

Abstract
Calponin is an extensively studied actin-binding protein but its function is not well understood. Among three isoforms of calponin, h2-calponin is found in both smooth muscle and non-muscle cells. The present study demonstrates that epidermal keratinocytes and fibroblast cells express significant amounts of h2-calponin. The expression of h2-calponin is cell anchorage-dependent. The levels of h2-calponin decrease when cells are rounded up and remain low when cells are prevented from adherence to culture dish. H2-calponin expression resumes after the floating cells are allowed to form a monolayer in plastic dish. Cell cultures on polyacrylamide gels of different stiffness demonstrated that h2-calponin expression is affected by the mechanical properties of the culture matrix. When cells are cultured on soft gel that applies less traction force to the cell and, therefore, lower mechanical tension in the cytoskeleton, the level of h2-calponin is significantly lower than that in cells cultured on hard gel or rigid plastic dish. Force-expression of h2-calponin enhanced actin filaments’ resistance to cytochalasin B treatment. Keratinocyte differentiation is accompanied by a mechanical tension related up-regulation of h2-calponin. Lowering the tension of actin cytoskeleton by inhibiting non-muscle myosin II ATPase decreased h2-calponin expression. In contrast to the mechanical tension regulation of endogenous h2-calponin, the expression of h2-calponin using a cytomegalovirus promotor was independent of the stiffness of culture matrix. The results suggest that h2-calponin represents a novel manifestation of mechanical tension responsive gene regulation that may modify cytoskeleton function.
Calponin is a family of actin-associated proteins first found in smooth muscle cells (1). Three calponin isoforms (h1-, h2-, and acidic calponins) are encoded by three homologous genes. H1-calponin (2, 3) is specifically expressed in differentiated smooth muscle cells and has been extensively studied for its role in the regulation of smooth muscle contractility (reviewed in 4-6). The acidic calponin is found in nervous tissues and implicates in neuronal regeneration and growth (7-8).
H2-calponin (3) is found in both smooth muscle and non-muscle cells such as human epidermal keratinocytes (9). H2-calponin mRNA has been also detected in endothelial cells (10) and fibroblasts (11). The gene regulation and function of h2-calponin are largely unknown. Previous studies suggested that h2-calponin may play a role in the organization of actin cytoskeleton (12) and in cytokinesis (13). This hypothesis is supported by the observation that h2-calponin is expressed at significant levels in growing and remodeling tissues (13). Forced expression of h2-calponin in cells lacking endogenous calponin results in an association with the actin stress fibers and a decrease in the rate of cell proliferation (13). These results suggest a microfilament-associated activity of h2-calponin, which may regulate the function of actin cytoskeleton.
The actin cytoskeleton has essential functions in many cellular activities, such as cell division (14), migration (15), and contraction (16). A major component of the actin filaments in higher eukaryotes is tropomyosin. Tropomyosin is known to participate in the regulation of striated muscle contraction (17) and to stabilize actin filaments in non-muscle (18) as well as muscle cells (19). Calponin binds actin and tropomyosin and is proposed to be an analog of striated muscle troponin (1, 20) and, therefore, may regulate the actin-myosin II-based cytoskeleton function.
Previous studies have shown that living cells respond to mechanical forces exerted through surrounding fluid, adhered beads, or cultural matrix (21-25). Physical properties of the adhesion substrate can profoundly affect cell locomotion, growth, and differentiation (26-28). The stiffness of cultural matrix has effects on cell motility (29, 30), phagocytosis (31) and differentiation (32, 33). Varying stiffness of the cultural matrix applies different traction force to the cell and, therefore, different mechanical tension to the cytoskeleton (34, 35). The regulation and function of actin cytoskeleton in the cellular responses to mechanical tension represent an important but little known subject of signal transduction.
In the present study, we investigated the regulation of h2-calponin by mechanical tension. We demonstrated that epidermal keratinocytes and fibroblast cells express significant amounts of h2-calponin, which decrease when cells are rounded up and remain low when cells are prevented from anchorage on the culture dish. H2-calponin expression resumes after the floating cells are allowed to attach to the plastic dish. When cells are cultured on soft gel matrix that applies lower mechanical tension to the cytoskeleton, the level of h2-calponin is significantly lower than that in cells cultured on hard gel or rigid plastic dish. Keratinocyte differentiation is accompanied by a mechanical tension related up-regulation of h2-calponin. Force-expression of h2-calponin increased actin filaments’ resistance to cytochalasin B treatment. Lowering the tension of actin cytoskeleton by inhibiting non-muscle myosin II ATPase decreased h2-calponin expression. In contrast to the mechanical tension regulation of endogenous h2-calponin, the expression of h2-calponin using a cytomegalovirus (CMV) promotor was independent of the stiffness of culture matrix. The results suggest that h2-calponin represents a novel manifestation of mechanical tension responsive gene regulation that may modify cytoskeleton function.
MATERIALS AND METHODS
Specific antibodies—A rabbit polyclonal antiserum (RAH2) raised against mouse h2-calponin with a weak cross-reaction to h1-calponin has been described previously (36).
Monoclonal antibody (mAb) 1D2 against human h2-calponin was developed as described previously for the production of anti-mouse h2-calponin mAbs (13). Briefly, human h2-calponin cDNA was cloned using reverse transcription-coupled polymerase chain reaction as described previously (36) and expressed in E. coli for protein preparation (37). Purified human h2-calponin was used to immunize 8-week-old female Balb/c mice in a short-term immunization protocol (38). Spleen cells were harvested from the immunized mouse for fusion with SP2/0- Ag14 mouse myeloma cells. Hybridoma colonies were screened by indirect enzyme-linked immunosorbant assay (ELISA) and subcloned three times to establish stable cell line. The mAbs were produced in the forms of hybridoma cultural supernatant and mouse ascites fluids. ELISA Immunoglobulin isotyping (Invitrogen) determined its subclass as IgG2b κ. Its specificity was determined by Western blotting on purified h1- and h2-calponins (37).
Mouse anti-h1-calponin mAb CP1 was prepared and characterized previously (39). Mouse anti-tropomyosin isoform mAbs, CG1 against hTM1, CGβ6 against hTM2 and hTM3, LC24 against hTM4 and CG3 against hTM5, were described previously (40-43). A rabbit anti-human involucrin antibody was produced as previously described (44, 45).
Western blotting—SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting were carried out as described previously (38) to examine the expression of calponin in mouse skin tissues and human keratinocyte and fibroblast cultures. The samples were homogenized in SDS gel electrophoresis sample buffer containing 2% SDS and analyzed on 12% gel with an acrylamide:bisacrylamide ratio of 29:1 in the Laemmli buffer system. After electrophoresis, the gels were fixed and stained with Coomassie blue R250 to confirm sample integrity and protein contents. Protein bands in unfixed duplicate gels were transferred to nitrocellulose membrane for Western blotting with the anti-calponin, anti-tropomyosin or anti-involucrin antibodies. The specific protein bands recognized by the first antibodies were revealed by using alkaline phosphatase-labeled anti-rabbit IgG or anti-mouse IgG second antibody (Sigma) and 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium chromogenic substrate reaction. Purified h2- and h1-calponins (37) was used as control.
Immunohistochemistry—To examine the in vivo expression of h2-calponin in epidermal keratinocytes, paraffin sections of epidermal scar (anonymously provided by the Skin Disease Research Center at Case Western Reserve University School of Medicine) were stained with anti-h2 calponin mAb 1D2 that has no cross-reaction to h1-calponin, followed by horseradish peroxidase-labeled anti-mouse IgG second antibody (Sigma) and H2O2-daminobenzidin substrate reaction using standard immunohistochemical method (46). To avoid background staining of tissue sections by immunosera and mouse ascites fluid (data not shown), 1D2 hybridoma culture supernatant was used. The morphology of tissue sections was outlined by counterstaining of the slides with 0.6 % hematoxylin for 20 sec.
Cell cultures—Primary keratinocytes were isolated from human foreskin samples (47) and cultured in keratinocyte serum-free media (KSFM, Invitrogen) with supplements (50 μg/ml bovine pituitary extract, 5 ng/ml human recombinant epidermal growth factor, Invitrogen) at 37 °C in 5% CO2. Culture media were replaced every 3 days. The monolayer cells around 70% confluence were passed at 1:5 ratio and cells at second or third passages were used for experiments. The methods for culturing human epidermal keratinocytes were described in detail previously (48, 49).
Human fibroblast cell line KD (American Type Culture Collection, CRL 1295) was cultured in DMEM (Dulbecco’s modified Eagle medium) containing 10% fetal bovine serum (FBS), penicillin (100 i.u./ml) and streptomycin (50 i.u./ml) at 37 °C in 5% CO2.
SM3 is an immortalized cell line derived from rabbit aortic smooth muscle (50) and has ceased endogenous calponin expression (13). Stable transfected SM3 cells force-expressing h2-calponin driven by CMV promoter in pcDNA3.1 vector (13) were used in the present study to investigate h2-calponin regulation and functional effect. The sense cDNA construct expresses non-fusion full-length mouse h2-calponin protein for authentic functional characterization and antisense cDNA transfected cell lines were used as negative control. The transfected SM3 cells were cultured in DMEM containing 10% FBS, penicillin (100 i.u./ml), streptomycin (50 i.u./ml), and G418 (100 μg/ml) at 37°C in 5% CO2.
Immunfluorescence microscopy—Precleaned glass cover slips were coated with 0.1% gelatin and dried under UV radiation for 3 hours before placed in the culture dish. Keratinocytes and KD cells were seeded to grow monolayer on the cover slips. After 3 days of culture, the cells grown on coverslips were washed with Dulbecco’s phosphate buffered saline (D-PBS, 136.89 mM NaCl, 2.68 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.2). The cells were then fixed with cold acetone for 20 minutes. Immunofluorescence microscopy was carried out as described previously (13) to examine the cellular localization of h2-calponin and tropomyosin using anti-calponin and anti-tropomyosin antibodies. Tetramethylrhodamine isothiocyanate (TRITC)- or fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit or anti-mouse IgG second antibodies (both from Sigma) were used to distinguish the rabbit and mouse first antibody staining representing the localization of calponin and tropomyosin, respectively, using a Zeiss Axiovert 100H phase contrast-epifluorescence microscope with two sets of filters (CZ915 and CZ909). Actin stress fibers in the cell were examined by rhodamine-labeled phalloidin (Sigma) staining. The co-localization of h2-calponin and tropomyosin in KD cells were also examined by confocal microscopy at the Neurobiology Core Facility at Case Western Reserve University School of Medicine.
