempting a template-assisted cyclization of a linear DNA precursor containing many short repeats. Our interest in telomere structure and function prompted us to use the circular telomeric repeat sequence d(CCCTAA)9. In order for the splint to hybridize only at the ends, the Dma-dA phosphoramidite was inserted at all A positions in the oligomer except within a distance of 10 bases from each end. Thus, in all, the 54mer contained 12 Dma-modified dAs. The automated solid phase assembly was straightforward and gave >98.5% average stepwise yield. The 5′ end was phosphorylated as required for enzyme-assisted ligation. Following an overnight K2CO3 treatment, the resulting semi-protected DNA was neutralized with 2 M tetramethylammonium acetate, then dialyzed and lyophilized.
Ligation was performed with 1.5 equivalents of an 18mer DNA template to align the reactive ends and T4 DNA ligase to achieve the ligation. The reaction was incubated at room temperature for 18 h, and the solution dialyzed and lyophilized. Gratifyingly, denaturing PAGE analysis confirmed significant conversion of precircle to circle (Fig. 2, lane 2). In a control experiment, ligation was attempted with unprotected DNA of the same sequence and length. As expected, this did not result in any observable formation of circular product (Fig. 2, lane 4). Finally, the remaining 12 Dma groups were removed by treatment with AMA for 8 h at room temperature to afford the deprotected circle of nine uninterrupted hexameric repeats (Fig. 2, lane 5). Typically, isolated yields were between 15 and 30% based on amounts of the linear precursor. Circularity was confirmed by S1 endonuclease cleavage (Fig. 3; see also Materials and Methods).
Figure 2.

Analysis of ligation reactions showing the efficiency of Dma protection in promoting template-assisted cyclization of a linear DNA precursor consisting of nine hexamer repeats of the human telomere sequence 5′-dCCCTAA (PAGE, 20%) (see also text). Lane 1, Dma- protected precircle; lane 2, ligation of Dma-protected precircle; lane 3, native precircle; lane 4, attempted ligation of native precircle; lane 5, DNA circle after final cleavage of Dma with AMA reagent.
Figure 3.

Gel analysis of S1 endonuclease cleavage of the circular product [cyclic (dCCCTAA)9] and the linear precursor (PAGE, 20%). Circularity is confirmed by the lack of banding between the bands corresponding to the circular and linear DNA (see text for details). Lane 1, circular DNA, no nuclease S1; lane 2, circular DNA, S1 reaction; lane 3, linear DNA, no S1; lane 4, linear DNA, S1 reaction.
In conclusion, we find that the Dma protecting group successfully resists hydrolysis when other standard groups are removed from oligonucleotides. In addition, we find that Dma-protected dA prevents unwanted hybridization where it would otherwise interfere with subsequent manipulation (such as in ligation). We expect that this same strategy may also be useful in another class of DNA that is refractory to ligation: namely, structured DNA. If one or both DNA ends to be ligated form a hairpin or other stable structure, this may well prevent binding of a complementary splint, and thus prevent ligation. The present orthogonal protection approach could prevent such undesired secondary structure, making ligations proceed where they were otherwise blocked. Finally, such a strategy may find special utility in chemical and enzymatic modifications of DNA, by blocking reactivity at unwanted bases, thus encouraging it at others.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the U.S. National Institutes of Health (RR15054 and GM62658) for partial support of this work. U.M.L. acknowledges the Swedish Research Council for a postdoctoral fellowship.
REFERENCES
- 1.Xu Y., Karalkar,N.B. and Kool,E.T. (2001) Nonenzymatic autoligation in direct three-color detection of RNA and DNA point mutations. Nat. Biotechnol., 19, 148. [DOI] [PubMed] [Google Scholar]
- 2.Nilsson M., Barbany,G., Antson,D., Gertow,K. and Landegren,U. (2000) Enhanced detection and distinction of RNA by enzymatic probe ligation. Nat. Biotechnol., 18, 791–793. [DOI] [PubMed] [Google Scholar]
- 3.Gunderson K.L., Huang,X.C., Morris,M.S., Lipshutz,R.J., Lockhart,D.J. and Chee,M.S. (1998) Mutation detection by ligation to complete n-mer DNA arrays. Genome Res., 8, 1142–1153. [DOI] [PubMed] [Google Scholar]
- 4.Landegren U., Samiotaki,M., Nilsson,M., Malmgren,H. and Kwiatowski,M. (1996) Detecting genes with ligases. Methods, 9, 84–90. [DOI] [PubMed] [Google Scholar]
