


Abstract
Illegitimate recombination (IR) is the process by which two DNA molecules not sharing homology to each other are joined. In Kluyveromyces lactis, integration of heterologous DNA occurred very frequently therefore constituting an excellent model organism to study IR. IR was completely dependent on the nonhomologous end-joining (NHEJ) pathway for DNA double strand break (DSB) repair and we detected no other pathways capable of mediating IR. NHEJ was very versatile, capable of repairing both blunt and non-complementary ends efficiently. Mapping the locations of genomic IR-events revealed target site preferences, in which intergenic regions (IGRs) and ribosomal DNA were overrepresented six-fold compared to open reading frames (ORFs). The IGR-events occurred predominantly within transcriptional regulatory regions. In a rad52 mutant strain IR still preferentially occurred at IGRs, indicating that DSBs in ORFs were not primarily repaired by homologous recombination (HR). Introduction of ectopic DSBs resulted in the efficient targeting of IR to these sites, strongly suggesting that IR occurred at spontaneous mitotic DSBs. The targeting efficiency was equal when ectopic breaks were introduced in an ORF or an IGR. We propose that spontaneous DSBs arise more frequently in transcriptional regulatory regions and in rDNA and such DSBs can be mapped by analyzing IR target sites.
INTRODUCTION
Living cells are challenged with a large number of DNA lesions in every cell cycle. Among these lesions, the DNA double strand break (DSB) is the most severe, leading to cell death if not repaired. Moreover, DSBs repaired in an incorrect manner can also be detrimental to the organism. Examples are the formation of oncogenic fusion genes or the juxtaposition of oncogenes to strong enhancers, both of which are a primary cause of malignancy. Studying how DSBs arise and how they are repaired is thus of vital interest. Cells have evolved two major pathways for DSB repair, the homologous recombination (HR) and the nonhomologous end-joining (NHEJ) pathways. The HR pathway (1,2) depends on the presence of a homologous chromosome or sister chromatid that is used as a template for homology-driven and mostly error-free repair. HR in fact refers to several different processes, including gene conversion and alternative pathways called break-induced replication (BIR) and single strand annealing (SSA). A key-protein in HR is Rad52, which forms a heptameric ring in vitro (3), that interacts with single stranded DNA and facilitates the strand invasion reaction (4,5). Rad52 is thought to act early during HR and strains lacking Rad52 are completely deficient for all types of HR in Saccharomyces cerevisiae. Another important protein is Rad51 (similar to bacterial RecA), which facilitates the strand exchange reaction between the damaged substrate and the undamaged template molecule (6).
In contrast, the NHEJ pathway requires little or no homology and simply fuses two free DNA-ends, often generating small deletions and insertions. Among the proteins required for NHEJ are the Ku70/Ku80 heterodimer (7), which has high affinity for DNA ends. The heterodimer binds to the DSB protecting the free ends from extensive degradation and holds the DNA ends in an appropriate configuration for subsequent end-processing and ligation (8). Binding of Ku70/Ku80 is required for the recruitment of a second complex to the DSB (9). This complex consists of a DNA ligase, Dnl4 (or Lig4) (10–12) and Lif1 (Ligase four interacting factor 1) (13), the latter being the yeast functional homologue of mammalian Xrcc4. Lif1 is required for the stability and full activity of Dnl4 (14). A third complex consisting of Mre11, Rad50 and Xrs2 (MRX), is also required for efficient NHEJ in S.cerevisiae (15,16), but not in mammals or fission yeast (17–20). Mre11 has an endo/exo nuclease activity, but this activity appears to be dispensable for NHEJ in vivo (21). The MRX-complex promotes the end-joining reaction in vitro (22), but the exact role of this complex in NHEJ remains unclear. In S.cerevisiae, cell-type regulates NHEJ, haploid MATa or MATα strains perform NHEJ efficiently, but diploid MATa/MATα strains perform NHEJ inefficiently (23,24). In diploids, the a1/α2 repressor inhibits the transcription of the NEJ1 gene. Nej1 interacts with Lif1 and constitutes an essential component of the NHEJ pathway (25–28). Diploid cells can rely on the HR pathway to repair DSBs even in the G1 phase of the cell cycle, since these cells contain a homologous chromosome. Because HR is more accurate than NHEJ this cell-type regulation may have evolved to promote genome stability in S.cerevisiae.
Despite the high degree of molecular conservation in NHEJ mechanisms between yeast and mammalian cells, there are some striking differences regarding the in vivo function. In mammals, NHEJ is the major pathway for DSB repair. In S.cerevisiae, the HR pathway for DSB repair is very active and the NHEJ pathway has largely a back-up function at least under laboratory conditions. Consequently, S.cerevisiae mutant strains deficient for NHEJ do not have any obvious growth disadvantages. In contrast, in mice some NHEJ proteins (Ligase IV and Xrcc4) are required for embryonic development (29,30). In addition, S.cerevisiae strains lacking proteins essential for NHEJ such as Lig4, Lif1 and Nej1 are not more sensitive to DNA damaging agents than a wild-type strain, whereas mammalian cells lacking NHEJ components are highly sensitive to DNA damage (31).
Since both the HR and NHEJ pathways use the same substrate, a DSB, a competition between them can be envisaged. Thus, inactivation of the NHEJ pathway would lead to an increase of homologous integration events upon introduction of a gene targeting cassette. This hypothesis was tested in mouse ES-cells, but NHEJ mutant cells did not have a more favorable gene targeting frequency (32). In S.cerevisiae, the reciprocal experiment showed that the NHEJ efficiency did not increase in the absence of HR-components (23,33). A moderate competition between NHEJ and HR has been observed, however, when genomic DSBs were introduced in vivo using inducible endonucleases (20,32,34,35).
NHEJ, the direct rejoining of DNA DSBs, is closely associated with illegitimate recombination (IR) and chromosomal rearrangements. Hence, IR between two genomic loci results in deletions, duplications, insertions or translocations. In S.cerevisiae, the frequency of IR is reduced in rad50, mre11, xrs2 and hdf1 (yku70) mutant strains (36,37), indicating that IR takes place through the NHEJ pathway. Target sites of IR correlate with consensus sites for Topoisomerase I (38) and are dependent on the TOP1 gene. In addition, overexpression of Top1 leads to increased IR, demonstrating that the nicking activity of Top1 promotes IR (38).
In this study, we investigated IR and NHEJ in Kluyveromyces lactis (milk yeast). Similarly to mammalian cells, milk yeast NHEJ was capable of repairing blunt and noncohesive ends efficiently. Integration of a nonhomologous DNA molecule into the genome was 1000-fold more frequent than in S.cerevisiae and ectopically introduced DSBs were hotspots for integration. We present evidence supporting a model in which IR takes place at spontaneous mitotic DSB and that these breaks occurred more frequently within promoter regions and rDNA.
MATERIALS AND METHODS
Plasmids
For the gene disruption of K.lactis NEJ1, two PCR-fragments corresponding to the nucleotides −166 to +250, (the ATG start codon corresponding to nucleotides +1 to +3) and +474 to +895 were digested with SacI–BamHI and BamHI–XhoI, respectively. Both PCR-fragments were combined with a SacI–XhoI digested pBluescript SK(+) in a three-factor cloning, generating plasmid pAKP68. Targeting constructs for two-step gene disruption of the RAD52, RAD51 and YKU80 genes were all generated using a similar approach. For RAD52, two PCR fragments corresponding to nucleotides −329 to +112 (SacI–BamHI) and +835 to +1353 (BamHI–XhoI) were combined with SacI–SalI digested pRS406 (39) generating vector p580. For RAD51, two PCR fragments corresponding to nucleotides −536 to +65 (XhoI–BamHI) and +435 to +1085 (BamHI–XbaI) were combined with XhoI–XbaI digested pRS406 generating vector p182. For YKU80, two PCR fragments corresponding to nucleotides +5 to +177 (XhoI–BamHI) and +1267 to +1866 (BamHI–XbaI) were ligated with XhoI–XbaI digested pRS406 generating plasmid pAKP120. Into the resulting BamHI site of pAKP68, p580, p182 and pAKP120, a 2.9 kb BglII-fragment containing the LEU2 gene from vector pCXJ20 (40) was cloned, generating plasmid pAKP69 (pBluescript SK(+)-nej1::LEU2), p582 (pRS406-rad52::LEU2), p183 (pRS406-rad51::LEU2) and pAKP122 (pRS406-yku80::LEU2), respectively.