Examination of cell plating time-dependence of h2-calponin expression—To investigate the expression of h2-calponin during the time course of monolayer cell culture, pre-confluent keratinocytes were trypsinized and seeded at high and low densities (initial density = 9 × 105 cells and 3 ×105 cells per 60 mm dish, respectively). The cells were suspended from the culture dishes after 1, 2, 3, 4 and 5 days using the Versene solution (in mM: 0.537 EDTA, 136.8 NaCl, 2.68 KCl, 8.1 Na2HPO4, pH 7.2) and washed 3 times with D-PBS. Omitting trypsin digestion during the cell harvest avoids artificial degradation of cellular proteins. SDS gel sample buffer containing 2% SDS was used to lyze the cells and total cellular protein was extracted by vortexing. Levels of h2-calponin in high and low density keratinocyte cultures at the post plating time points were examined by Western blot analysis using anti-h2-calponin antibody RAH2. KD fibroblasts and stable transfected SM3 cells were examined similarly for the plating time effect on the levels of h2-calponin.
Examination of matrix adhesion-dependent expression of h2-calponin—To examine the effects of matrix attachment on the expression of h2-calponin in cultured cells, continuous ibration was applied to keratinocytes, KD fibroblasts and SM3 cells to prevent cell attachment to tissue culture dishes (51). Incubated on a humidified CO2 incubator at 37 °C, the cultural dishes were placed on an orbital shaker (Bel-Art Products, Pequannock, NJ) driven by a magnetic stirrer at 80 rpm. The cells were harvested after 3 days of culture in vibration to examine the levels of h2-calponin. After washed with PBS, the cells were lyzed in SDS-gel sample buffer and examined by Western blotting analysis as described above. The viability of the floating cell aggregates was confirmed by growing into monolayers after re-seeding on tissue culture dishes and incubated without vibration. The expression of h2-calponin in the cells after re-seeding was examined as described above.
Examination of culture matrix stiffness-dependent expression of h2-calponin—To compare with the rigid plastic cultural dish, a thin layer of polyacrylamide gel was prepared on glass cover slips to provide soft matrix for cell culture as described previously (30, 35, 52). The cover slips were passed briefly through the inner flame of a Bunsen burner. A drop of 0.1N NaOH was smeared across the surface of the cover slip and air-dried. Thereafter, the cover slips were treated with 3-aminopropyltrimethoxysilane and then with 0.5% glutaraldehyde. Polyacrylamide gels of different stiffness (very hard, 10% gel with an acrylamide:bisacrylamide ratio of 40:1; hard, 3% gel with an acrylamide:bisacrylamide ratio of 14:1; and soft, 3% gel with an acrylamide:bisacrylamide ratio of 74:1) were polymerized in between the treated cover slip at the bottom and an untreated cover slip on the top. After polymerization, the untreated cover slip was removed to expose the ∼100 μm thin layer of gel to be used as cell culture matrix. Previous studies have shown that the stiffness of the polyacrylamide gel matrix is directly proportional to the concentration of bis-acrylamide cross-linker (30, 34, 52).
After treating the gel with a bifunctional cross-linker (1 mM sulfo-SANPAH, Pierce, Rockford, IL) in 50 mM HEPES, pH 8.5 (30), Type I collagen was applied to the gel surface, followed by gently shaking overnight at 4°C for cross linking to the gel. Gels were then washed with D-PBS and sterilized with UV irradiation for 3 hours. The gel was soaked in culture media at 37 °C for 1 hour before plating cells.
To examine the effect of the tension that the culture matrix applied to the cell through traction force on the expression of h2-calponin, keratinocytes were seeded on plastic culture dish or polyacrylamide gels of different stiffness. The cells were harvested after 3 days of culture by directly lysis in SDS gel sample buffer after D-PBS washes. The level of h2-calponin was examined by Western blot analysis with the anti-h2-calponin antibody RAH2 as described.
Measurement of cell spreading area—Monolayer keratinocytes cultured on plastic dish or polyacrylamide gels of different stiffness were photographed at 2, 4, 8, 12, 24, 48, and 72 hrs of culture. The NIH Image program version 1.61 was used to measure the two-dimensional spreading area of randomly selected non-overlapping cells.
Ca2+ induction of keratinocyte differentiation—To examine the expression of h2-calponin during keratinocyte differentiation in vitro, cells were plated on plastic culture dishes or soft polyacrylamide gel (3% with an acrylamide:bisacrylamide ratio of 74:1). One day after plating, the concentration of CaCl2 in culture media was increased to 0.3 mM. The cells were harvested after 48 hours in the high Ca2+ media for Western blotting examination of h2-calponin as described above. The expression of involucrin, an established cell differentiation marker, in epidermal keratinocytes was examined by Western blot to monitor the induction of differentiation (53-55).
Cytochalasin B treatment—To investigate the effect of h2-calponin on actin cytoskeleton, stable transfected SM3 cells expressing h2-calponin were plated on gelatin-coated cover slips. After 3 days of culture, the cells were treated with 0.5, 0.75 and 1.0μg/ml of cytochalasin B (Sigma) at 37 °C for 30 min. h2-calponin-negative SM3 cells were examined as control. The cytochalasin B stock was prepared in DMSO, so a 0.2 % DMSO-treated control group of cells was also examined. The cells were washed with PBS and fixed with cold acetone. The structure of stress fibers stained with rhodamine-labeled phalloidin was examined as above and evaluated for the resistance to cytochalasin B treatment.
Blebbistatin treatment—To examine the effect of reducing cytoskeleton tension which is mainly built up by myosin II motor activity on the expression of h2-calponin in cells, NIH 3T3 cells were cultured on plastic dish and gelatin-coated cover slips (3 × 104 cells/35 mm dish). After 3 days of monolayer culture, the cells were treated with 100 μM blebbistatin, a myosin II ATPase inhibitor (56), for 3 days. Parallel cultures in normal media or containing 0.2 % DMSO (solvent for making the blebbistatin stock) were examined as controls. The cells in culture dish were harvested by lysis in SDS-PAGE sample buffer after PBS washes and the levels of h2-calponin were determined by Western blot analysis as above. The cells on the cover slips were fixed with cold acetone and the actin stress fibers were stained with rhodamine-conjugated phalloidin. The fluorescence and phase contrast images were viewed under a Zeiss Axiovert 100H fluorescence microscope.
Data analysis—Densitometry analysis of SDS-gel and Western blots was done on digital images scanned at 600 dpi and the NIH Image program version 1.61 was used to quantify the levels of h2-calponin and tropomyosin expression. The calponin and tropomyosin bands detected in Western blots were normalized by the amounts of actin in equally loaded SDS-gel. The quantitative data of cell area, h2-calponin and tropomyosin levels are presented as mean ± SD. Statistical analysis was done using the Microsoft Excel computer program.
RESULTS
H2-calponins expression in epidermal keratinocytes—The expression of h2-calponin in mouse and human skin tissues was examined by Western blot and immunohistochemistry. The results in Fig. 1A show significant amounts of h2-calponin in mouse skin. Normalized by actin contents, mouse footpad has more h2-calponin expression than that in the abdominal skin, in agreement with the thickness of keratinocyte layer. No h1-calponin was detected in the mouse skin. Immunohistochemical staining detected h2-calponin expression in the keratinocytes of human skin enriched in the basal layer and the early suprabasal layers (Fig. 1B). Western blots with either polyclonal (RAH2) or monoclonal (1D2) antibody confirmed the high level expression of h2-calponin in primary cultures of human epidermal keratinocytes (Fig.1 C). H1-calponin was not detected.
Fig. 1.
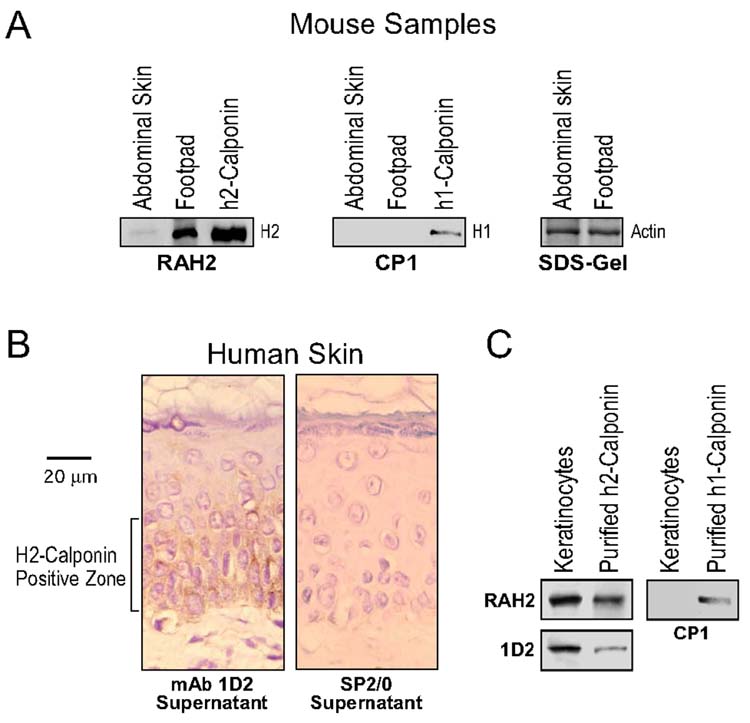
H2-calponin expression in epidermal keratinocytes. (A) Total protein extracts from mouse abdominal and footpad skin tissues were analyzed by Western blots using the anti-h2-calponin polyclonal antibody RAH2 and anti-h1-calponin mAb CP1. Purified mouse h1- and h2-calponin proteins were used as control. Normalized by the level of actin, the blots show a significant level of h2-, but not h1-, calponin in the mouse skin, especially footpad. (B) Thin paraffin sections of human epidermal scar tissues were examined by immunocytochemistry with anti-h2-calponin mAb 1D2 and SP2/0 myeloma cultural supernatant control. The results show h2-calponin expression in the keratinocyte layers. (C) Western blots using anti-h2-calponin polyclonal antibody RAH2 and anti-h1-calponin mAb CP1 on total protein extracts from human keratinocytes cultured 3 days on plastic dish detected high levels of h2- but not h1-calponin. The sample loading was normalized by the level of actin and purified mouse h1- and h2-calponins were included as control.