- 5.Samiotaki M., Kwiatkowski,M., Parik,J. and Landegren,U. (1994) Dual-color detection of DNA sequence variants by ligase-mediated analysis. Genomics, 20, 238–242. [DOI] [PubMed] [Google Scholar]
- 6.Nickerson D.A., Kaiser,R., Lappin,S., Stewart,J., Hood,L. and Landegren,U. (1990) Automated DNA diagnostics using an ELISA-based oligonucleotide ligation assay. Proc. Natl Acad. Sci. USA, 87, 8923–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barringer K.J., Orgel,L., Wahl,G. and Gingeras,T.R. (1990) Blunt-end and single-strand ligations by Escherichia coli ligase: influence on an in vitro amplification scheme. Gene, 89, 117–122. [DOI] [PubMed] [Google Scholar]
- 8.Wu D.Y. and Wallace,R.B. (1989) The ligation amplification reaction (LAR)-amplification of specific DNA sequences using sequential rounds of template-dependent ligation. Genomics, 4, 560–569. [DOI] [PubMed] [Google Scholar]
- 9.Landegren U., Kaiser,R., Sanders,J. and Hood,L. (1988) A ligase-mediated gene detection technique. Science, 241, 1077–1080. [DOI] [PubMed] [Google Scholar]
- 10.Shabarova Z.A., Merenkova,I.N., Oretskaya,T.S., Sokolova,N.I., Skripkin,E.A., Alexeyeva,E.V., Balakin,A.G. and Bogdanov,A.A. (1991) Chemical ligation of DNA: the first non-enzymatic assembly of a biologically active gene. Nucleic Acids Res., 19, 4247–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferretti L., Karnik,S.S., Khorana,H.G., Nassal,M. and Oprian,D.D. (1986) Total synthesis of a gene for bovine rhodopsin. Proc. Natl Acad. Sci. USA, 83, 599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J.H. and Seeman,N.C. (1991) The synthesis from DNA of a molecule with the connectivity of a cube. Nature, 350, 631–633. [DOI] [PubMed] [Google Scholar]
- 13.Seeman N.C. (1998) DNA nanotechnology: novel DNA constructions. Annu. Rev. Biophys. Biomol. Struct., 27, 225–248. [DOI] [PubMed] [Google Scholar]
- 14.Yan H., Zhang,X., Shen,Z. and Seeman,N.C. (2002) A robust DNA mechanical device controlled by hybridization topology. Nature, 415, 62–65. [DOI] [PubMed] [Google Scholar]
- 15.Kovtun I.V., Goellner,G. and McMurray,C.T. (2001) Structural features of trinucleotide repeats associated with DNA expansion. Biochem. Cell Biol., 79, 325–336. [PubMed] [Google Scholar]
- 16.Bowater R.P. and Wells,R.D. (2001) The intrinsically unstable life of DNA triplet repeats associated with human hereditary disorders. Progr. Nucleic Acids Res. Mol. Biol., 66, 159–202. [DOI] [PubMed] [Google Scholar]
- 17.Cummings C.J. and Zoghbi,H.Y. (2000) Trinucleotide repeats: mechanisms and pathophysiology. Annu. Rev. Genom. Hum. Genet., 1, 281–328. [DOI] [PubMed] [Google Scholar]
- 18.Usdin K. and Grabczyk,E. (2000) DNA repeat expansions and human disease. Cell. Mol. Life Sci., 57, 914–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cummings C.J. and Zoghbi,H.Y. (2000) Fourteen and counting: unraveling trinucleotide repeat diseases. Hum. Mol. Genet., 9, 909–916. [DOI] [PubMed] [Google Scholar]
- 20.Singer R.H. (1998) Triplet-repeat transcripts: a role for RNA in disease. Science, 280, 696–697. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y. and Patel,D.J. (1993) Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure, 1, 263–282. [DOI] [PubMed] [Google Scholar]
- 22.Wright W.E., Tesmer,V.M., Huffman,K.E., Levene,S.D. and Shay,J.W. (1997) Normal human chromosomes have long G-rich telomeric overhangs at one end. Genes Dev., 11, 2801–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkinson G.N., Lee,M.P.H. and Neidle,S. (2002) Crystal structure of parallel quadruplexes from human telomeric DNA. Nature, 417, 876–880. [DOI] [PubMed] [Google Scholar]
- 24.Pirrung M.C., Fallon,L., Lever,D.C. and Shuey,S.W. (1996) Inverse phosphotriester DNA synthesis using photochemically-removable dimethoxybenzoin phosphate protecting groups. J. Org. Chem., 61, 2129–2136. [Google Scholar]
- 25.Hayakawa Y., Kato,H., Uchiyama,M., Kajino,H. and Noyori,R. (1986) Allyloxycarbonyl group: a versatile blocking group for nucleotide synthesis. J. Org. Chem., 51, 2400–2402. [Google Scholar]
- 26.McBride L.J., Kierzek,R., Beaucage,S.L. and Caruthers,M.H. (1986) Amidine protecting groups for oligonucleotide synthesis. J. Am. Chem. Soc., 108, 2040–2048. [Google Scholar]
- 27.Ti G.S., Gaffney,B.L. and Jones,R.A. (1982) Transient protection: efficient one-flask syntheses of protected deoxynucleosides. J. Am. Chem. Soc., 104, 1316–1319. [Google Scholar]