Introduction of the I-SceI-endonuclease recognition site within the VMR1 promoter region and within the VMR1 coding sequence was accomplished as follows. Two PCR fragments corresponding to nucleotides +1455 to −189, and to nucleotides −187 to −974 with respect to VMR1 start codon were digested with XhoI–HindIII and HindIII–NotI, respectively and ligated into XhoI–NotI digested pRS406, generating plasmid pAKP175. Two oligonucleotides (5 pmol/µl) (5′-agctacgctagggataacagggtaataca-3′) and (5′-agctgtattaccctgttatccctagcgt-3′) were annealed in 2 mM Tris–HCl (pH7.4), 0.2 mM MgCl2 and 5 mM NaCl and cloned into the HindIII site of pAKP175, generating plasmid pAKP179. A PCR fragment corresponding to nucleotide +2190 to +5097 with respect to the VMR1 start codon was digested with SacI–XhoI and ligated into SacI–XhoI digested pRS406, creating plasmid pPMB19. Two oligonucleotides, (5′-gcgccgctagggataacagggtaatac-3′) and (5′-gcgcgtattaccctgttatccctagcg-3′), were annealed and cloned into the VMR1 endogenous KasI site at position +3871, creating plasmid pPMB20. To express the I-SceI endonuclease in K.lactis, an XhoI–SalI fragment containing PGAL1-I-SCEI from plasmid pTW468 (kindly provided by T. E. Wilson) was ligated into SalI digested pCXJ18 (40), generating plasmid pAKP164. To measure IR at a specific DSB the promoter regions of two S.cerevisiae genes ADH1 (−1/−479) and TEF2 (−1/−1000) were PCR-amplified and digested with XbaI–BamHI and XhoI–BamHI, respectively. Both PCR-fragments were combined with SpeI–XhoI digested pRS405 in a three-factor cloning, generating plasmid pPMB18. The oligonucleotide sequences used in this study are available upon request.
Yeast strains
The strains used in this study are listed in Table 1. All null alleles generated were confirmed by genomic DNA-blots or locus specific PCR. Crossing the strain SAY119 with CK213-4c generated a diploid MATa/MATα strain (SAY681). Strain SAY572 (nej1::LEU2) was generated by a one-step gene disruption procedure (41) by transforming CK213-4c with a PCR-fragment using pAKP69 as template. Strain SAY573 (ku80::LEU2) was generated by a two-step gene disruption procedure (42) transforming CK213-4c with NheI-linearized pAKP122. A segregant from the cross of strain SAY119 with SAY572 generated a MATα nej::LEU2 strain (SAY509). SAY509 was crossed to SAY573 and from this cross the double mutant nej1::LEU2 ku80::LEU2 segregant (SAY574) was recovered by screening for a non-parental ditype (NPD) for Leu+. Strain SAY516 (rad51::LEU2) and strain SAY507 (rad52::LEU2) were generated in a two-step gene disruption procedure transforming CK213-4c with PmlI-linearized p183, and BglII-linearized p582, respectively. Crossing strain SAY516 with SAY509 and SAY507 with SAY509 generated the double mutant strains SAY510 (rad52::LEU2 nej1::LEU2) and SAY517 (rad51::LEU2 nej1::LEU2). The strains SAY545 (lig4::KANMX nej1::LEU2), SAY554 (rad50::KANMX nej1::LEU2) and SAY555 (mre11::KANMX nej1::LEU2) were generated from the parental SAY572 strain (MATa nej1::LEU2) using the PCR-based gene-targeting method described below. Strains SAY545, SAY554 and SAY555 were further crossed to SAY119 to obtain the wild-type NEJ1 segregants SAY683 (lig4::KANMX), SAY559 (mre11::KANMX) and SAY557 (rad50::KANMX).
Table 1.
Yeast strains used in this study
| K.lactis | Genotype | Reference |
|---|---|---|
| CK213-4c | MATaleu2 lysA1 metA1 trp1 uraA1 | (73) |
| SAY102 | MATaleu2 lysA1 metA1 trp1 uraA1 sir2::URA3 | (74) |
| SAY119 | MATα ade2 leu2 trp1 metA1 uraA1 | (56) |
| SAY186 | MATaleu2 lysA1 metA1 trp1 uraA1 hmlαΔp | (56) |
| SAY189 | MATaleu2 trp1 uraAI sir2::URA3 hmlαΔp | (56) |
| SAY507 | CK213-4C rad52::LEU2 | This study |
| SAY509 | MATα ade2 leu2 metA1 trp1 uraA1 nej1::LEU2 | This study |
| SAY510 | CK213-4C nej1::LEU2 rad52::LEU2 | This study |
| SAY516 | CK213-4C rad51::LEU2 | This study |
| SAY517 | CK213-4C nej1::LEU2 rad51::LEU | This study |
| SAY545 | CK213-4C nej1::LEU2 lig4::KANMX | This study |
| SAY554 | CK213-4C nej1::LEU2 rad50::KANMX | This study |
| SAY555 | CK213-4C nej1::LEU2 mre11::KANMX | This study |
| SAY557 | MATaleu2 lysA1 metA1 trp1 uraA1 rad50::KANMX | This study |
| SAY559 | MATaleu2 lysA1 metA1 trp1 uraA1 mre11::KANMX | This study |
| SAY572 | CK213-4C nej1::LEU2 | This study |
| SAY573 | CK213-4C ku80::EU2 | This study |
| SAY574 | MATα ade2 leu2 metA1 trp1 uraA1 ku80::LEU2 nej1::LEU2 | This study |
| SAY681 | MATa/MATα lysA1/LYSA1 leu2/leu2 uraA1/uraA1 ade1/ADE1 | This study |
| SAY683 | MATaleu2 lysA1 metA1 trp1 uraA1 lig4::KANMX | This study |
| SAY684 | CK213-4C PVMR1 (−189 I-SceI) | This study |
| SAY685 | MATaleu2 metA1 trp1 uraA1 nej1::LEU2 PVMR1 (−189 I-SceI) | This study |
| SAY686 | MATaleu2 lysA1 metA1 trp1 uraA1 rad52::LEU2 PVMR1 (−189 I-SceI) | This study |
| SAY687 | MATα ade2 leu2 metA1 trp1 uraA1 rad52::LEU2 | This study |
| PMY2 | MATa/MATα lysA1/LYSA1 trp1/trp1 leu2/leu2 metA1/metA1 uraA1/uraA1 ade2/ADE2 rad52::LEU2/rad52::LEU2 | This study |
| PMY34 | CK213-4C VMR1 (+ 3871 I-SceI) | This study |
Strain SAY684 and PMY34 were generated in a two-step gene replacement procedure transforming CK213-4c with either ClaI-linearized pAKP179 or AgeI-linearized pPMB20, respectively. Crossing strain SAY684 either with SAY509 or SAY687 generated the mutant segregants SAY685 (nej1::LEU2) and SAY686 (rad52::LEU2) both containing the I-SceI site in the VMR1 promoter. Strain SAY687 (MATα rad52::LEU2) is a segregant from the cross of strains SAY119 and SAY507. Strain PMY2 (MATa/MATα rad52::LEU2/rad52::LEU2 mutant strain was generated by crossing SAY687 and SAY507.
Growth media and standard methods
Media for growth of yeast and bacteria, protocols for RNA and DNA-blots, DNA and RNA preparation, yeast and bacterial transformations were carried out as described elsewhere (43–45). For detecting the NEJ1 transcript a [α32-P]dCTP-labeled PCR-fragment corresponding to the NEJ1 open reading frame (ORF) was used. The RNA-blot was analyzed using a phosphorimager. NHEJ efficiency was tested in a plasmid re-circularization assay (16). The plasmid pCXJ18 was cleaved either with HindIII, SmaI, PstI or HindIII and PstI and the linearized vector (0.5 µg) was used to transform the yeast strains as indicated. In parallel, the cells of the same strain were transformed with the same amount of super-coiled plasmid to normalize for differences in transformation efficiency. Cells were plated on SC-medium lacking uracil (SC-Ura), incubated at 30°C and colonies were counted after 48 to 72 h.
Gene targeting in K.lactis
A general method for performing accurate gene targeting was developed in K.lactis, similar to a previously described method for S.cerevisiae (46), but using the modifications described below. For each side of the target gene two 70mer oligonucleotides were designed sharing ∼50 bp of homology to the target locus (LIG4, MRE11 and RAD50). The stretches of homology were chosen so that the resulting deletions were from the start codon to the stop codon. On the 3′ end of the oligonucleotides 20 bp of homology to the KANMX gene was included (5′ n50-ccagcgacatggaggcccag-3′ and 5′ n50-ggatggcggcgttagtatcg-3′). PCR amplifications using these oligonucleotides and pFA6a-KANMX (47) as template were performed. The resulting fragments were cloned into the pGEM-T Easy vector. An aliquot of 4–10 µg of the resulting plasmid was digested with EcoRI and SpeI and the linear DNA fragments used to transform strain SAY572 (nej1::LEU2). Large colonies, indicating the integration of the KANMX gene into the genome, were picked after 48 to 72 h of incubation at 30°C. For confirmation of the correct genotypes, a gene-specific oligonucleotide homologous to a region 300–500 bp upstream of the target gene and an oligonucleotide (5′-cggcctcgaaacgtgagtc-3′) complementary to the KANMX gene were used in PCR amplifications, using genomic DNA as template.