Expression of h2-calponin is dependent on the time of culture and independent of cell density—Human epidermal keratinocytes from three-day old cultures were passed on plastic culture dishes. Western blots on samples taken at a series of time points of culture showed that the level of h2-calponin was low 24 hours after plating and up-regulated thereafter (Fig. 2). The levels increased significantly after 2 days of culture (P < 0.001) and reached the maximum at day 3. This culture time-dependent up-regulation of h2-calponin is independent of cell density since high and low density cultures showed similar expression patterns. Similar results were obtained following collection of cells by trypsin digestion or the EDTA-based Versene solution (data not shown). The results suggest that cell round up during passages down-regulates the level of h2-calponin that is resumed during monolayer culture, independent of cell-cell contacts.
Fig. 2.
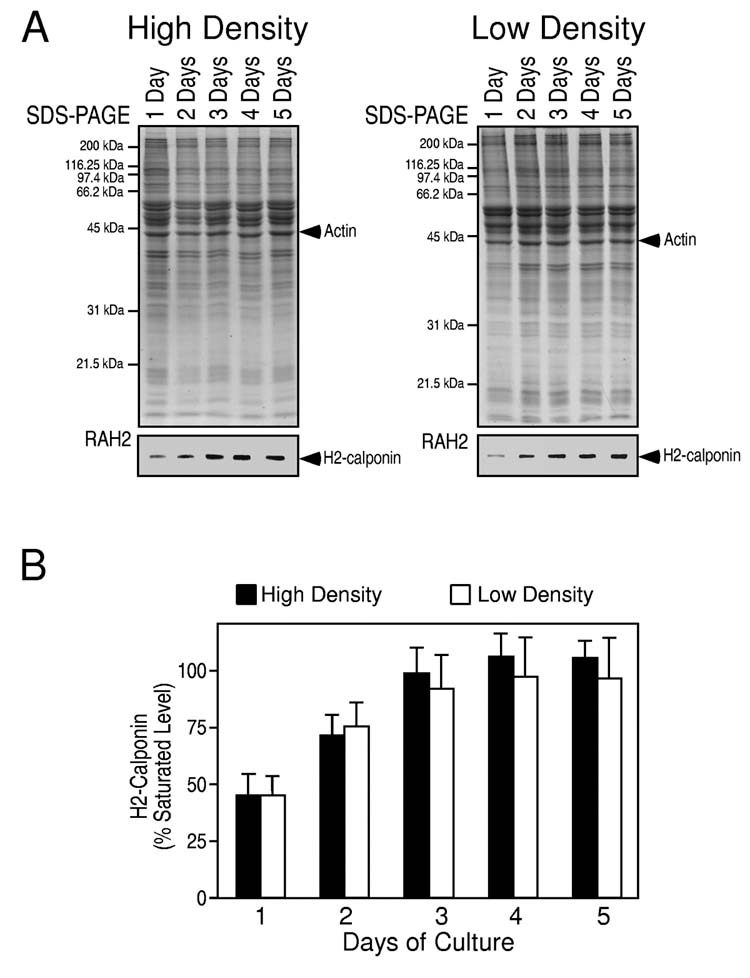
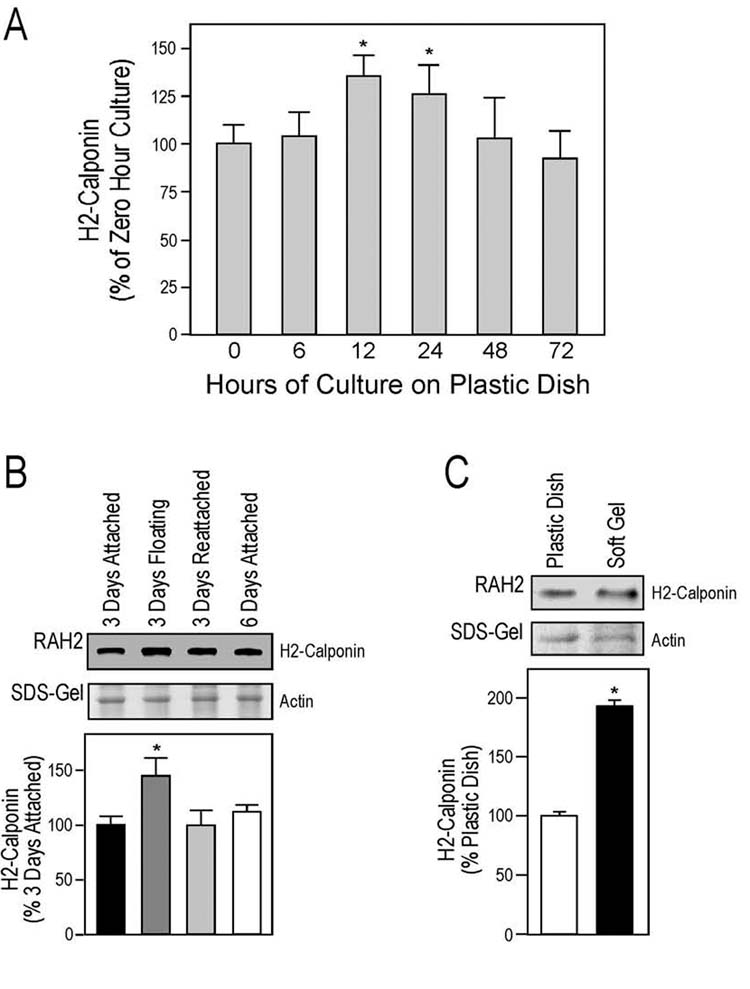
Culture time-dependent expression of h2-calponin in keratinocytes. From three-day old pre-confluence cultures, human epidermal keratinocytes were passed on plastic dishes at low or high densities. The cells were harvested at 1, 2, 3, 4 and 5 days after plating and the total protein extracts were examined by SDS-PAGE and Western blot using anti-h2-calponin polyclonal antibody RAH2 (A). Normalized against the actin band in the accompanying SDS-gel, densitometry quantification of the Western blots was used to compare the levels of h2-calponin expression (B). The results show that independent of the high (solid column) or low (open column) cell densities (the cells were at 100% and 60% confluence at three days in culture, respectively), the expression of h2-calponin was low at one day after plating (P < 0.001) and returned to the maximum level three days after plating. The results are summarized from 6 individual experiments.
Expression of h2-calponin is dependent on cell anchorage to the culture dish—Two factors may confer the recover of h2-calponin expression during the 3 days of culture after cell passage. One is the culture time after passage and the other is the re-attachment to the culture dish. We investigated which factor determines h2-calponin expression by producing floating cell cultures. The results in Fig. 3 A and 3B show that when keratinocytes were prevented from attachment to the culture dish by continuous vibration, the cells grew in multi-cell aggregates with significantly decreased h2-calponin expression.
Fig. 3.
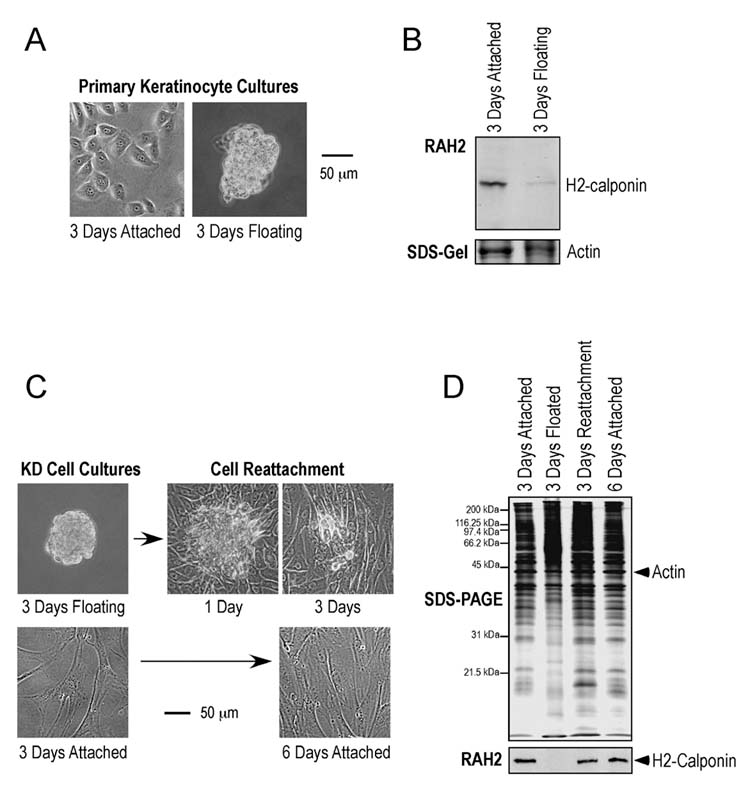
Matrix anchorage-dependent expression of h2-calponin in keratinocytes. Human epidermal keratinocytes were cultured on plastic cultural dishes steadily as a monolayer or with continuous vibration that prevented the cells from anchoring on the dish. After 3 days of culture, the cells were photographed for phase contrast images (A) and harvested to examine the total protein extracts by Western blotting with anti-h2-calponin polyclonal antibody RAH2 (B). Normalized by the actin level, the results show that the floating keratinocytes growing in multi-cell aggregates had significantly reduced h2-calponin expression. Similarly, phase contrast images (C) show continuous vibration prevented KD human fibroblasts from anchoring to the cultural dish and produced multi-cell aggregates. When the floating cell aggregates were re-seeded on plastic dish and cultured without vibration, they attached to the dish to form a monolayer similar to the steady culture control of the same age. The result verifies the viability of the cells in the floating aggregates. (D) Total protein extracts from the KD cells were examined by SDS-PAGE and Western blotting using anti-h2-calponin antibody RAH2. Normalized by the level of actin, the results show that KD cells lost h2-calponin expression when growing as floating aggregates. The expression of h2-calponin resumed when the floating cells anchored to the cultural dish.
