

Abstract
Levodopa reportedly inhibits insulin action in skeletal muscle. Here we show that C2C12 myotubes produce levodopa and that insulin-stimulated glucose transport is enhanced when endogenous levodopa is depleted. Exogenous levodopa prevented the stimulation of glucose transport by insulin (P < 0.05) and increased cAMP concentrations (P < 0.05). The decrease in insulin-stimulated glucose transport caused by levodopa was attenuated by propranolol (a β-adrenergic antagonist) and prevented by NSD-1015 (NSD), an inhibitor of DOPA decarboxylase (DDC; converts levodopa to dopamine). Propranolol and NSD both prevented levodopa-related increases in [cAMP]. However, the effects of levodopa were unlikely to be dependent on the conversion of levodopa to catecholamines because we could detect neither DDC in myotubes nor catecholamines in media after incubation of myotubes with levodopa. The data suggest the possibility of novel autocrine β-adrenergic action in C2C12 myotubes in which levodopa, produced by myotubes, could have hormone-like effects that impinge on glucose metabolism.
Keywords: excitatory amino acid, DOPA, GLUT4, myotubes, tyrosine hydroxylase
Introduction
It has been reported that levodopa causes hyperglycemia in laboratory animals and humans (1-4). Furthermore, it has recently been shown that levodopa decreases insulin-stimulated glucose transport, glycogen accumulation, and glycogen synthase activity in skeletal muscle (5). These data suggest that levodopa, or one of its metabolites, could contribute to skeletal muscle insulin resistance. Consistent with this idea, it has been demonstrated that dopamine, for which levodopa is a precursor, stimulates a β-adrenergic–dependent increase in cAMP in muscle cells (6). In addition, β-adrenergic effects of levodopa that are independent of catecholamine synthesis have been described for neural cells (7-9), although there are currently no data available for muscle cells in this regard. β-adrenergic action has previously been shown to attenuate insulin effects (e.g., stimulation of glucose transport and/or glycogen synthase activity) in skeletal muscle (5, 10-12). Because skeletal muscle is responsible for the majority of plasma glucose that is absorbed in response to insulin (13, 14), muscle is an important tissue for the study of factors that contribute to decreased and/or increased insulin action.
We hypothesized that insulin action would be improved after the depletion of endogenous levodopa stores. We also treated cells with exogenous levodopa to examine candidate mechanisms for the effects of levodopa on insulin action. We hypothesized that endogenous levodopa would inhibit insulin-stimulated glucose transport in a manner prevented both by a β-adrenergic antagonist and an inhibitor of DOPA decarboxylase (DDC), which catalyzes the conversion of levodopa to dopamine.
Materials and Methods
Materials. Purified porcine insulin was obtained from Eli Lilly & Co. (Indianapolis, IN) and 3H-labeled 2-deoxyglucose (DG) from American Radiolabeled Chemicals (St. Louis, MO). Cytochalasin B, levodopa, propranolol, NSD-1015 (NSD), α-methyl-L-tyrosine (L-AMPT), 2DG, tyrosinase, and Besthorn's hydrazone were obtained from Sigma Chemical Co. (St. Louis, MO).
Cell Culture. Mouse C2C12 myoblasts were purchased from the American Type Culture Collection (Rockville, MD) and cultured and differentiated as previously described (15, 16). Despite some reports that C2C12 myotubes are unresponsive to insulin (17), in our hands insulin stimulates GLUT4-dependent glucose transport in C2C12 myotubes (16). To verify that the myotubes contained GLUT4, we performed Western blots for GLUT4 with myoblasts (grown in media containing 10% fetal bovine serum), myotubes differentiated for 3 days in medium containing 2% horse serum (HS) with 100 nM insulin and then an additional day with 2% HS without insulin, or myotubes differentiated for 4 days with medium containing just 2% HS. All subsequent work was performed after differentiation of myotubes with medium containing 2% HS and insulin, as previously described (16).