Plasmid rescue and inverse PCR
To analyze the genomic target sites of IR of pFA6a-KANMX, the vector was digested with SacII and the enzyme was heat inactivated at 65°C for 20 min. The linearized pFA6a-KANMX was transformed either into wild-type cells CK213-4c or PMY2. Cells were plated on rich medium and incubated at 30°C for 24 h, replica-printed on rich medium containing 200 µg/ml G418 and incubated for 72 h. Approximately 250 colonies were re-streaked on fresh 200 µg/ml G418 plates. Chromosomal DNA was prepared and 1–5 µg of genomic DNA was digested with EcoRI for 3 h at 37°C in a total volume of 20 µl in plasmid rescue experiments. The samples were heated at 65°C for 20 min and then diluted to 100 µl. Following addition of T4 DNA ligase the samples were incubated at 16°C for 12 h. After adding 0.5 vol of 7.5 M NH4Ac and 3 vol of 2-propanol, the DNA was recovered by centrifugation. The DNA pellet was washed once in 70% ethanol, air-dried and resuspended in 10 µl of sterile water. For plasmid rescue, 1–5 µl of DNA was transformed into Escherichia coli DH5α by electroporation. Plasmid DNA was prepared from a single bacterial colony and the SacII junction was DNA sequenced (Macrogen-South Korea) using a custom primer (5′-ctaacgccgccatccagtgtcg-3′). For sequencing of both ends of the target sites an identical procedure was used, but the restriction enzymes used for plasmid rescue were HpaI, XbaI or XhoI and an additional sequencing primer (5′-ccccgcgcgttggccgattc-3′) was included. The data obtained were compared with the Genolevures database (48) to assign a genomic coordinate to the insertions.
The PMY2 transformants were analyzed by inverse PCR. Genomic DNA was prepared and digested with either HpaII or MspI and following intramolecular ligation, inverse PCR was performed with primer PMO5-F (5′-cgtatgtgaatgctggtcgct-3′) and PMO6-R (5′-tgacgcatgatattactttct-3′) using standard conditions. The SacII junction was sequenced and analyzed as described above.
Statistical methods
To identify any unusual clusters of insertions (or regions with higher insertion intensity), χ2-tests and scan statistics were used. Under the null hypothesis, insertions occurred independently, completely at random and were equally likely to occur anywhere on the chromosomes. χ2-tests were used to investigate if there were differences in insertion intensity between the six chromosomes and between other divisions of the genome (rDNA/not rDNA, ORF/not ORF). To discover any unexpected clusters within a chromosome we looked at the maximum number of insertions on a fixed proportion of the chromosome length. This number is a scan statistic, and the probability to find k or more insertions in any such subinterval of the chromosome was calculated with an approximate formula (49). To calculate the probability for the observed smallest distance between any of the insertions on a chromosome an exact formula (50) was used. All protein coding gene sequences from K.lactis were downloaded from http://cbi.labri.fr/Genolevures/raw/seq/K_lactis.rc2.nt. The codon adaptation index (CAI) values were calculated with the web-based software ‘The CAI calculator version 2’ (http://www.evolvingcode.net/codon/CalculateCAIs.php) using the ribosomal protein genes as the reference set. The ribosomal protein coding genes were determined based on the genome annotation.
RESULTS
Identification of K.lactis NHEJ genes
Based on sequence conservation, we identified the potential MRE11 (KLLA0C06930g), RAD50 (KLLA0C02915g), YKU80 (KLLA0B12672g), NEJ1 (KLLA0F20339g) and LIG4 (KLLA0D01089g) genes from K.lactis using the Genolevures database (51). The predicted proteins encoded by these genes shared homology with their S.cerevisiae orthologs; Yku80 was 29% identical and 55% similar, Mre11 (53/72), Rad50 (50/71), Lig4 (41/63) and Nej1 (18/42). To investigate the role of these genes in maintaining genome stability we generated yku80::LEU2, nej1::LEU2, rad50::KANMX, mre11::KANMX and lig4::KANMX null mutant strains. We also generated strains compromised for HR carrying rad51::LEU2 and rad52::LEU2 null alleles (52,53). Strains carrying the above mentioned mutations were crossed to each other and following tetrad analysis different double mutant combinations were recovered.
The molecular requirements for NHEJ were conserved in K.lactis
We investigated the molecular requirements for NHEJ in K.lactis. In particular, we wanted to see if the genes identified by us were required for efficient NHEJ, like their S.cerevisiae orthologs. Furthermore, we wanted to investigate whether haploids and diploids performed NHEJ with different efficiency. For this purpose, we performed a plasmid rejoining assay (16) using a centromeric plasmid linearized in the multiple cloning cassette, a region of the plasmid not sharing homology with the milk yeast genome. To obtain stable transformants the plasmid must be re-circularized using the NHEJ pathway, and the number of transformants using the linear vector was normalized to parallel transformations using circular plasmid. The ratio of linearized/circular transformants is a measure of NHEJ efficiency (16). In contrast to S.cerevisiae, the NHEJ efficiency in a MATa haploid and a MATa/MATα diploid strain was similar (Figure 1A), suggesting that none of the proteins required for NHEJ was regulated by cell-type. Strains lacking the genes encoding Nej1, Yku80, Lig4, Mre11 and Rad50 were severely deficient for NHEJ. The nej1 yku80 double mutant strain was not more deficient than either single mutant strain indicating that Nej1 and Yku80 acted in the same pathway. As expected, Rad51 and Rad52 were not required for efficient NHEJ.
Figure 1.

K.lactis NHEJ was versatile and cell-type independent. (A) Molecular requirements for K.lactis NHEJ measured in a plasmid re-circularization assay. Strains CK213-4c (wt), SAY681 (MATa/MATα), SAY572 (nej1::LEU2), SAY573 (ku80::LEU2), SAY574 (nej1::LEU2 ku80::LEU2), SAY683 (lig4::KANMX), SAY559 (mre11::LEU2), SAY557 (rad50::KANMX), SAY516 (rad51::LEU2) and SAY507 (rad52::LEU2) were tested for end-joining efficiency by parallel transformation of super-coiled or HindIII-linearized pCXJ18. The data are expressed as the relative ratio between the number of transformants obtained with the linearized and the super-coiled plasmid, normalized to the value obtained for the wild-type strain that was defined as 100%. (B) Versatility of K.lactis NHEJ. Wild-type strain CK213-4c transformed with linearized pCXJ18 bearing blunt (SmaI), 5′ cohesive (HindIII) 3′ cohesive (PstI) or noncohesive (HindIII + PstI) overhangs were analyzed for transformation efficiency. The results represent the transformation efficiency obtained with the linearized plasmid relative to the efficiency obtained with super-coiled (ccc) pCXJ18, defined as 100%. The data shown in (A) and (B) correspond to the average value from two or three independent experiments. (C) Repair events from NHEJ of noncohesive overhangs. Plasmids were rescued from 13 independent K.lactis colonies transformed with HindIII/PstI double digested pCXJ18. The DNA sequences of the junctions (black boxes indicating the bases remaining from the overhangs) were determined, and the type of alteration and how many times they were obtained is indicated on the right. (D) RNA blot hybridization of K.lactis total RNA from CK213-4c (MATa), SAY102 (MATa sir2), SAY189 (MATa sir2 hmlαΔp) and SAY186 (MATa hmlαΔp) strains. The blot was hybridized with a 500 bp [α32P]-labeled PCR fragment corresponding to NEJ1 gene (top), and then the blot was stripped and re-probed with an actin probe (bottom).
NHEJ was efficient with a wide variety of DNA ends and NEJ1 transcription was not regulated by cell-type
In S.cerevisiae, utilization of either NHEJ or HR is regulated by the nature of the DSB and by cell-type. In S.cerevisiae, NHEJ is efficient in rejoining ends with cohesive overhangs, but ∼40-fold less efficient in rejoining blunt ends (13,54) and substantially less efficient at joining non-cohesive ends (55). To test if K.lactis had a similar specificity, we compared the transformation efficiency of supercoiled plasmid pCXJ18 with the same plasmid digested with various restriction enzymes (Figure 1B). No significant differences were detected for blunt ends (SmaI digested), 5′ (HindIII) or 3′ (PstI) cohesive ends. Strikingly, the rejoining efficiency was high even for ends with opposite overhangs (HindIII PstI double digest), reduced only about 2-fold compared to the efficiency for cohesive ends. To analyze how the 3′ protruding single strand (3′ PSS) and 5′ PSS were joined we rescued the plasmid from 13 independent transformants and sequenced the joint. All events recovered had deletions of the 3′PSS and several of the 5′PSS or both (Figure 1C). In all events except one that had a 17 bp deletion, at least one base from the 3′ PSS was present in the repair product.
In S.cerevisiae, transcription of NEJ1 is regulated by the a1/α2 repressor, leading to an absence of Nej1 in MATa/MATα diploids (25–28). Diploid cells are therefore inefficient at NHEJ. This regulation is likely to be conserved among closely related yeasts, since a multiple sequence alignment of the promoter sequences of the NEJ1 genes from S.cerevisiae, Saccharomyces paradoxus, Saccharomyces mikitae, Saccharomyces bayanus and Saccharomyces kudriavzevii revealed the presence of the a1/α2 operator (data not shown). We previously showed that K.lactis contains a regulator similar to the a1/α2 repressor (56), but an a1/α2 operator site could not be identified in the KlNEJ1 promoter region. This could be due to either that KlNEJ1 was not regulated by cell-type or that the a1/α2 operator site was different compared to S.cerevisiae and thus not found using our search criteria. To distinguish between these possibilities, we performed an RNA-blot analysis on total RNA from the appropriate K.lactis strains. We could detect the NEJ1 transcript in all cell-types investigated (Figure 1D), both in the presence (MATa sir2) and in the absence (MATa, MATa hmlαΔp and MATa sir2 hmlαΔp) of the a1/α2 repressor. Hence, NEJ1 transcription was not regulated by cell-type in K.lactis, consistent with the observation that both haploid and diploid cells performed NHEJ with similar efficiency (Figure 1A). We concluded that NHEJ was very versatile in K.lactis, capable of handling a wide variety of ends and insensitive to cell-type.