Since primary human epidermal keratinocytes may not survive well in the floating culture, the observed phenotype after passages often vary between experiments. We therefore verified this observation using the human fibroblast cell line KD. The Western blots in Fig. 3 C and 3D show a significant expression of h2-calponin in the KD fibroblast monolayer cultured on plastic dishes. When KD cells were cultured with continuous vibration, they also grow in multi-cell aggregates (Fig. 3C). Western blot analysis showed that the floating KD cells completely lost h2-calponin in sharp contrast to the high level expression in monolayer cells attached on plastic dish (Fig. 3D). After the floating cells were re-plated and cultured without vibration, they attached to the plastic dishes and regained high level expression of h2-calponin (Fig. 3D). The results verify the viability of the floating cells cultured under vibration and further demonstrate that h2-calponin expression is dependent on the anchorage to the plastic culture dish other than the culture time after passage. Consistent with the independence of cell-cell contacts as shown in the cell density comparisons (Figs. 2), the aggregation of floating cells does not induce h2-calponin expression.
Matrix stiffness determines h2 calponin expression—Plastic dish provides a rigid culture matrix that allows the attached cell to exert a high traction force, applying a high mechanical tension to the cellular structure. To investigate a hypothesis that the high mechanical tension in monolayer cells growing on plastic tissue culture dish induces h2-calponin expression, we used polyacrylamide gel to provide cell culture matrices of different stiffness. It has been documented that the different stiffness of the gel matrix will produce different strength of traction forces to the cell (29). The results in Fig. 4 show that hard gel matrices produced high level of h2-calponin expression similar to that in cells cultured on plastic dishes, indicating that the difference in matrix material (collagen coated polyacrylamide gel or plastic) has little effect on h2-calponin expression. On the other hand, cells cultured on the soft gel had significantly lower level of h2-calponin expression in comparison to that of the hard gel or plastic controls (P<0.001). The results support a novel observation that mechanical tension determines the expression of h2-calponin.
Fig. 4.
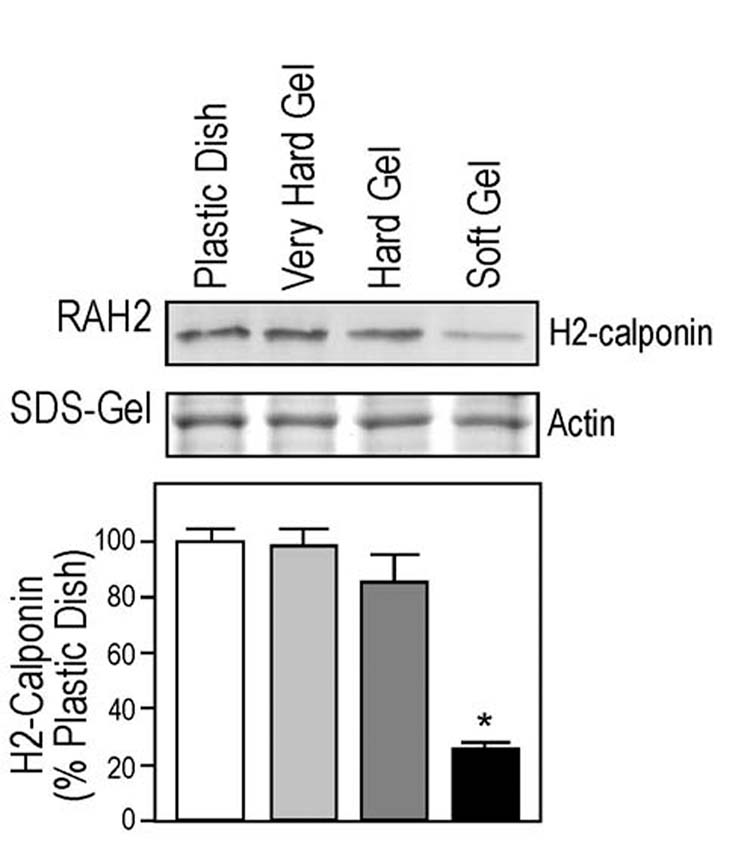
Matrix stiffness dependent expression of h2-calponin in keratinocytes. Keratinocytes were plated on plastic surface or polyacrylamide gels of different stiffness. After 3 days of culture, the levels of h2-calponin were determined by Western blot analysis using RAH2 antibody. Normalized by the amount of actin, densitometry quantification results in show a significantly lower expression of h2-calponin in keratinocytes grown on soft polyacrylamide gel in comparison with that of the hard gel and plastic dish cultures (* P < 0.001). The results were summarized from 5 individual experiments.
It has been established that centripetal directed traction forces exerted on cells and cell areas are increased with increasing in stiffness of culture matrix (29). The spreading area of keratinocytes at 4 hours after plating was smaller when cultured on soft gel than that of the very hard gel or plastic controls (Fig. 5A and 5B). The less spreading morphology of the cells cultured on soft matrix implies less tension applied to the cytoskeleton than that in the more spreading cells cultured on hard matrices. Nonetheless, the cell area is increasing at similar rates during culture under these conditions (Fig. 5C), indicating adjusted cytoskeleton activity.
Fig. 5.
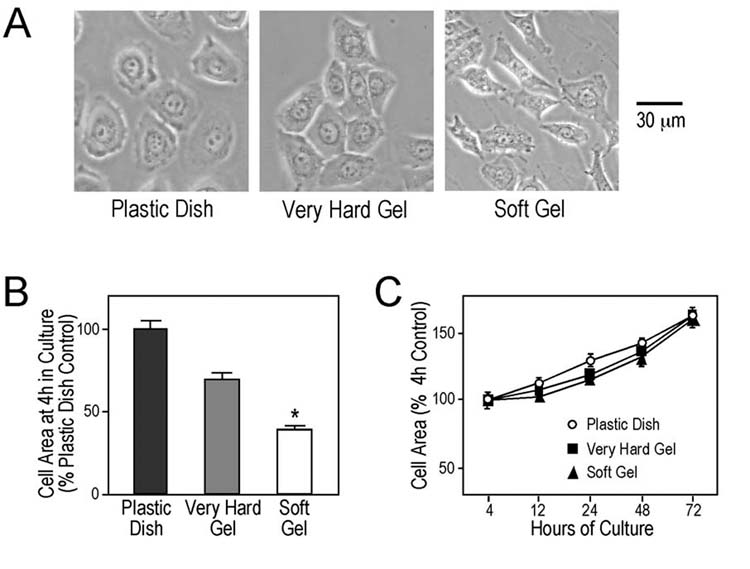
Cell spreading areas when cultured on matrices of different stiffness. Keratinocytes were cultured on plastic surface or polyacrylamide gels of different stiffness. (A) Phase contrast images at 3 days of culture show that the cells attached and grew in monolayers on the plastic and gel matrices. The cell spreading areas 4 hr after plating were significantly lower (* P < 0.001) in the soft gel culture as compared with the very hard gel and plastic controls (B). However, the cell spreading areas under the three matrix stiffness conditions increased at similar rates during culture (C).
Up-regulation of h2-calponin during Ca2+-induced differentiation of keratinocytes—Keratinocyte is the major cell type of the continually renewing epidermis and undergoes a choreographed program of differentiation. An increase in extracellular Ca2+ level is thought to induce the keratinocyte differentiation in the skin (57). To evaluate the functional significance of mechanical tension-related expression of h2-calponin in epidermal keratinocytes, its relationship with phenotypic differentiation was investigated. The results in Fig. 6 show that when keratinocytes were cultured on soft gel matrix, Ca2+ induced differentiation as shown by the expression of involucrin. The low level expression of h2-calponin on soft matrix was significantly increased during differentiation, suggesting a role in the phenotype transition. In contrast, keratinocytes grown on plastic matrix already had high level expression of h2-calponin before differentiation, which did not show further increase after differentiation (Fig. 6). The observation that the Ca2+-induced differentiation and high mechanical tension did not have additive effects on the level of h2-calponin suggests that h2-calponin gene regulation is a common target of these two very distinct cell signaling mechanisms.
Fig. 6.
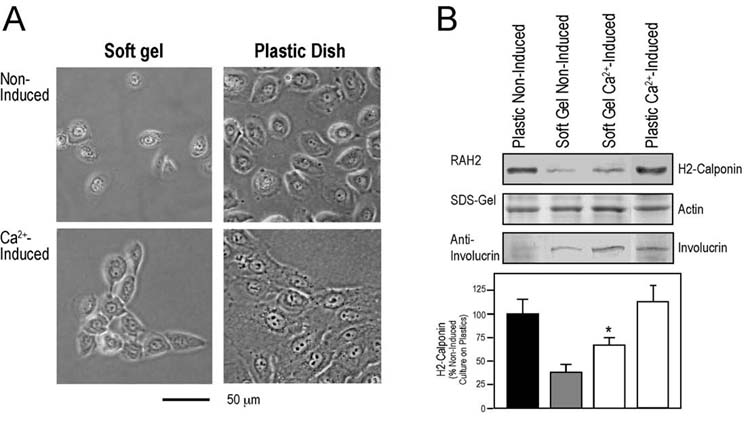
Increased h2-calponin expression during Ca2+-induced differentiation of keratinocytes grown on soft matrix. Human epidermal keratinocytes were plated on soft polyacrylamide gel or plastic dish and induced with 0.3mM CaCl2 at 40-70 % confluence for 48 hours. (A) Phase contrast images show that in contrast to non-induced spreading cell culture, the cells in both soft and rigid matrix cultures migrated together upon Ca2+ induction. (B) Normalized by the levels of actin, total protein extracted from the cells was analyzed by Western blot to examine the expression of h2-calponin and involucrin. The increase in involucrin level upon Ca2+ induction verified the differentiation of keratinocytes in cultures on soft and rigid matrix. While the expression of h2-calponin was very low in keratinocytes cultured on soft gel, it is significantly up-regulated upon Ca2+ induction (* P < 0.001). Different from the soft matrix cultures, the expression of h2-calponin in the cells cultured on rigid matrix was already high and did not further increase during differentiation. Five individual experiments each were performed for the soft and rigid matrix differentiation experiments.