Effects of L-AMPT on Levodopa Content of Cells and Insulin Action. Myotubes were incubated for 3 hrs in minimal essential medium (MEM) containing 2% horse serum in the absence or presence of 30 μM L-AMPT, an inhibitor of tyrosine hydroxylase (TH), an enzyme that catalyzes the conversion of tyrosine to levodopa. After rinsing in Hepes-buffered saline (HBS; described in the next section), cells were scraped in ice-cold 0.3 M perchloric acid and gently homogenized in Kontes ground-glass tubes. A portion of each homogenate was stored at −80°C for bicinchoninic acid (BCA) protein assays. Homogenates were centrifuged for 10 mins at 14,000 g at 4°C, known volumes of the supernatants were neutralized with KOH, the neutralized samples were centrifuged again, and levodopa concentrations were assayed with a sensitive spectrophotometric method based on a previously described TH assay (18). The assay relies on the conversion of levodopa to dopaquinone by tyrosinase and subsequent reaction with Besthorn's hydrazone to form a pink-colored product with maximal absorbance at 505 nm. With this assay in our hands, levodopa yields a product with an extinction coefficient of ∼33,000·M−1·cm−1. L-AMPT does not interfere with the assay.
To determine whether the prevention of endogenous levodopa production would be associated with increased insulin action, myotubes were incubated for 3 hrs in the absence or presence of 30 μM L-AMPT as previously described. Following the 3-hr incubation, myotubes were incubated for 20 mins in the absence or presence of 10 nM insulin (with L-AMPT present if it had been present in the previous step) before glucose transport assays (as described next). In a separate experiment, glucose transport was assayed in the absence or presence of 10 nM, 50 nM, and 100 nM of insulin. The lowest concentration of insulin for this experiment, 10 nM, has previously been shown for these cells to be an insulin concentration that is near the threshold for stimulation of glucose transport (16) and, thus, is an appropriate insulin concentration for determining whether L-AMPT potentiates insulin action. Myotubes were not serum starved for this experiment because serum starvation itself increases insulin sensitivity (19) and could potentially confound the detection of changes in insulin sensitivity caused by the experimental treatment.
Levodopa Effects on Insulin-Stimulated Glucose Transport. For the following experiments in which inhibition of insulin action was expected (as opposed to the expectation of increased insulin effects in the previous experiment), myotubes were serum starved 3 hrs before the assay of 2DG uptake, as previously described (15, 16). To determine the effects of levodopa on insulin action and whether the effects of levodopa were preventable with propranolol, myotubes were first incubated for 30 mins in HBS solution (20 mM Hepes, pH 7.4; 140 mM NaCl; 5 mM KCl; 2.5 mM MgSO4; 1 mM CaCl2) containing 5 mM glucose in the absence or presence of 10 μM propranolol. Next, myotubes were incubated in the glucose-containing HBS in the absence or presence of 15 μM levodopa and with propranolol, if it had been present in the previous step. Then, the cells were incubated for 20 mins in HBS containing 5 mM glucose in the absence or presence of 100 nM insulin and in the presence of levodopa and/or propranolol (if they had been present in the previous step). Thus, there were five treatment groups: control, insulin, levodopa + insulin, levodopa + insulin + propranolol, and propranolol. Cells were rinsed twice with glucose-free HBS and incubated at room temperature for 10 mins in 200 μl of glucose-free HBS containing 3H-labeled 2DG (3 μCi/ml); 10 μM 2DG; and insulin, levodopa, or propranolol, if they had been present for the previous step. Nonspecific 2DG uptake was determined by quantitation of cell-associated radioactivity in the presence of 10 μM cytochalasin B. After transport incubations, cells were rapidly rinsed three times with ice-cold 0.9% saline and lysed in 0.2 N NaOH containing 0.2% sodium dodecyl sulfate. Protein content of lysates was determined by the BCA assay (Pierce, Rockford, IL), and samples were neutralized before scintillation counting.