IR and NHEJ had identical genetic requirements
IR is defined as the joining of two DNA molecules not sharing extensive homology to each other. To test the genetic requirements for IR wild-type strain CK213-4C and mutant strains compromised for NHEJ (nej1, ku80, nej1 ku80, lig4, mre11 and rad50) or HR (rad51 and rad52) were transformed with linearized plasmid pRS406. Plasmid pRS406 has no homology to the K.lactis genome and is unable to replicate as an episome. Thus, Ura+ stable transformants only arise as a result of integrative IR in the genome. IR occurred with a frequency of ∼1 × 103 transformants/µg of plasmid DNA, which can be compared to 1–5 × 100 in S.cerevisiae using a plasmid of similar size (57). Integrative transformation by IR thus occurred ∼1000-fold more frequently in K.lactis than in S.cerevisiae. No transformants were obtained with NHEJ mutant strains, but mutations in the HR pathway did not affect the IR frequency (Table 2). In addition, all strains investigated had approximately the same transformation efficiencies using a circular centromeric plasmid. The results clearly showed that IR was completely dependent on the NHEJ pathway and independent of the HR pathway.
Table 2.
IR was completely dependent on the NHEJ pathway
| Strains | Number of Ura+ colonies/µg | |
|---|---|---|
| pRS406 | pCXJ18 | |
| Wild-type | 1.2 × 103 | 8.7 × 104 |
| nej1 | 0 | 7.4 × 104 |
| yku80 | 0 | 6.6 × 104 |
| nej1 yku80 | 0 | 5.8 × 104 |
| lig4 | 0 | 4.3 × 104 |
| mre11 | 0 | 7.8 × 104 |
| rad50 | 0 | 5.5 × 104 |
| rad51 | 1.1 × 103 | 7.1 × 104 |
| rad52 | 1.1 × 103 | 8.1 × 104 |
| nej1 rad51 | 0 | 4.6 × 104 |
| nej1 rad52 | 0 | 5.2 × 104 |
One µg of plasmid pRS406 was linearized with SacII and transformed into the indicated strains. The number of Ura+ colonies was determined by growth on SC-Ura plates representing IR events. Parallel transformations of supercoiled pCXJ18 (1 µg) into the strains revealed no significant differences in transformation efficiency.
Target sites of IR
Since the NHEJ pathway repairs DSBs and IR in K.lactis completely depended on NHEJ, we reasoned that the mechanism of IR in K.lactis involved DSB repair events during which the plasmid becomes captured in-between two DNA ends. Consistent with this model, K.lactis strains transformed with circular pFA6a-KANMX resulted in very low transformation efficiency (0–2 transformants/µg of plasmid DNA), indicating that free DNA ends were necessary for efficient integration. If this model was correct, then mapping the locations of the IR-events should reveal the loci where mitotic DSBs repaired by NHEJ occur. The linearized pFA6a-KANMX was transformed into a wild-type strain. To explore the target sites, we carried out plasmid rescue from several independent integrants and sequenced the flanking genomic regions (see Materials and Methods). For 38 out of 164 successful plasmid rescue events, we could not determine an exact chromosomal location. Twenty-three were insertions in the rDNA loci on chromosome IV, three were in pKD1, an endogenous episomal plasmid, and four insertions were in repetitive DNA other than rDNA (subtelomeric repeats or transposons). For eight of the plasmids we did not find homology to K.lactis chromosomal DNA, but two of these showed significant homology to K.lactis mitochondrial DNA. By crossing the strains containing these insertions, followed by tetrad analysis, we determined that the insertions segregated as nuclear single copy genes. Our interpretation of these events was that fragmented mitochondrial DNA was captured between the ends of the plasmid and the genomic target locus during IR, thus leading to transfer of mitochondrial DNA into the nucleus. Regarding the remaining six events, we speculate that salmon sperm DNA introduced during the transformation procedure was inserted at the junctions, as observed previously in Schizosaccharomyces pombe (58). For 126 insertions we could determine an exact location in the genome using the newly released K.lactis genome sequence, and these insertions are schematically shown in Figure 2 (exact locations are available on request).
Figure 2.
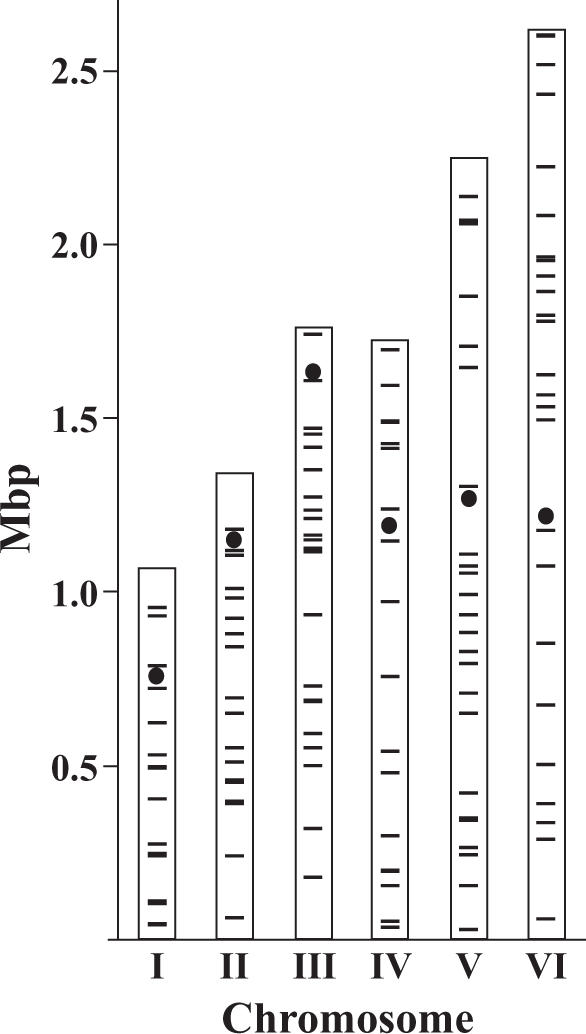
Global distribution of 126 independent target sites of IR. K.lactis contains six chromosomes ranging in size from 1 to 2.6 Mb. Black lines represent insertions and circles represent centromeres. The rDNA cluster on chromosome IV with 23 insertions was omitted.
The target sites were largely randomly distributed with similar insertion intensities on all chromosomes (Table 3). The distances between the insertions ranged from 180 bp to 343 kb, with a mean distance of 84.4 kb and a median distance of 62 kb. A statistical calculation showed that the smallest distance of 180 bp was not unexpected (P = 0.25) assuming uniformly distributed insertions. Only 31% of the insertions (39) were in ORFs, an unexpectedly low number given that 71.6% of the K.lactis genome is occupied by protein encoding genes (48). The difference in insertion intensity (Insertions/Mbp) between ORFs and intergenic regions (IGRs) was highly significant (P << 0.001). In addition, the number of insertions observed in the rDNA locus on chromosome IV was higher than would be expected by chance (χ2-test, P = 0.0014). We noted that the insertion intensity in IGRs and rDNA was similar (Table 4), suggesting that they may arise from a similar mechanism. In summary, IR-target sites were overrepresented in IGRs and in the rDNA loci, but did not have a preference for a particular chromosome.
Table 3.
Insertion distribution on K.lactis chromosomes
| Chromosomes | Length (Mbp) | Wild-type | rad52/rad52 | ||
|---|---|---|---|---|---|
| Insertions | Insertions/Mbp | Insertions | Insertions/Mbp | ||
| I | 1.06 | 16 | 15.1 | 2 | 1.9 |
| II | 1.32 | 18 | 13.6 | 8 | 6.0 |
| III | 1.75 | 22 | 12.5 | 5 | 2.9 |
| IV | 2.52 | 42 | 16.7 | 9 | 3.6 |
| V | 2.23 | 25 | 11.2 | 10 | 4.5 |
| VI | 2.6 | 26 | 10.0 | 12 | 4.6 |
| Total | 11.49 | 149 | 13.0 | 46 | 4.0 |
Table 4.
Insertion distribution in rDNA versus non-rDNA and in ORFs versus IGRs
| Sequence analyzed | Length (Mbp) | Wild-type | rad52/rad52 | |||
|---|---|---|---|---|---|---|
| Insertions | Insertions/Mbp | Insertions | Insertions/Mbp | Wt/rad52/rad52 insertions/Mbpa | ||
| Chr IV non rDNA | 1.72 | 19 | 11.1 | 6 | 3.5 | Not determined |
| Chr IV rDNA | 0.8b | 23 | 28.8 | 3 | 3.75 | Not determined |
| ORFs | 7.65 | 39 | 5.1 | 12 | 1.6 | 4.05/3.7 |
| IGRs | 3.04 | 87 | 28.7 | 31 | 10.2 | 22.8/23.7 |
aThe values were normalized to a total of 100 insertions.
bEstimated size of the rDNA locus in K.lactis made by the Genolevures consortium.