Association of h2-calponin with actin cytoskeleton—Immunofluorescence staining of human epidermal keratinocytes using the anti-h2-calponin antibody RAH2 revealed a pattern similar to that obtained with phalloidin staining (Fig. 7A). Although the primary human epidermal keratinocyte cultures did not show clear stress fibers, the similar staining patterns indicate an association of h2-calponin with the actin filaments. Typical actin stress fibers were seen in KD fibroblasts and anti-h2-calponin antibody produced a staining pattern similar to that of phalloidin (Fig. 7B). The localization of tropomyosin isoforms hTM4 and hTM5 was examined as representatives to compare the cellular localizations of h2-calponin and tropomyosin. The results in Fig. 7B show similar distribution of h2-calponin and tropomyosin. Tropomyosin is an actin associate protein and the results further demonstrate the association of h2-calponin to the actin cytoskeleton. The colocalization of h2-calponin and tropomyosin in actin stress fibers was further demonstrated by confocal microscopy on the double-stained KD fibroblast cells (Fig. 7C). The results suggest that h2-calponin may function in actin cytoskeleton activities together with tropomyosin.
Fig. 7.
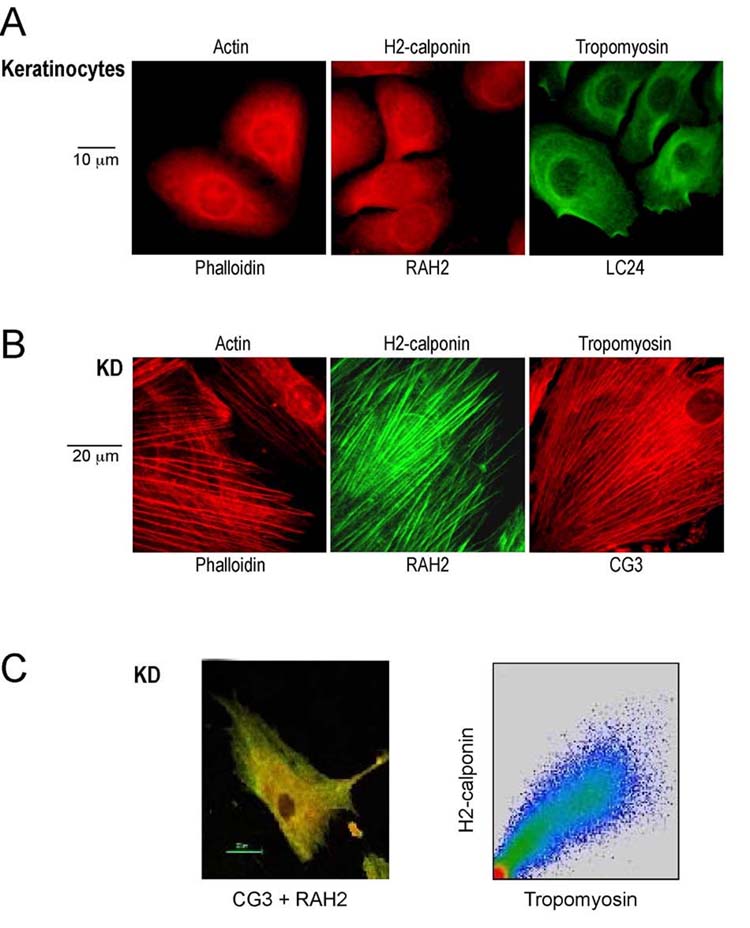
Association of h2-calponin with actin cytoskeleton. Human keratinocytes and KD fibroblasts were cultured on gelatin-coated glass cover slips. (A) Pre-confluent monolayer keratinocyte cultures were examined by immunofluorescence microscopy with the rabbit anti-h2-calponin antibody RAH2 or anti-tropomyosin (hTM4) mAb LC24 in comparison with TRITC-phalloidin stained actin filaments. (B) Immunofluorescence assay on KD fibroblast culture was carried out using anti-h2-calponin antibody RAH2 and anti-tropomyosin (hTM5) mAb CG3 together with TRITC-phalloidin control. (C) Confocal microscopy of the double-stained KD cells demonstrates the co-localization of h2-calponin (green) and Tm (red) in actin stress fibers (the yellow color). Plotting the intensity of co-localized h2-calponin and Tm stains (right panel) shows a positive correlation.
H2-calponin increases actin filaments’ resistance to cytochalasin B—By binding to the barbed ends of actin filament and thus inhibiting polymerization, cytochalasins disrupt actin cytoskeleton and can be used to evaluate the stability of actin filaments (58). We investigated the role of h2-calponin in the function of actin cytoskeleton by examining the effects of cytochalasin B on actin stress fibers. SM3 cell provides an endogenous h2-calponin null background to investigate the functional effect of transfective expression of h2-calponin. Normalized against actin, the h2-calponin contents in stable transfected SM3 lines, keratinocytes, mouse and human fibroblast cell lines showed a wide range of variance (up to ∼2.4 folds differences, data not shown) where the levels of h2-calponin expressed in the transfected SM3 cells was not an outsider, indicating that the h2-calponin force-expressed in SM3 cells is within the natural range. The rhodamine-labeled phalloidin stain in Fig. 8A shows that the actin stress fibers in h2-expressing SM3 cells tolerated higher concentrations (>0.75 g/ml) of cytochalasin B than that of the calponin null control SM3 cells. Tropomyosin has been demonstrated to contribute to the stability of actin filament in cytochalasin assays (59). Four isoforms of tropomyosin are detected in SM3 cells (hTM2, hTM3, hTM4 and hTM5) and all of them were increased in the presence of forced expression of h2-calponin (Fig. 8B and C). Moreover, the increases in tropomyosin amount parallel to the force-expressed levels of h2-calponin (r2 = 0.888, P < 0.05). These results suggest that h2-calponin may regulate the dynamics and function of actin filaments together with the function of tropomyosin. It is worth noting that the tropomyosin contents in non-transfected SM3 cells, keratinocytes, KD and 3T3 fibroblast cell lines vary significantly (up to ∼3 folds differences, data not shown). Therefore, it is not simply the level of Tm that confers the functional effect of h2-calponin. The contribution of Tm to the cell phenotype changes corresponding the presence or absence of h2-calponin remains to be investigated.
Fig. 8.
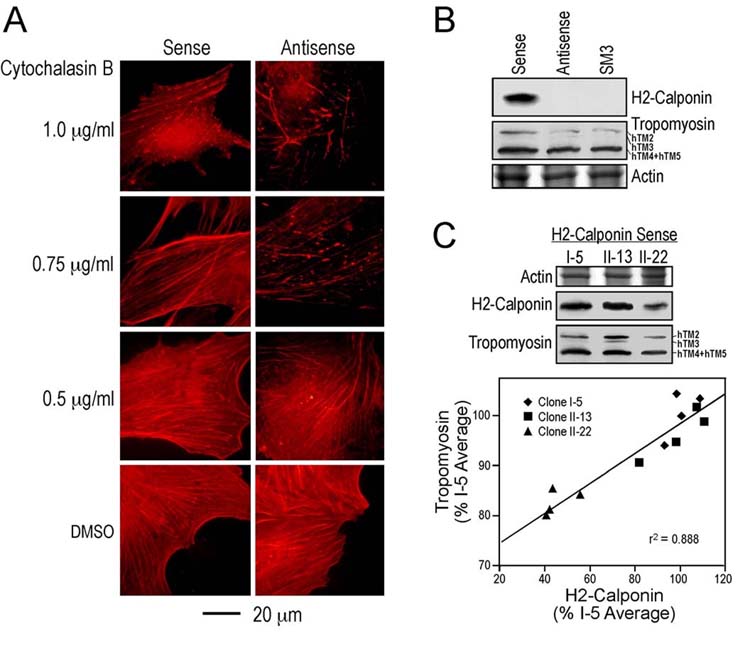
H2-calponin stabilizes actin cytoskeleton. (A) Three-day old monolayer cultures of h2-sense and antisense cDNA stable transfected SM3 cells were treated with serial concentrations of cytochalasin B (0.5, 0.75 and 1.0 μg/ml) at 37°C for 30 minutes. The cells were then fixed with cold acetone and actin stress fibers were visualized by staining with TRITC-conjugated phalloidin. The fluorescence microscopic images show that the presence of h2-calponin produced a higher tolerance of the actin cytoskeleton to cytochalasin B. (B) A significant level of h2-calponin was expressed in the sense cDNA transfected SM3 cells as shown by the RAH2 Western blot in contrast to the non-transfected and antisense cDNA transfected controls. Normalized by the level of actin in SDS-gel, Western blots using a mixture of mAbs CGβ6, LC24 and CG3 showed that the level of tropomyosin (hTm2 + hTM3 + hTM4 + hTM5) was higher in the sense h2-calponin cDNA transfectedSM3 cells than that in antisense and non-transfected controls. (C) Correlation analysis on three original h2-calponin sense cDNA-transfected SM3 cell lines showed a positive relationship between the levels of h2-calponin and tropomyosin (P < 0.05).