Propranolol experiments were repeated with a lower concentration of propranolol (100 nM). In some experiments that followed the same design as the previously described propranolol experiments, cells were incubated in the absence or presence of levodopa, the absence or presence of insulin, and the absence or presence of 250 μM NSD, a DDC inhibitor, to assess whether or not NSD would prevent the levodopa-mediated inhibition of insulin-stimulated glucose transport. This yielded five treatment groups: control, insulin, levodopa + insulin, levodopa + insulin + NSD, and NSD. Glucose transport assays were performed as previously described.
cAMP Concentrations. Myotubes were incubated for 30 mins in HBS containing 5 mM glucose (control) or with the addition of 15 μM levodopa, levodopa with 10 μM propranolol, levodopa with 250 μM NSD, propranolol, or NSD. Cells were rinsed with ice-cold HBS before scraping in ice-cold 0.5 M perchloric acid (20). Cell samples were homogenized, assayed for protein content, and neutralized as previously described for levodopa assay. Neutralized supernatants were assayed for cAMP with a cycling fluorometric method (5, 20).
Western Blot Analyses. Myotubes were rinsed with HBS and scraped in ice-cold HES buffer containing 20 mM Hepes (pH 7.4), 250 mM sucrose, and 1 mM EDTA. Western blots for GLUT4 were performed as previously described (21), using an antibody generously provided by Dr. Mike Mueckler (Washington University School of Medicine, St. Louis, MO). A BALB/c mouse was killed with an overdose of pentobarbital, after which its kidneys were removed and clamp frozen with tongs cooled in liquid nitrogen and stored at −80°C. The animal procedure was approved by the Saint Louis University Animal Care Committee. For TH and DDC Western blots, myotubes and kidney samples in HES buffer were homogenized in Kontes ground-glass tubes and centrifuged for 10 mins at 14,000 g. After determination of protein concentration using a BCA assay, samples were subjected to Western blot and enhanced chemiluminescence detection procedures (5) using antibodies against TH (described next) or DDC (obtained from Chemicon International, Temecula, CA). The TH antibody was developed by Nicole Le Douarin and Catherine Ziller and was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development of the National Institutes of Health and maintained by The University of Iowa, Department of Biological Sciences (Iowa City, IA). TH and DDC are both expressed in kidney (22, 23), so this tissue served as a positive control for the assays. Peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibodies were obtained from Pierce Biotechnology, Inc. (Rockford, IL).
To assess the potential effects of levodopa on insulin signaling, we performed Western blots for phosphorylated Akt for myotubes incubated for 30 mins in the absence or presence of 15 μM levodopa before a 20-min incubation in the absence or presence of 100 nM insulin with the continued presence of levodopa, if it had been present at the first step. These myotubes were scraped and homogenized in buffer-containing phosphatase and protease inhibitors (50 mM Hepes [pH 7.4], 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EDTA, 10 mM sodium pyrophosphate, 100 mM NaF, 2 mM Na3VO4, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 0.5 μg/ml pepstatin, and 2 mM PMSF; Ref. 24) and centrifuged for 10 mins at 14,000 g before the resulting supernatants were subjected to Western blotting. Antibodies against Akt and Akt phosphorylated on T308 were obtained from Cell Signaling Technology (Beverly, MA).
Voltametric Detection of Catecholamines. Myotubes in six-well plates were rinsed with HBS, provided with fresh serum-free αMEM, and incubated for 3 hrs in the absence or presence of 15 μM levodopa. After the 3-hr incubation, the cell media were removed and immediately assayed for the presence of catecholamines by cyclic voltametry, a technique that has previously been used by a member of this group to assay dopamine in vivo on a subsecond time scale (25). Dopamine standards were prepared in αMEM and were identified by previously established electrophysiologic criteria (25). The working, reference, and counter electrodes were equilibrated in the samples, controls, and standards before testing. The MEM cell medium acted as the electrolyte for the system. Data were collected in three-electrode mode. The system was probed at a scan rate of 0.1 V/sec. CH Instruments (Austin, TX) software was used to acquire and analyze the voltametric data from a CH700A potentiostat (CH Instruments).