IR was associated with insertions and deletions, but not with microhomology and topoisomerase I cleavage sites
Next we investigated if sequence alterations could be found close to the sites of integration. We performed plasmid rescues of 31 independent insertions using restriction enzymes that did not cleave pFA6a-KANMX, thus rescuing genomic DNA from both sides of the insertions. The flanking sequences were then compared to the undisrupted wild-type sequence in the Genolevures database. Several different mutagenic events occurred close to the breakpoints and 10 representative examples are shown in Figure 3. Small deletions or insertions (less than 5 bp) or a combination of both were the most common sequence alterations representing 15 out of 31 samples. In four cases, we observed a target site duplication of 2 bp, an event resulting in a 2 bp insertion. In five cases the alterations were large deletions or insertions (more than 100 bp). Seven of the samples revealed no sequence alteration on either side of the insertion.
Figure 3.

Sequence analyses of genomic IR target sites. Plasmid pFA6a-KANMX was linearized with SacII resulting in 3′ overhangs (top). The type of alteration of the target site is indicated on the right. Insertions and deletions at the genomic target site or at the SacII restriction end are indicated with boldface and small letters, respectively. Integration by microhomology was only found in integrant 101. A deletion of more than 100 bp was observed for integrant 81, where the flanking sequences were homologous to 18S rDNA and 26S rDNA, respectively. The most common sequence alterations were small deletions or insertions as seen in integrant 14, 59, 11, 22, 73, 85 and 94-1. The numbers for each insertion are given according to their order of preparation during the plasmid rescue experiment.
In S.cerevisiae, the most common IR events involve base pairing between the target sequence and the terminal few base pairs of the transforming DNA (57). This microhomology mediated IR requires at least two contiguous bases of homology between the target and the plasmid end. In addition, IR in S.cerevisiae often occurs close to consensus cleavage sites for topoisomerase I (Top1). Top1 has a degenerate recognition motif (C/G T/A T) shared by all eukaryotes investigated (38) and thus probably also shared by K.lactis. For one of the integration events (3%) we found microhomology between both ends of the integrating plasmid and the target site. In two cases (6%), we found microhomology between one of the ends of the integrating plasmid and the target site. In 28 cases (90%), no microhomology between either ends was found, suggesting that base pairing between the integrating plasmid and the genomic target site was of little relevance during K.lactis IR. Furthermore, only five (16%) of the integration events analyzed had an adjacent Top1 site, a number close to what would be expected on a random basis. Thus, IR was highly mutagenic, but did not correlate with microhomology or Top1 sites.
IR-target sites were linked with transcriptional regulatory regions
To further explore the intergenic insertions we investigated the transcriptional orientation of the ORFs flanking the insertion points. There were three possibilities, (type I) IGRs representing the 5′ ends of the flanking genes, (type II) IGRs representing one 5′ end and one 3′ end of the flanking genes and (type III) IGRs representing the 3′ ends of the flanking genes. To determine the genome wide size of each type of IGR, we analyzed the total length of type I, II and III on six randomly selected 100 kb segments of the genome (one on each chromosome). Out of 298 analyzed IGRs, type I represented 38.5%, type II 45.4% and type III 16.0% (Figure 4A, white bars). The average length of type I was significantly longer than type III (770 and 324 bp, respectively). Analyzing the IGRs in which IR had occurred (Figure 4A, black bars) revealed an obvious preference for insertions into the upstream regions of genes. Remarkably, not a single insertion in an IGR representing the 3′ ends of two flanking genes was observed.
Figure 4.

The genomic target site distribution of IR events is similar in the wild-type and rad52/rad52 strains and occurred preferentially in promoter regions independently of transcriptional activity. (A) IGRs are located either between the 5′ ends of two genes, between the 5′ and the 3′ ends of two similar oriented genes or between the 3′ ends of two opposite oriented genes (type I, type II and type III, respectively). The K.lactis genome wide size of type I, II and III IGRs was calculated by analyzing a random 100 kb DNA segment from each chromosome. The sum of the length of each type of IGR was divided by the total length of all IGR types (298 IGRs analyzed). These values (white bars) thus represent the theoretical fraction each type of IGR occupies in the genome. The SacII-linearized pFA6a-KANMX was transformed into the wild-type CK213-4c strain (black bars) and in the homozygous rad52/rad52 mutant PMY2 strain (grey bars). The genomic target sites of IR were determined as described in Materials and Methods. The percent of type I, II and III IGR insertions with respect to the total number of IGR insertions in each strain was compared to the theoretical expected genome wide value. (B) The CAI values of the genes in which pFA6a-KANMX had integrated in CK213-4c strain within the ORF (blue) and in their 5′-promoter region (red) are shown together with CAI values of all K.lactis protein coding genes (grey) and ribosomal protein genes (green). CAI, codon adaptation index; GC3, the third codon position GC content.
The high frequency of IR-events in the promoter regions prompted us to examine whether they correlated with transcription levels. We therefore determined the CAI of the genes targeted by IR. The CAI is a single value measurement that summarizes the codon usage of a gene relative to the codon usage of a reference set of genes. This value ranges from 0 to 1. A high CAI value suggests that the gene of interest has similar codon usage compared to the reference genes. Previously, this value was suggested as a useful predictor of gene expression level when highly expressed genes were used as reference genes (59). We computed the CAI values of all protein coding genes in the K.lactis genome (N = 5330) using ribosomal protein genes, assumed to be highly expressed, as the reference set. The CAI values were plotted against the GC content at the third codon position (GC3) (Figure 4B). Indeed, most of the ribosomal protein coding genes exhibit high CAI values and cluster on top of the plot, consistent with the assumption that they are highly expressed. Moreover, a lack of correlation between the genome CAI values and GC3 suggested that CAI values were not dependent upon the GC composition due to mutational bias (60), further confirming that CAI values were valid predictors of gene expression levels in K.lactis.
To explore whether the target sites of IR correlated with highly expressed genes, the CAI values of the genes in which the insertion of pFA6a-KANMX occurred either within the ORF or in their 5′-promoter region were determined and compared with the genome global CAI value distribution. When the insertion target site corresponded to two different promoter regions, both ORFs were included in the calculations. The mean CAI values of the ORFs with intragenic insertions (0.204, 0.156 SD) and ORFs with insertions in the promoter region (0.256, 0.179 SD) were close to the global genome mean (0.206, 0.13 SD) and significantly lower than the mean value of the ribosomal protein genes (0.798, 0.156 SD). Hence, IR target sites did not correlate with highly expressed genes.
Ploidy or a rad52 mutation did not change the target site preference for IGRs
The IR-target site preference in IGRs and rDNA could be an artifact due to the use of a haploid strain in these experiments. Insertions within ORFs could result in lethality more often than insertions into IGRs or rDNA. Furthermore, it was possible that spontaneous DSBs occurred with equal frequency throughout the genome, but that DSBs in ORFs were preferentially repaired by the less error prone HR pathway. To test these possibilities we examined IR target sites in a rad52/rad52 diploid strain, a strain defective for HR and insensitive to recessive lethal insertions. If either of the above hypotheses were valid, then we would expect an altered target site distribution in the rad52/rad52 diploid strain. Forty-six targets sites were investigated using an inverse PCR approach. The insertion intensity varied somewhat between chromosomes, probably due to the smaller sample size (Table 3). Significantly, 31 insertions were in IGRs and only 12 were in ORFs (Table 4). The insertion intensity into IGRs was 5.6-fold (28.7/5.1) higher than into ORFs in the wild-type haploid strain and 6.4-fold (10.2/1.6) in the rad52/rad52 diploid strain. In addition, the rad52/rad52 diploid strain maintained the target site preference for transcriptional regulatory regions (Figure 4B). For insertions into rDNA, however, the rad52/rad52 diploid strain no longer showed a preference for this region. The insertion intensity into the rDNA loci was similar to the insertion intensity into the non rDNA portion of chromosome IV (Table 4).
IR at ectopic DSBs
It is well known that chromatin is a dynamic structure and that histone modification patterns differ across the genome. The observed preference of IR target sites within IGRs corresponding to promoter regions prompted us to examine whether the NHEJ machinery was more efficient at repairing DSBs in these regions compared to coding regions. Alternatively, more spontaneous DSBs may arise in promoters than in ORFs. To address whether IR could be directed to DSBs and whether the NHEJ efficiency was dependent on the genomic location (ORF versus promoter region) we introduced ectopic I-SceI recognition sites either within or upstream of ORF E00462g, generating strains SAY648 and PMY34, respectively (Figure 5A). Based on sequence conservation with S.cerevisiae we named this ORF VMR1. VMR1 encodes a predicted multiple drug resistance pump. Insertion of a strong promoter upstream of VMR1 results in strains resistant to high levels of G418 (E. Barsoum and S. U. Åström, unpublished data), suggesting that Vmr1 may rid cells of this compound. The strains were transformed with either pCXJ18 or pCXJ18 containing the I-SCEI gene controlled by the galactose-inducible GAL1 promoter. The effect of the expression of the endonuclease was examined on DNA-blots and on glucose and galactose containing plates (Figure 5B and C). The I-SceI endonuclease cut the two sites with similar efficiency in vivo; ∼11% efficiency was observed after 3 h in galactose containing medium in both PMY34 and SAY684 (Figure 5B). Both strains containing an ectopic I-SceI site and expressing the I-SceI endonuclease grew slowly on galactose compared to glucose (Figure 5C). However, plating efficiency was not severely affected probably because the DSBs were efficiently repaired. Endonuclease expression in a strain lacking the I-SceI site did not affect growth. In addition, SAY648 and PMY34 containing pCXJ18 alone had no growth defect on galactose. Both HR and NHEJ could repair the I-SceI induced break, since strains lacking either Rad52 (SAY686) or Nej1 (SAY685) and containing the I-SceI site in the VMR1 promoter, were sensitive to increased endonuclease expression. This sensitivity was seen as lowered plating efficiency on galactose plates. It should be noted that the GAL1 promoter was leaky in K.lactis (data not shown). This explained the reduced plating efficiency of the rad52 strain on glucose plates.