Inhibition of myosin II results in decreases in h2-calponin—Blebbistatin is a small molecule inhibitor that selectively binds non-muscle myosin II with high affinity, slows down its ATPase rate, and blocks myosin II in an actin-detached state (60). Blebbistatin provides a tool to reduce mechanical tension built into the actin cytoskeleton (61). When three-day old monolayer cultures of NIH 3T3 fibroblasts that express significant levels of h2-calponin (Fig. 9A) were treated with 100 μM blebbistatin, the morphology of the cells started to change within 1 hour and remained at a “slack” state (Fig. 9B). H2-calponin expression was determined by Western blot analysis with anti-h2-calponin antibody RAH2. Densitometry quantification results show a significantly lower level of h2-calponin in blebbistatin treated cells in comparison with that of non-treated (P < 0.01) and 0.2 % DMSO (P < 0.001) treated cells (Fig. 9C). H2-calponin expression in DMSO-treated cells was slightly increased (P <0.01) in comparison to that in non-treated NIH 3T3 cells, suggesting that DMSO may induce h2-calponin expression. Nonetheless, the opposite change indicates that the trace amount of DMSO carried over from the blebbistatin stock is not responsible for the decrease in h2-calponin in the blebbistatin treated group. The results support the hypothesis that mechanical tension in the actin filaments regulates h2-calponin expression. This mechanism is plausible for calponin’s function in regulating actin-myosin interaction and worth further investigation.
Fig. 9.
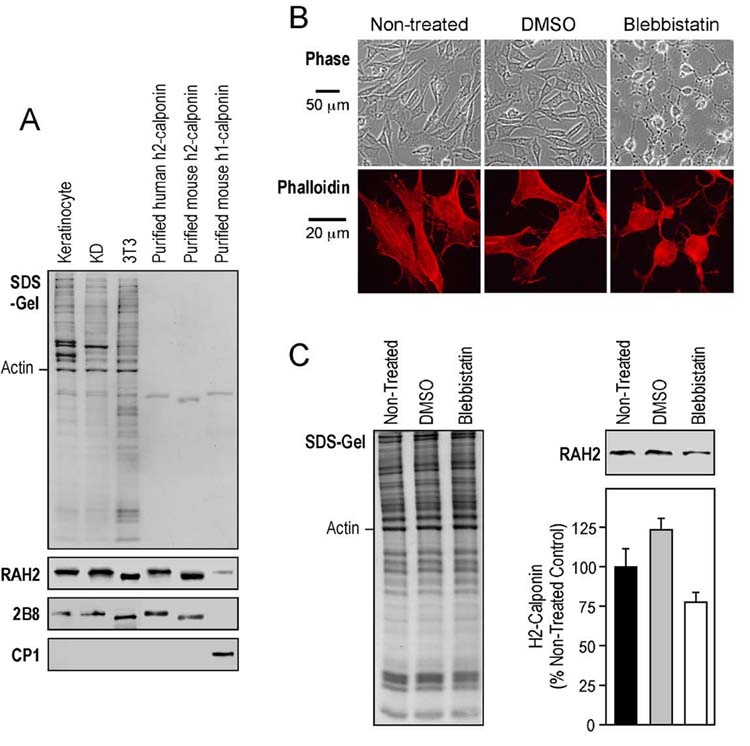
Myosin II inhibitor reduces h2-calponin in NIH 3T3 cells. (A) Western blots using RAH2 antibody detected a significant amount of h2, but not h1, calponin in NIH 3T3 mouse fibroblast, comparable with that in human epidermal keratinocytes and KD fibroblasts. The results were confirmed by Western blots using ant-h2-calponin mAb 2B8 (13) and anti-h1-calponin mAb CP1 (39). The protein loading was normalized by the level of actin. Purified human h2-, mouse h1- and h2-calponins were used as controls. (B) Three-day old monolayer 3T3 cell cultures on plastic dishes or gelatin-coated cover slips were treated with 100 μM blebbistatin for 3 days. Phase contrast images of the cells were taken before fixation. The cells on the cover slips were examined for actin stress fibers by TRITC-conjugated phalloidin staining after acetone fixation. The results show that the blebbistatin treated cells became slack with diminished actin stress fibers. (C) H2-calponin levels were determined by Western blot analysis with RAH2 antibody. Normalized by the amount of actin, densitometry quantification results show a significantly decrease in h2-calponin in blebbistatin-treated cells in comparison with that of non-treated (P < 0.01) and DMSO (P < 0.001) controls. An increase in h2-calponin in the DMSO control was observed (P < 0.01). The results were summarized from 4 individual experiments.
Viral promoter-directed expression of h2-calponin is independent of cell attachment or matrix stiffness—The regulation of h2-calponin by mechanical tension applied to the actin cytoskeleton and h2-calponin’s role in stabilizing actin filaments suggested two possible mechanisms of cellular regulation. One is that the mechanical signals regulate the expression of h2-calponin gene. The other is that a reorganization of cytoskeleton in response to mechanical tension alters the accumulation of h2-calponin. To investigate these potential mechanisms, we examined forced expression of h2-calponin by a viral promoter in SM3 cells, an immortalized cell line derived from rabbit aortic smooth muscle that has lost endogenous calponin expression. With a calponin-positive origin implying a compatible cellular environment, SM3 cell provides a calponin null background for investigating the function of exogenous h2-calponin. CMV promoter produced a high level expression of h2-calponin in monolayer SM3 cultures. The results in Fig. 10A show that in contrast to the post-cell roundup decreases of endogenous h2-calpon seen in keratinocytes (Fig. 2) and fibroblasts (data not shown), the level of CMV promoter force-expressed h2-calponin in SM3 cells was not decreased but slightly increased at 12 and 24 h after cell passage (P <0.01).
Fig. 10.
Mechanical tension independent expression of viral promoter-directed h2-calponin transgene. (A) Time course of h2-calponin levels in h2-calponin sense cDNA transfected SM3 cells after passage. The results are summarized from densitometry of multiple Western blots using RAH2 antibody on total protein extracts from two original stable transfected cell lines. In contrast to the post cell roundup decrease of endogenous h2-calpon in keratinocytes and fibroblasts, the level of CMV promoter force-expressed h2-calponin in SM3 cells was not decreased but slightly increased at 12 and 24 h after cell passage (P < 0.01). (B) H2-calponin sense cDNA transfected SM3 cells were cultured on plastic dishes with or without continuously vibration that prevented cell attachment on the dish. Three days old floating cells were further cultured without vibration to form a monolayer. The cells were harvested at each of the states for Western blot analysis using RAH2 antibody to examine the levels of h2-calponin. The sample loading was normalized by their actin contents. Densitometry quantification of the blots shows no decrease but an increase in h2-calponin in the floating cells (*P < 0.05). (C) Transfected SM3 cells cultured for 3 days on plastic dishes or soft polyacrylamide gels were examined by SDS-gel and Western blot using RAH2 antibody for the level of h2-calponin. Quantified by densitometry and normalized by the actin bands on parallel gel, the results showed a higher level of h2-calponin in the soft gel cultures than that of the plastic dish control (*P < 0.001). The results for the cell anchorage and matrix stiffness experiments were each summarized from 4 individual experiments.
Similar to keratinocytes and fibroblasts (Fig. 3), SM3 cells grew in aggregates under vibration and the cell aggregates can reattach to the dish to form monolayer when reseeded in steady culture dish (data not shown). Fig. 10B shows that in contrast to the dramatic decrease in endogenous h2-calponin in floating keratinocytes and fibroblasts (Fig. 3), the level of force-expressed h2-calponin was even higher in the floating cells than that in the attached cells. After re-seeding and cultured without vibration, the transfected SM3 cell aggregates anchored to the plastic dish to form monolayer and the level of h2-calponin returned to the initial steady level. The distinct responses of endogenous gene and viral promoter driven expression of h2-calponin to mechanical tension suggest that mechanical tension regulates h2-calponin gene expression rather than affects cytoskeleton accumulation of the protein.
Consistently, when the transfected SM3 cells were cultured on soft gel matrix, the level of h2-calponin did not decrease (Fig. 10C). Instead, like in the floating culture, there was an increase in h2-calponin level in cultures on the soft gel matrix. These results further support that the mechanical tension-dependent expression of endogenous h2-calponin in keratinocytes and fibroblasts is via gene regulation. On the other hand, the low tension-caused increase in h2-calponin expressed by the presumably mechanical tension-independent CMV promoter may reflect an increased accumulation of calponin protein in the actin filaments due to cytoskeleton reorganization, in agreement with h2-calponin’s proposed function in cytoskeleton remodeling.
DISCUSSION
Abundant h2-calponin in fibroblast and epithelial cells—A previous study using an antibody raised against h1-calponin detected an association of calponin with actin in activated platelets and along stress fibers of both fibroblasts and smooth muscle cells (62). Using h1- and h2-calponin-specific mAbs, we detected only h2-calponin in epidermal keratinocytes and fibroblast (Figs. 1 and 3). Therefore, the previously reported fibroblast calponin might be h2-calponin detected by antibody cross reactions.
An established activity of h1 calponin is the inhibition of smooth muscle actomyosin ATPase (4-6). Structural conservation suggests that h1, h2 and acidic calponins may function via similar molecular mechanisms to regulate actin-myosin interactions. We previously showed that forced expression of h2-calponin in SM3 cells inhibits cytokinesis and the rate of cell proliferation (13). The association of h2-calponin to the actin filament in keratinocytes and fibroblasts (Fig. 7) is consistent with a role in the regulation of actin-based cell motility and other cellular activities (12). Similar to the fact that smooth muscle can contract normally without h1-calponin (36, 63, 64), not all tissues or cultured cells express h2-calponin. Therefore, h2-calponin seems not an essential protein for epithelial cells or fibroblasts to grow and function but may represent a regulatory mechanism that fine tunes the function of actin cytoskeleton.