Statistical Analysis. A one-way ANOVA among experimental groups was performed in each study. Post-hoc comparisons were performed with Fisher's protected least significance difference tests. A level of P < 0.05 was set for significance for all tests, and all values are expressed as means ± SE. Statistics were performed with SPSS software (version 10.0; Chicago, IL).
Results
GLUT4 Expression. Under the differentiation conditions of the current study (media containing 2% HS and 100 nM insulin), GLUT4 content per mg of myotube protein was ∼60% greater than GLUT4 content in myoblasts (P < 0.05) and more than 2-fold greater than GLUT4 content of myotubes differentiated with HS, but not insulin (P < 0.005; Fig 1). The differentiation medium containing 2% HS and 100 nM insulin was used to obtain all data (shown next) to optimize the expression of GLUT4 (the insulin-responsive glucose transporter) on differentiation.
Figure 1.
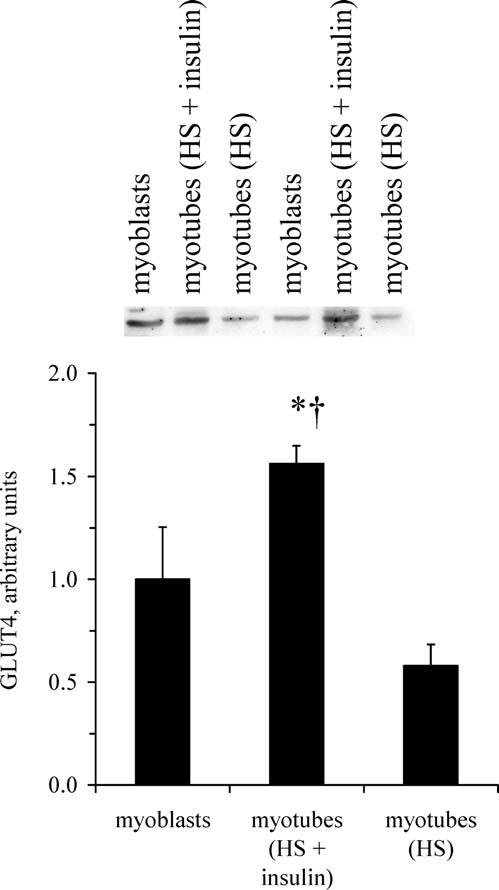
GLUT4 content of C2C12 myotubes. Under the differentiation conditions for this study (media containing 2% HS and 100 nM insulin), C2C12 myotubes contain GLUT4, the insulin-responsive glucose transporter. Values are means with SEs (n = 4 per group). *Greater GLUT4 content than myoblasts (P < 0.05); †greater GLUT4 content than myotubes differentiated with HS, but without insulin (P < 0.005).
L-AMPT Depletes Endogenous Levodopa Concomitant With Improved Insulin Action. Myotubes incubated with the TH inhibitor L-AMPT for 3 hrs contained ∼90% less levodopa than control myotubes (P < 0.05; Fig. 2A). The levodopa contents of control myotubes were within ranges previously reported for levodopa-producing cells in culture (26). Pretreatment of myotubes with L-AMPT had a permissive effect on the stimulation of glucose transport insulin, allowing about a 65% increase in glucose transport above baseline (Fig. 2B). The insulin concentration (10 nM) used to obtain the data in Fig. 2B was deliberately chosen to be near the threshold for insulin to stimulate glucose transport in these cells (16) and, therefore, enhance the ability to detect increased insulin action. Treatment with L-AMPT increases both insulin sensitivity and insulin responsiveness (Fig. 2C). As shown in Figure 2C, 50 nM and 100 nM insulin both stimulate an increase in glucose transport above the basal level (P < 0.05 and P < 0.005, respectively), although 10 nM insulin did not stimulate glucose transport. L-AMPT had no effect on glucose transport in the absence of insulin. For myotubes incubated with 10 nM, 50 nM, and 100 nM insulin, glucose transport values for myotubes treated with L-AMPT were greater (P < 0.001) than the corresponding values for cells not exposed to L-AMPT. For myotubes treated with L-AMPT, each increment in insulin concentration produced a corresponding increase (P < 0.05) in glucose transport. The increase in both insulin sensitivity and insulin responsiveness caused by L-AMPT is reminiscent of the effects of lithium, which increase sensitivity and the responsiveness of glucose transport to insulin and other stimuli (27). In contrast, contraction of muscle (i.e., exercise) stimulates sensitivity to insulin but does not increase the maximal glucose response to insulin (28).