Figure 5.

Genomic DSBs constituted the target sites for IR events. (A) Schematic illustration of the VMR1 chromosomal locus. The I-SceI recognition site was introduced either in the VMR1 coding region (at nucleotide +3871, strain PMY34) or in its promoter region (at nucleotide −183, strain SAY684). In the experiment described in (D), cells were simultaneously transformed with an episomic plasmid containing the I-SCEI endonuclease under the GAL1 promoter (p164) and the BamHI-linearized pPMB18 that contains the ADH1 and TEF2 promoters, which directs transcription in opposite directions (indicated by arrows). The position of oligonucleotides (A, B, C, D and E) used for PCR analysis of the location of IR integration events and relevant restriction sites (BHI, BamHI; EV, EcoRV) are indicated. (B) The I-SceI endonuclease cleaves in the VMR1 coding sequence and in its promoter region with similar efficiency. Exponentially growing PMY34 and SAY684 cells containing either pCXJ18 or p164 in SC-Ura-glucose were washed, resuspended in SC-Ura-galactose and incubated for 3 h. Genomic DNA from PMY34 and SAY684 cells digested with BamHI or EcoRV, respectively, was separated, blotted and the membrane was hybridized with a 273 bp [α32P]-labeled PCR fragment corresponding to VMR1 gene as shown in (A). The fragment sizes are indicated on both sides of the blot and the I-SceI cutting efficiency is indicated below the blot with a standard deviation from two independent experiments. (C) Genomic DNA cleavage at the ectopic I-SceI sites results in a slow growth phenotype and is highly deleterious in combination with nej1 and rad52 mutations. Ten-fold serial dilutions of the indicated strains containing either pCXJ18 or p164 were spotted on SC-Ura containing either glucose or galactose as a carbon source and incubated at 30°C for 48 h. The relevant genotypes are: CK213-4c (WT), PMY34 (VMR1 +3871 I-SceI), SAY684 (pVMR1 −189 I-SceI), SAY686 (pVMR1 −189 I-SceI rad52) and SAY685 (pVMR1 −189 I-SceI nej1). (D) Strains CK213-4c, SAY684 and PMY34 were co-transformed either with pCXJ18 or p164 and BamHI-linearized pPMB18. Transformants were selected on SC-Ura-Leu containing galactose as the carbon source. IR insertion events of pPMB18 at both I-SceI sites were analyzed by PCR using the oligonucleotides indicated in (A). An additional phenotypic selection was carried out in those strains having the I-SceI site in the VMR1 promoter since insertion of a strong promoter in front of the VMR1 gene results in G418 resistance. The results are expressed as the percent of I-SceI site targeted events with respect to the total number of Ura+ Leu+ transformants and correspond to the average of three independent experiments. For control experiments 8 to 12 transformants were PCR analyzed. The sample size analyzed by PCR corresponding to the co-transformation of pPMB18 and p164 into PMY34 and SAY684 was 77 and 39, respectively. Phenotypic analysis for G418 resistance was performed with approximately 150 transformants in each case.
To explore if IR in the VMR1 ORF and in the VMR1 promoter region was equally efficient, a linearized LEU2 plasmid (pPMB18) with the break close to the ADH1 and TEF2 promoters and the plasmid expressing the endonuclease were co-introduced into SAY648 and PMY34 (Figure 5A). The resulting integration events were analyzed by PCR using the primers indicated in Figure 5A. The results showed that there was efficient targeting of pPMB18 to both ectopic DSBs. No significant difference in targeting efficiency between the strains was observed (Figure 5D). In addition, there was a perfect correlation between G418 resistance and isolates that the PCR analysis showed had an insertion of pPMB18 into the VMR1 promoter. Targeted integration required both the I-SceI site and the endonuclease, since none of the Leu+ transformants from a strain (CK213-4c) lacking the I-SceI site was G418 resistant. No I-SceI site targeted events were observed when pCXJ18 was co-introduced with pPMB18 into both SAY648 and PMY34. Targeted integration was also dependent on the NHEJ pathway since an isogenic nej1 mutant strain did not render any integrative transformants (data not shown). These results demonstrated that IR could be efficiently targeted to a specific locus by introducing a DSB. Moreover, when ectopic DSBs were generated using an endonuclease, a promoter region and an ORF were equally good targets for IR.
DISCUSSION
In this study we showed that IR depended entirely on the NHEJ pathway and present very strong evidence that IR occurred at spontaneous mitotic DSB in milk yeast. In contrast to S.cerevisiae, K.lactis performed IR very efficiently and therefore constituted an excellent model organism to analyze the molecular mechanisms underlying NHEJ. Indeed, IR in K.lactis was 1000-fold more efficient than in S.cerevisiae (Table 2). The molecular requirements for NHEJ were identical in both organisms. K.lactis mutant strains lacking Nej1, Ku80, Lig4, Mre11 and Rad50 were unable to rejoin a linearized episomic plasmid or integrate a non-homologous DNA molecule into the genome (Figure 1A and Table 2).
In similarity with mammalian cells, the K.lactis NHEJ pathway was capable of repairing linearized plasmids containing complementary, blunt and non-complementary ends (Figure 1B). Importantly, it was highly efficient at rejoining a plasmid linearized with restriction endonucleases generating a 5′ and a 3′ overhang. This observation may partly explain the efficient IR observed in K.lactis. The capability of efficient ligation of blunt and noncohesive ends is also a feature of S.pombe and mammalian NHEJ (17,20,61,62). However, in S.cerevisiae efficient NHEJ only takes place with complementary overhangs (54). K.lactis must be able to form a stable NHEJ-complex capable of aligning the two ends without the additional energy provided by base pairing between the severed ends. By analyzing the repair events from PstI–HindIII double digested plasmids, we found that the 3′ PSS was not completely removed in most repair events (Figure 1C). Provided that DNA polymerases only synthesize DNA in the 5′–3′ direction, two alternative models for the molecular mechanism underlying the repair of 3′ PSS-5′ PSS can be envisaged. First, a single stranded DNA ligation event takes place followed by fill-in DNA synthesis. Second, an alignment event stabilizing the interaction between the ends takes place, followed by fill-DNA synthesis and dsDNA ligation. In both cases DNA synthesis is primed by the recessed strand of the 5′ PSS end. Among eukaryotic DNA ligases only a viral ligase has been reported to possess ssDNA ligase activity (63) and there is no evidence of such an activity for DNA ligase IV. Therefore, we favor the latter model, in which milk yeast NHEJ can efficiently align the non-complementary DNA ends. The same model has been proposed for NHEJ of mismatched non-complementary ends in human lymphoblasts (61).
Another interesting difference between both yeasts was that NHEJ was not regulated by cell-type in K.lactis and that NEJ1 transcription was not repressed by the a1/α2 heterodimer (Figure 1D). In contrast to S.cerevisiae, K.lactis vegetative growth is mostly restricted to the haplophase, since diploids are rather unstable and spontaneously enter meiosis. The unstable nature of the diplophase may have prevented the evolution of cell-type regulation of NHEJ in K.lactis, given that milk yeast cells seldom linger in the diploid state. The NEJ1 gene was nevertheless essential for NHEJ in K.lactis, indicating that Nej1 did not evolve in S.cerevisiae as a function to impart cell-type regulation of NHEJ. We suggest that NHEJ required the NEJ1 gene in a common ancestor, and that Saccharomyces sensu stricto species later acquired a specific regulation of this gene. We cannot exclude, however, that the NEJ1 gene was regulated by cell-type in the common ancestor and that K.lactis lost this regulation.
In nature, IR must repair DNA lesions in the context of highly condensed chromatin fibers. Although plasmid rejoining assays have provided useful information regarding NHEJ mechanisms, it is important to note that the substrate for NHEJ in this assay is unlikely to be chromatin. The fact that K.lactis very efficiently integrates a non-homologous linear DNA fragment in the genome allowed us to further study the IR events taking place genome-wide and in chromatin. We showed that IR entirely depended upon the NHEJ pathway (Table 2). This observation stands in contrast to the situation in mouse ES-cells where NHEJ proteins are not required for many illegitimate integration events (32). In mammalian cells, there is a poorly characterized second pathway for NHEJ that is independent of the previously characterized NHEJ proteins (64). We could not observe IR events in strains compromised for NHEJ. Therefore, if there is such an alternative pathway in K.lactis, it must be extremely inefficient. The construction of K.lactis mutant strains is tedious due to the high IR efficiency. We found that K.lactis strains lacking Nej1, Yku80 or Lig4 were useful tools for improving gene targeting. Using strains compromised for NHEJ led to the recovery of integrants that were invariably targeted to the homologous locus (data not shown). The use of yku80 mutant strains for gene targeting purposes has also been proposed by others (65). Given that yku80 mutations in S.cerevisiae show pleiotrophic phenotypes, including strain-dependent temperature sensitivity and telomere shortening (66), we suggest that nej1 or lig4 mutations may be a better choice for large-scale gene targeting projects. We have evidence that yku80 mutant K.lactis strains are partly compromised for telomere capping (S. D. Carter and S. U. Åström, unpublished data).