Mechanical tension regulates h2-calponin gene expression—Living cells can respond to both the magnitude and distribution of adhesion force (65). In the present study, we demonstrated that the expression of h2-calponin in cell culture is determined by cell anchorage to the culture matrix other than cell-cell contacts. The fact that the floating culture cells by continuously shaking do not induce h2-calponin expression (Figs. 3) suggests that the regulation is different from the fluid shear stress-related cell signaling (66). The down-regulation of h2-calponin in soft gel cultures is similar to that in floating keratinocytes. Although the two culture conditions are rather different, a common effect on the cytoskeleton is a lowered mechanical tension in comparison to the monolayer cells anchored on rigid culture matrix. Therefore, the similar down regulation of h2-calponin under these two conditions suggests that mechanical tension is the key signaling mechanism that determines h2-calponin gene expression.
The steady tension in actin stress fibers depends on the motor function of non-muscle myosin II. Inhibition of myosin II ATPase by blebbistatin reduces the mechanical tension of actin cytoskeleton (61). The data that blebbistatin treatment resulted in a decreased level of h2-calponin together with diminished stress fibers in NIH 3T3 cells cultured on plastic dish supports the hypothesis that h2-calponin is regulated by actomyosin based cytoskeletal tension. This novel mechanism is consistent with the potential role of h2-calponin in regulating the function of actin filaments in non-muscle cells similar to h1-calponin’s regulatory effect on acitn-myosin interaction in smooth muscle.
In contrast to the endogenous h2-calponin in keratinocyte and fibroblasts, no post passage transient decrease in level was found for the transfectively expressed h2-calponin in SM3 cells (Fig. 10A). The viral promoter-driven expression of h2-calponin was independent of matrix stiffness (Fig. 10 B and C), indicating a primary control at the level of gene regulation. However, the decrease in h2-calponin level after cell round up during passages is a rapid process (data not shown). Proteolytic degradation may play a critical role in this process. The lack of post round up decrease of h2-calponin in SM3 cells suggest that the proteolytic mechanism is not a ubiquitous mechanism in all cells and the mechanisms remain to be investigated. Although keratinocytes and KD fibroblasts reattach to the culture dish within 30 min after re-plating, 3 days are needed for their h2-calponin level to return to the high level prior to the passage. This slow recovery suggests that rapid degradation may have removed most of the h2-calponin protein and de novo synthesis in response to high mechanical tension is required to restore h2-calponin level in the cell. The lack of rapid degradation of exogenous h2-calponin in SM3 cells indicates that the proteolytic regulation may be cell type specific.
Up-regulation of h2-calponin during epidermal keratinocyte differentiation—Increases in extracellular Ca2+ concentration result in termination of cell growth and induction of terminal differentiation phenotype of keratinocytes in culture (57, 67). An increasing gradient of Ca2+ concentration is present from the basal to the cornified layers of epidermis in the skin (68). The expression of h2-calponin in Ca2+-induced keratinocytes is significantly up-regulated when cells were cultured on soft gel matrix (Fig. 6). Although the keratinocytes grown on plastic dish had high levels of h2-calponin expression which did not further increase upon differentiation, the epidermal keratinocytes in vivo do not reside on such rigid matrix and their anchoring environment is soft tissue to which the soft gel matrix may resemble. Our previous study showed that h2-calponin is expressed at higher levels in growing and remodeling tissues while over-expression of h2-calponin inhibited cytokinesis and resulted in decreases in the rate of cell proliferation (13). The hypothesis is that the more rapidly remodeling cytoskeleton may need a more active h2-calponin regulation. Therefore, the higher level of h2-calponin in the basal layer of skin (Fig. 1) may reflect such regulatory role during epidermal keratinocyte differentiation (67).
Nonetheless, the high level of h2-calponin expression in keratinocytes cultured on rigid plastic dish, which was not changed upon Ca2+ induction, indicates that mechanical properties of extracellular matrix and Ca2+ induction may input into a common signaling pathway to regulate the differentiation and growth of keratinocytes in the skin. The notion that tension-induced expression of h2-calponin may override the Ca2+ based physiological control of keratinocyte differentiation suggests that h2-calponin may play a role in the effects of mechanical tension on normal skin growth and wound healing (69, 70). Further studies on this direction will contribute to our understanding of epidermal keratinocyte biology and diseases.
The role of h2-calponin in the function of actin cytoskeleton—H2-calponin’s association to the actin filaments and its regulation by the mechanical tension of the cytoskeleton indicate functional significance. A previous study observed that transfective expression of GFP-fusion h1-calponin produced a higher resistance to cytochalasin B than that by transfection of GFP-fusion h2-calponin in NIH 3T3 fibroblast cells (12). Based on the assumption that 3T3 cells had no endogenous h2-calponion, untransfected 3T3 control was not included in that study for comparison (12). However, we have found that NIH 3T3 cells express a significant level of h2-calponin (Fig. 9A). In the presence of endogenous h2-calponin, the cells over-expressing GFP-fusion h2-calponin retained most stress fibers after treatment with 2.5 M (1.2 g/ml) cytochalasin B for 15 min (Fig. 5 in ref. 12). This actually reflects a significant resistance and, therefore, the previous results may indicate that both h1 and h2 calponins could confer a resistance to cytochalasin B treatment (Fig. 8). The notion that h1 and h2 calponns may have conserved function is consistent with another previous study that transfective expression of h1-calponin had effects on the proliferation rate of NIH 3T3 cells (71), similar to the results of forced expression of h2-calponin in SM3 cells (13).
Tropomyosin is a major component in the actin cytoskeleton (43) and a protein that binds calponin (1, 20). The co-localization of h2-calponin and tropomyosin in the actin stress fibers (Fig. 7) and the positive correlation between the levels of force-expressed h2-calponin and endogenous tropomyosin in SM3 cells (Fig. 8C) suggest a functional relationship. Tropomyosin has been shown to play a role in actin filament stability (42). Therefore, the effect of h2-calponin in stabilizing actin stress fibers as shown by the resistance to cytochalasin B treatment (Fig. 8A) may be through enhancing the function of Tm. Like the effect of C-terminal fragment of caldesmon on actin filament stability (59), the relationship between h2-calponin and Tm in the structure and function of non-muscle cytoskeleton deserves further investigation. The finding that mechanical tension regulates the expression of h2-calponin with potential functional significance lays a foundation for understanding the physiological function of calponin.
Acknowledgements
We thank Dr. Paul A. Janmey, University of Pennsylvania, for advices on the polyacrylamide gel culture matrix and Dr. Anita C. Gilliam, Case Western Reserve University, for providing human skin paraffin sections.
Footnotes
This study was supported by grants from the National Institutes of Health to J-PJ (AR039750-130037, AR048816 and HD044824), RLE (AR046494 and AR045357) and JJ-CL (HD018577). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- BCIP
- 5-bromo-4-chloro-3-indolyl phosphate
- BSA
- bovine serum albumin
- CMV
- cytomegalovirus
- DMEM
- Dulbecco’s modified Eagle medium
- D-PBS
- Dulbecco’s phosphate buffered saline
- ELISA
- enzyme-linked immunosorbant assay
- FBS
- fetal bovine serum
- mAb
- monoclonal antibody
- NBT
- nitroblue tetrazolium
- PAGE
- polyacrylamide gel electrophoresis
- pI
- isoelectric point
- TBS
- Tris-buffered saline
- TRITC
- Tetramethylrhodamine isothiocyanate.