Exploring the genomic target sites for IR revealed a globally random distribution (Figure 2) and an absence of both microhomology and consensus Top1 sites (Figure 3). Interestingly, an obvious over-representation of IGRs and the rDNA locus compared to ORFs was observed (Table 4). The analysis of the IGR events showed a striking preference for 5′ promoter regions compared to 3′ non-coding regions (Figure 4A). However, no correlation with transcriptional activity could be made (Figure 4B). The insertions into the rDNA did not show a hot spot for a particular region of the rDNA repeats (data not shown), which would be expected if a particular feature of rDNA such as a replication fork block sequence would facilitate the increased level of IR. Loci with two divergently directed promoters and rDNA are expected to experience negative super-coiling that is relieved by topoisomerase activities. In S.cerevisiae, there are no reports that IR occurs preferentially in IGRs, but a preference for the rDNA locus is observed after human Top1 over expression (67). Similar studies in Candida glabrata also revealed a preference for IGRs, absence of Top1 sites and limited presence of microhomology (68). However, in this study a smaller number of IR-events were analyzed, and the transformants were partly selected for their inability to adhere to human epithelial cells. This selection may have biased the genome wide IR target site distribution.
The IR target site distribution in a diploid rad52/rad52 strain was similar to the distribution in the wild-type haploid strain with respect to the preference for IGRs and excess of 5′ promoter regions (Table 4 and Figure 4a). These results ruled out the possibility that recessive lethal insertions significantly biased the observed distribution in haploid wild-type cells. In addition, we analyzed 32 independent wild-type diploid strains with random insertions of the pFA6a-KANMX plasmid using tetrad analysis. We found only two strains (6%) in which the insertions co-segregated with lethality. Furthermore, a compromised HR pathway did not change the target site preference for IGRs, showing that DSBs arising in ORFs were not preferentially repaired by HR. However, in the rad52/rad52 diploid strain, the IR target site preference for rDNA was lost, suggesting that HR somehow facilitated high levels of IR in the rDNA loci. To firmly establish this notion, a larger number of IR target sites from the rad52/rad52 diploid strain should be analyzed.
The observation that IR could be targeted very efficiently to an ectopically induced DSB demonstrated that spontaneous genomic DSBs likely constituted the target sites for illegitimate integration events. A model for the observed target site preference for IGRs and rDNA was that DSBs arise more frequently in IGRs and rDNA compared to ORFs. Alternatively, the NHEJ-proteins could be more efficiently recruited to these loci as a result of specific protein–protein interactions or a generally more accessible chromatin structure. Our results showed that I-SceI induced breaks in a promoter region or in an ORF were equally efficient targets for IR, strongly supporting the first model. In bacteria, it has also been proposed an IR pathway mediated by DSBs followed by end-joining (69).
Studies of the location of meiotic DSBs in S.cerevisiae demonstrate that most hotspots are intergenic rather than intragenic (70,71). In addition, among 20 intergenic DSBs representing meiotic hotspots, 13 were between the 5′ ends of two genes and only two were between the 3′ ends of two genes (72). Our tentative mapping of mitotic DSBs thus showed similarities with the location of meiotic DSBs in S.cerevisiae, suggesting that chromosomal sites prone to experience DSBs are similar during both vegetative growth and meiosis.
In summary, our data strongly suggest that mitotic DSBs arise in IGRs ∼6-fold more frequently than in ORFs. To our knowledge, this report is the first comprehensive attempt to study the locations of mitotic DSBs in a eukaryote. We propose that spontaneous mitotic DSBs can be mapped by analyzing sites of IR integration events, providing a molecular tool for understanding how these DSBs arise. Since mitotic DSBs contribute to genomic instability in all organisms, these findings should be of significant interest.
Acknowledgments
The authors thank Jan-Olov Persson for help with statistical calculations and Monique Bolotin for generously supplying K.lactis DNA sequences prior to the release of the genome. The authors acknowledge Wu Gang and Stephen Freeland for CAI calculations and for fruitful discussions and Thomas Wilson for the gift of the plasmid expressing I-SceI. This work was supported by grants from the Swedish Cancer Society (4592-B03-03XAB) and the Swedish Research Council (621-2004-1942) to S.U.Å. Funding to pay the Open Access publication charges for this article was provided by the Swedish Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1.Symington L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paques F., Haber J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Dyck E., Stasiak A.Z., Stasiak A., West S.C. Visualization of recombination intermediates produced by RAD52-mediated single-strand annealing. EMBO Rep. 2001;2:905–909. doi: 10.1093/embo-reports/kve201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.New J.H., Sugiyama T., Zaitseva E., Kowalczykowski S.C. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–410. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 5.Shinohara A., Ogawa T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature. 1998;391:404–407. doi: 10.1038/34943. [DOI] [PubMed] [Google Scholar]
- 6.Nishinaka T., Shinohara A., Ito Y., Yokoyama S., Shibata T. Base pair switching by interconversion of sugar puckers in DNA extended by proteins of RecA-family: a model for homology search in homologous genetic recombination. Proc. Natl Acad. Sci. USA. 1998;95:11071–11076. doi: 10.1073/pnas.95.19.11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milne G.T., Jin S., Shannon K.B., Weaver D.T. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol. Cell Biol. 1996;16:4189–4198. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldmann E., Schmiemann V., Goedecke W., Reichenberger S., Pfeiffer P. DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res. 2000;28:2585–2596. doi: 10.1093/nar/28.13.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nick McElhinny S.A., Snowden C.M., McCarville J., Ramsden D.A. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol. Cell Biol. 2000;20:2996–3003. doi: 10.1128/mcb.20.9.2996-3003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teo S.H., Jackson S.P. Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double-strand break repair. EMBO J. 1997;16:4788–4795. doi: 10.1093/emboj/16.15.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson T.E., Grawunder U., Lieber M.R. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- 12.Schär P., Herrmann G., Daly G., Lindahl T. A newly identified DNA ligase of Saccharomyces cerevisiae involved in RAD52-independent repair of DNA double-strand breaks. Genes Dev. 1997;11:1912–1924. doi: 10.1101/gad.11.15.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrmann G., Lindahl T., Schär P. Saccharomyces cerevisiae LIF1: a function involved in DNA double-strand break repair related to mammalian XRCC4. EMBO J. 1998;17:4188–4198. doi: 10.1093/emboj/17.14.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teo S.H., Jackson S.P. Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr. Biol. 2000;10:165–168. doi: 10.1016/s0960-9822(00)00317-1. [DOI] [PubMed] [Google Scholar]
- 15.Moore J.K., Haber J.E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boulton S.J., Jackson S.P. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–1828. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson S., Warr N., Taylor D.L., Watts F.Z. The role of Schizosaccharomyces pombe Rad32, the Mre11 homologue, and other DNA damage response proteins in non-homologous end joining and telomere length maintenance. Nucleic Acids Res. 1999;27:2655–2661. doi: 10.1093/nar/27.13.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamaguchi-Iwai Y., Sonoda E., Sasaki M.S., Morrison C., Haraguchi T., Hiraoka Y., Yamashita Y.M., Yagi T., Takata M., Price C., et al. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999;18:6619–6629. doi: 10.1093/emboj/18.23.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harfst E., Cooper S., Neubauer S., Distel L., Grawunder U. Normal V(D)J recombination in cells from patients with Nijmegen breakage syndrome. Mol. Immunol. 2000;37:915–929. doi: 10.1016/s0161-5890(01)00008-6. [DOI] [PubMed] [Google Scholar]
- 20.Manolis K.G., Nimmo E.R., Hartsuiker E., Carr A.M., Jeggo P.A., Allshire R.C. Novel functional requirements for non-homologous DNA end joining in Schizosaccharomyces pombe. EMBO J. 2001;20:210–221. doi: 10.1093/emboj/20.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis L.K., Storici F., Van Komen S., Calero S., Sung P., Resnick M.A. Role of the nuclease activity of Saccharomyces cerevisiae Mre11 in repair of DNA double-strand breaks in mitotic cells. Genetics. 2004;166:1701–1713. doi: 10.1534/genetics.166.4.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L., Trujillo K., Ramos W., Sung P., Tomkinson A.E. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol. Cell. 2001;8:1105–1115. doi: 10.1016/s1097-2765(01)00388-4. [DOI] [PubMed] [Google Scholar]
- 23.Lee S.E., Paques F., Sylvan J., Haber J.E. Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and non-homologous repair paths. Curr. Biol. 1999;9:767–770. doi: 10.1016/s0960-9822(99)80339-x. [DOI] [PubMed] [Google Scholar]