REFERENCES
- 1.Takahashi K, Hiwada K, Kokubu T. Biochem. Biophys. Res. Commun. 1986;141:20–26. doi: 10.1016/s0006-291x(86)80328-x. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Nadal-Ginard B. J. Biol. Chem. 1991;266:13284–13288. [PubMed] [Google Scholar]
- 3.Strasser P, Gimona M, Moessler H, Herzog M, Small JV. FEBS. Lett. 1993;330:13–18. doi: 10.1016/0014-5793(93)80909-e. [DOI] [PubMed] [Google Scholar]
- 4.Winder SJ, Allen BG, Clement-Chomienne O, Walsh MP. Acta. Physiol. Scand. 1998;164:415–426. doi: 10.1111/j.1365-201x.1998.tb10697.x. [DOI] [PubMed] [Google Scholar]
- 5.Small JV, Gimona M. Acta. Physiol. Scand. 1998;164:341–348. doi: 10.1046/j.1365-201X.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- 6.Morgan KG, Gangopadhyay SS. J. Appl. Physiol. 2001;91:953–962. doi: 10.1152/jappl.2001.91.2.953. [DOI] [PubMed] [Google Scholar]
- 7.Ferhat L, Charton G, Represa A, Ben-Ari Y, der Terossian E, Khrestchatski M. Eur. J. Neurosci. 1996;8:1501–1509. doi: 10.1111/j.1460-9568.1996.tb01612.x. [DOI] [PubMed] [Google Scholar]
- 8.Ferhat L, Rami G, Medina I, Ben-Ari Y, Represa A. J. Cell Sci. 2001;114:3899–3904. doi: 10.1242/jcs.114.21.3899. [DOI] [PubMed] [Google Scholar]
- 9.Fukui Y, Masuda H, Takagi M, Takahashi K, Kiyokane K. J. Dermatol. Sci. 1997;14:29–36. doi: 10.1016/s0923-1811(96)00545-2. [DOI] [PubMed] [Google Scholar]
- 10.Sakihara C, Nishimura J, Kobayashi S, Takahashi S, Kanaide H. Biochem. Biophys. Res. Commun. 1996;222:195–200. doi: 10.1006/bbrc.1996.0721. [DOI] [PubMed] [Google Scholar]
- 11.Masuda H, Tanaka K, Takagi M, Ohgami K, Sakamaki T, Shibata N, Takahashi K. J. Biochem. (Tokyo) 1996;120:415–424. doi: 10.1093/oxfordjournals.jbchem.a021428. [DOI] [PubMed] [Google Scholar]
- 12.Danninger C, Gimona M. J. Cell Sci. 2000;113:3725–3736. doi: 10.1242/jcs.113.21.3725. [DOI] [PubMed] [Google Scholar]
- 13.Hossain MM, Hwang D-Y, Huang Q-Q, Sasaki Y, Jin J-P. Am. J. Physiol. 2003;284:156–167. doi: 10.1152/ajpcell.00233.2002. [DOI] [PubMed] [Google Scholar]
- 14.Han EKH, Guadagno TM, Dalton SL, Assoian RK. J. Cell Biol. 1993;122:461–471. doi: 10.1083/jcb.122.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Science. 2004;305:1782–1786. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- 16.Filipe JA, Nunes JP. Auton. Autacoid. Pharmacol. 2002;22:155–159. doi: 10.1046/j.1474-8673.2002.00255.x. [DOI] [PubMed] [Google Scholar]
- 17.Smillie LB. Tropomyosin. In: Barany M, editor. Biochemistry of smooth muscle contraction. Academic Press; NY: 1996. pp. 63–75. [Google Scholar]
- 18.Broschat KO, Weber A, Burgess DR. Biochemistry. 1989;28:8501–8506. doi: 10.1021/bi00447a035. [DOI] [PubMed] [Google Scholar]
- 19.Broschat KO. J. Biol. Chem. 1990;265:21323–21329. [PubMed] [Google Scholar]
- 20.Nakamura F, Mino T, Yamamoto J, Naka M, Tanaka T. J. Biol. Chem. 1993;268:6194–6201. [PubMed] [Google Scholar]
- 21.Wang N, Butler JP, Ingber DE. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 22.Ingber DE. J. Cell Sci. 1993;104:613–627. doi: 10.1242/jcs.104.3.613. [DOI] [PubMed] [Google Scholar]
- 23.Girard PR, Nerem RM. J. Cell Phys. 1995;163:179–193. doi: 10.1002/jcp.1041630121. [DOI] [PubMed] [Google Scholar]
- 24.Davies PF. Physiol. Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choquet D, Felsenfeld DP, Sheetz MP. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 26.Harris A. Exp. Cell Res. 1973;77:285–297. doi: 10.1016/0014-4827(73)90579-x. [DOI] [PubMed] [Google Scholar]
- 27.Folkman J, Moscona A. Nature (London) 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- 28.Ingber DE, Folkman J. J. Cell Biol. 1989;109:317–330. doi: 10.1083/jcb.109.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo CM, Wang HB, Dembo M, Wang YL. Biophys. J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelham RJ, Wang Y-L. Proc. Natl. Acad. Sci. USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beningo KA, Wang YL. J. Cell Sci. 2002;115:849–856. doi: 10.1242/jcs.115.4.849. [DOI] [PubMed] [Google Scholar]
- 32.Cukierman E, Pankov R, Stevens DR, Yamada KM. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 33.Deroanne CF, Lapiere CM, Nusgens BV. Cardiovasc. Res. 2001;49:647–658. doi: 10.1016/s0008-6363(00)00233-9. [DOI] [PubMed] [Google Scholar]
- 34.Engler A, Bacakova L, Newman C, Hategan A, Griffn M, Discher D. Biophys. J. 2004;86:617–628. doi: 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 36.Nigam R, Triggle CR, Jin J-P. J. Muscle Res. Cell. Motil. 1998;19:695–703. doi: 10.1023/a:1005389300151. [DOI] [PubMed] [Google Scholar]
- 37.Jin J-P, Wu D, Gao J, Nigam R, Kwong S. Protein Exp. Purif. 2003;31:231–239. doi: 10.1016/s1046-5928(03)00185-2. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Jin J-P. Biochemistry. 1998;37:14519–14528. doi: 10.1021/bi9812322. [DOI] [PubMed] [Google Scholar]
- 39.Jin J-P, Walsh MP, Resek ME, McMartin GA. Biochem. Cell Biol. 1996;74:187–196. doi: 10.1139/o96-019. [DOI] [PubMed] [Google Scholar]
- 40.Lin JJ, Hegmann TE, Lin JL. J. Cell Biol. 1988;107:563–572. doi: 10.1083/jcb.107.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novy RE, Sellers JR, Liu LF, Lin JJ. Cell Motil. Cytoskeleton. 1993;26:248–261. doi: 10.1002/cm.970260308. [DOI] [PubMed] [Google Scholar]
- 42.Warren KS, Lin JLC, McDermontt JP, Lin JJC. J. Cell Biol. 1995;129:697–708. doi: 10.1083/jcb.129.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin JJ, Warren KS, Wamboldt DD, Wang T, Lin JL. Int. Rev. Cytol. 1997;170:1–38. doi: 10.1016/s0074-7696(08)61619-8. [DOI] [PubMed] [Google Scholar]
- 44.Robinson NA, LaCelle PT, Eckert RL. J. Invest. Dermatol. 1996;107:101–107. doi: 10.1111/1523-1747.ep12298323. [DOI] [PubMed] [Google Scholar]
- 45.LaCelle PT, Lambert A, Ekambaram MC, Robinson NA, Eckert RL. Skin Pharmacol. Appl. Skin Physiol. 1998;11:214–226. doi: 10.1159/000029830. [DOI] [PubMed] [Google Scholar]
- 46.Jin J-P, Brotto MA, Hossain MM, Huang Q-Q, Brotto LS, Nosek TM, Morton DH, Crawford TO. J. Biol. Chem. 2003;278:26159–26165. doi: 10.1074/jbc.M303469200. [DOI] [PubMed] [Google Scholar]
- 47.Welter JF, Crish JF, Agarwal C, Richard L, Eckert RL. J. Biol. Chem. 1995;270:12614–12622. doi: 10.1074/jbc.270.21.12614. [DOI] [PubMed] [Google Scholar]
- 48.Efimova T, LaCelle P, Welter JF, Eckert RL. J. Biol. Chem. 1998;273:24387–24395. doi: 10.1074/jbc.273.38.24387. [DOI] [PubMed] [Google Scholar]
- 49.Efimova T, Broome AM, Eckert RL. Mol. Cell Biol. 2004;24:8167–8183. doi: 10.1128/MCB.24.18.8167-8183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sasaki Y, Uchida T, Sasaki Y. J. Biochem. 1989;106:1009–1018. doi: 10.1093/oxfordjournals.jbchem.a122956. [DOI] [PubMed] [Google Scholar]
- 51.Furukawa KS, Ushida T, Sakai Y, Suzuki M, Tanaka J, Tateishi T. Cell Transplant. 2001;10:441–445. [PubMed] [Google Scholar]
- 52.Pelham RJ, Wang Y-L. Biol. Bull. 1998;194:348–350. doi: 10.2307/1543109. [DOI] [PubMed] [Google Scholar]
- 53.Rice RH, Green H. Cell. 1979;18:681–694. doi: 10.1016/0092-8674(79)90123-5. [DOI] [PubMed] [Google Scholar]
- 54.Deucher A, Efimova T, Eckert RL. J. Biol. Chem. 2002;277:17032–17040. doi: 10.1074/jbc.M109076200. [DOI] [PubMed] [Google Scholar]
- 55.Eckert RL, Crish JF, Efimova T, Dashti SR, Deucher A, Bone F, Adhikary G, Huang G, Gopalakrishnan R, Balasubramanian S. J. Invest. Dermatol. 2004;123:13–22. doi: 10.1111/j.0022-202X.2004.22723.x. [DOI] [PubMed] [Google Scholar]
- 56.Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- 57.Eckert RL, Crish JF, Robinson NA. Physiol. Rev. 1997;77:397–424. doi: 10.1152/physrev.1997.77.2.397. [DOI] [PubMed] [Google Scholar]
- 58.Cooper JA. J. cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Warren KS, Lin JL, Wamboldt DD, Lin JJ. J. Cell Biol. 1994;125:359–368. doi: 10.1083/jcb.125.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kovacs M, Toth J, Hetenyi C, Malnasi-Csizmadia A, Sellers JR. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 61.Griffin MA, Sen S, Sweeney HL, Discher DE. J. Cell Sci. 2004;117:5855–5863. doi: 10.1242/jcs.01496. [DOI] [PubMed] [Google Scholar]
- 62.Takeuchi K, Takahashi K, Abe M, Nishida W, Hiwada K, Nabeya T, Maruyama K. J. Biochem. (Tokyo) 1991;109:311–316. [PubMed] [Google Scholar]
- 63.Matthew JD, Khromov AS, McDuffie MJ, Somlyo AV, Somlyo AP, Taniguchi S, Takahashi K. J. Physiol. 2000;529:811–824. doi: 10.1111/j.1469-7793.2000.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takahashi K, Yoshimoto R, Fuchibe K, Fujishige A, Mitsui-Saito M, Hori M, Ozaki H, Yamamura H, Awata N, Taniguchi S, Katsuki M, Tsuchiya T, Karaki H. Biochem. Biophys. Res. Commun. 2000;279:150–157. doi: 10.1006/bbrc.2000.3909. [DOI] [PubMed] [Google Scholar]
- 65.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 66.Shyy JY, Chien S. Circ Res. 2002;91:769–775. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 67.Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Cell. 1980;19:245–254. doi: 10.1016/0092-8674(80)90406-7. [DOI] [PubMed] [Google Scholar]
- 68.Menon GK, Elias PM, Lee SH, Feingold KR. Cell Tissue Res. 1992;270:503–512. doi: 10.1007/BF00645052. [DOI] [PubMed] [Google Scholar]
- 69.Pickett BP, Burgess LP, Livermore GH, Tzikas TL, Vossoughi J. Arch. Otolaryngol. Head Neck. 1996;122:565–568. doi: 10.1001/archotol.1996.01890170097017. [DOI] [PubMed] [Google Scholar]
- 70.Burgess LP, Morin GV, Rand M, Vossoughi J, Hollinger JO. Arch. Otolaryngol. Head Neck Surg. 1990;116:798–802. doi: 10.1001/archotol.1990.01870070046008. [DOI] [PubMed] [Google Scholar]
- 71.Jiang Z, Grange RW, Walsh MP, Kamm KE. FEBS. Lett. 1997;413:441–445. doi: 10.1016/s0014-5793(97)00944-7. [DOI] [PubMed] [Google Scholar]