- 24.Åström S.U., Okamura S.M., Rine J. Yeast cell-type regulation of DNA repair. Nature. 1999;397:310. doi: 10.1038/16833. [DOI] [PubMed] [Google Scholar]
- 25.Frank-Vaillant M., Marcand S. NHEJ regulation by mating type is exercised through a novel protein, Lif2p, essential to the ligase IV pathway. Genes Dev. 2001;15:3005–3012. doi: 10.1101/gad.206801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kegel A., Sjöstrand J.O., Åström S.U. Nej1p, a cell type-specific regulator of nonhomologous end joining in yeast. Curr. Biol. 2001;11:1611–1617. doi: 10.1016/s0960-9822(01)00488-2. [DOI] [PubMed] [Google Scholar]
- 27.Ooi S.L., Shoemaker D.D., Boeke J.D. A DNA microarray-based genetic screen for nonhomologous end-joining mutants in Saccharomyces cerevisiae. Science. 2001;294:2552–2556. doi: 10.1126/science.1065672. [DOI] [PubMed] [Google Scholar]
- 28.Valencia M., Bentele M., Vaze M.B., Herrmann G., Kraus E., Lee S.E., Schär P., Haber J.E. NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature. 2001;414:666–669. doi: 10.1038/414666a. [DOI] [PubMed] [Google Scholar]
- 29.Frank K.M., Sharpless N.E., Gao Y., Sekiguchi J.M., Ferguson D.O., Zhu C., Manis J.P., Horner J., DePinho R.A., Alt F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 30.Barnes D.E., Stamp G., Rosewell I., Denzel A., Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 31.Moshous D., Callebaut I., de Chasseval R., Corneo B., Cavazzana-Calvo M., Le Deist F., Tezcan I., Sanal O., Bertrand Y., Philippe N., et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 32.Pierce A.J., Hu P., Han M., Ellis N., Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karathanasis E., Wilson T.E. Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics. 2002;161:1015–1027. doi: 10.1093/genetics/161.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clikeman J.A., Khalsa G.J., Barton S.L., Nickoloff J.A. Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKU-dependent and -independent mechanisms. Genetics. 2001;157:579–589. doi: 10.1093/genetics/157.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank-Vaillant M., Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol. Cell. 2002;10:1189–1199. doi: 10.1016/s1097-2765(02)00705-0. [DOI] [PubMed] [Google Scholar]
- 36.Tsukamoto Y., Kato J., Ikeda H. Budding yeast Rad50, Mre11, Xrs2 and Hdf1, but not Rad52, are involved in the formation of deletions on a dicentric plasmid. Mol. Gen. Genet. 1997;255:543–547. doi: 10.1007/s004380050527. [DOI] [PubMed] [Google Scholar]
- 37.Schiestl R.H., Zhu J., Petes T.D. Effect of mutations in genes affecting homologous recombination on restriction enzyme-mediated and illegitimate recombination in Saccharomyces cerevisiae. Mol. Cell Biol. 1994;14:4493–4500. doi: 10.1128/mcb.14.7.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J., Schiestl R.H. Topoisomerase I involvement in illegitimate recombination in Saccharomyces cerevisiae. Mol. Cell Biol. 1996;16:1805–1812. doi: 10.1128/mcb.16.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christianson T.W., Sikorski R.S., Dante M., Shero J.H., Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 40.Chen X.J. Low- and high-copy-number shuttle vectors for replication in the budding yeast Kluyveromyces lactis. Gene. 1996;172:131–136. doi: 10.1016/0378-1119(96)00125-4. [DOI] [PubMed] [Google Scholar]
- 41.Rothstein R.J. One-step gene disruption in yeast. Meth. Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 42.Scherer S., Davis R.W. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc. Natl Acad. Sci. USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiestl R.H., Gietz R.D. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr. Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 44.Ausubel F.M. Short Protocols in Molecular Biology:A Compendium of Methods From Current Protocols in Molecular Biology. 4th edn. NY: Wiley; 1999. [Google Scholar]
- 45.Sambrook J., Russell D.W. Molecular Cloning:A Laboratory Manual. 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 46.Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 47.Wach A., Brachat A., Pohlmann R., Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 48.Dujon B., Sherman D., Fischer G., Durrens P., Casaregola S., Lafontaine I., De Montigny J., Marck C., Neuveglise C., Talla E., et al. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- 49.Glaz J., Naus J.I., Wallenstein S. Scan Statistics. NY: Springer-Verlag Inc.; 2001. [Google Scholar]
- 50.Parzen E. Modern probability theory and its applications. NY: John Wiley & Sons Inc.; 1960. [Google Scholar]
- 51.Sherman D., Durrens P., Beyne E., Nikolski M., Souciet J.L. Genolevures: comparative genomics and molecular evolution of hemiascomycetous yeasts. Nucleic Acids Res. 2004;32:D315–D318. doi: 10.1093/nar/gkh091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milne G.T., Weaver D.T. Dominant negative alleles of RAD52 reveal a DNA repair/recombination complex including Rad51 and Rad52. Genes Dev. 1993;7:1755–1765. doi: 10.1101/gad.7.9.1755. [DOI] [PubMed] [Google Scholar]
- 53.Donovan J.W., Milne G.T., Weaver D.T. Homotypic and heterotypic protein associations control Rad51 function in double-strand break repair. Genes Dev. 1994;8:2552–2562. doi: 10.1101/gad.8.21.2552. [DOI] [PubMed] [Google Scholar]
- 54.Boulton S.J., Jackson S.P. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson T.E., Lieber M.R. Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway. J. Biol. Chem. 1999;274:23599–23609. doi: 10.1074/jbc.274.33.23599. [DOI] [PubMed] [Google Scholar]
- 56.Åström S.U., Kegel A., Sjöstrand J.O., Rine J. Kluyveromyces lactis Sir2p regulates cation sensitivity and maintains a specialized chromatin structure at the cryptic alpha-locus. Genetics. 2000;156:81–91. doi: 10.1093/genetics/156.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schiestl R.H., Dominska M., Petes T.D. Transformation of Saccharomyces cerevisiae with nonhomologous DNA: illegitimate integration of transforming DNA into yeast chromosomes and in vivo ligation of transforming DNA to mitochondrial DNA sequences. Mol. Cell Biol. 1993;13:2697–2705. doi: 10.1128/mcb.13.5.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Decottignies A. Capture of extranuclear DNA at fission yeast double-strand breaks. Genetics. 2005;171:1535–1548. doi: 10.1534/genetics.105.046144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharp P.M., Li W.H. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knight R.D., Freeland S.J., Landweber L.F. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001;2:RESEARCH 0010.1-0010.13. doi: 10.1186/gb-2001-2-4-research0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith J., Baldeyron C., De Oliveira I., Sala-Trepat M., Papadopoulo D. The influence of DNA double-strand break structure on end-joining in human cells. Nucleic Acids Res. 2001;29:4783–4792. doi: 10.1093/nar/29.23.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roth D.B., Wilson J.H. Relative rates of homologous and nonhomologous recombination in transfected DNA. Proc. Natl Acad. Sci. USA. 1985;82:3355–3359. doi: 10.1073/pnas.82.10.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Odell M., Kerr S.M., Smith G.L. Ligation of double-stranded and single-stranded [oligo(dT)] DNA by vaccinia virus DNA ligase. Virology. 1996;221:120–129. doi: 10.1006/viro.1996.0358. [DOI] [PubMed] [Google Scholar]
- 64.Wang H., Perrault A.R., Takeda Y., Qin W., Iliakis G. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2003;31:5377–5388. doi: 10.1093/nar/gkg728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kooistra R., Hooykaas P.J., Steensma H.Y. Efficient gene targeting in Kluyveromyces lactis. Yeast. 2004;21:781–792. doi: 10.1002/yea.1131. [DOI] [PubMed] [Google Scholar]
- 66.Boulton S.J., Jackson S.P. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 1996;24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu J., Schiestl R.H. Human topoisomerase I mediates illegitimate recombination leading to DNA insertion into the ribosomal DNA locus in Saccharomyces cerevisiae. Mol. Genet. Genomics. 2004;271:347–358. doi: 10.1007/s00438-004-0987-7. [DOI] [PubMed] [Google Scholar]
- 68.Cormack B.P., Falkow S. Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics. 1999;151:979–987. doi: 10.1093/genetics/151.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ikeda H., Shiraishi K., Ogata Y. Illegitimate recombination mediated by double-strand break and end-joining in Escherichia coli. Adv. Biophys. 2004;38:3–20. [PubMed] [Google Scholar]
- 70.Baudat F., Nicolas A. Clustering of meiotic double-strand breaks on yeast chromosome III. Proc. Natl Acad. Sci. USA. 1997;94:5213–5218. doi: 10.1073/pnas.94.10.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu T.C., Lichten M. Meiosis-induced double-strand break sites determined by yeast chromatin structure. Science. 1994;263:515–518. doi: 10.1126/science.8290959. [DOI] [PubMed] [Google Scholar]
- 72.Gerton J.L., DeRisi J., Shroff R., Lichten M., Brown P.O., Petes T.D. Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 2000;97:11383–11390. doi: 10.1073/pnas.97.21.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen X.J., Clark-Walker G.D. sir2 mutants of Kluyveromyces lactis are hypersensitive to DNA-targeting drugs. Mol. Cell Biol. 1994;14:4501–4508. doi: 10.1128/mcb.14.7.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Åström S.U., Rine J. Theme and variation among silencing proteins in Saccharomyces cerevisiae and Kluyveromyces lactis. Genetics. 1998;148:1021–1029. doi: 10.1093/genetics/148.3.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]