Figure 2.
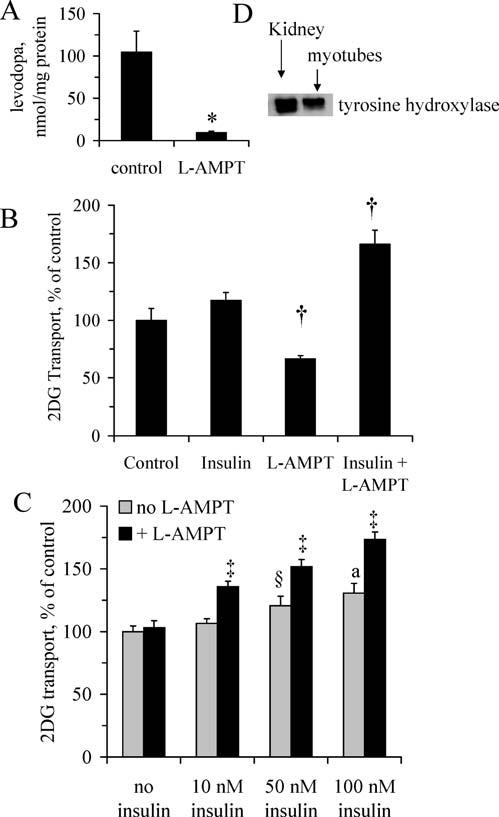
Depletion of endogenous levodopa enhances insulin-stimulated glucose transport. Myotubes were incubated in the absence or presence of L-AMPT, an inhibitor of levodopa synthesis, for 3 hrs before (A) levodopa assays; (B) assessment of glucose transport rates in the absence or presence of 10 nM insulin (n=5 per group); or (C) glucose transport assays in the absence or presence of 10 nM, 50 nM, or 100 nM insulin (n = 7–12 per group). (D) Western blot confirms the presence of TH, which catalyzes the conversion of tyrosine to levodopa and is inhibited by L-AMPT. Kidney samples were included as a positive control for TH expression. * Significant difference (P < 0.05); †significantly different from means for control and insulin groups (P < 0.05); ‡significantly greater than corresponding control group not treated with L-AMPT (P < 0.001), and greater than groups with L-AMPT but with a lesser concentration of insulin (P < 0.05); § ,a greater than control without insulin (§P < 0.05;aP < 0.005). Values are means ± SE.
As shown in Figure 2D, myotubes contain TH, the enzyme that catalyzes the conversion of tyrosine to levodopa and is inhibited by L-AMPT. These data appear to be the first to demonstrate levodopa production in muscle and, together with the glucose transport data (Fig. 2B and C), they suggest a role for endogenous levodopa in attenuation of insulin-stimulated glucose transport.
Levodopa Inhibits Insulin Action. Figure 3 illustrates that levodopa prevents the increase in glucose transport stimulated by insulin (P < 0.05). Insulin action in the presence of levodopa was restored by propranolol. These data suggest that the inhibition of insulin action by levodopa is at least in part a β-adrenergic effect. Insulin action in the presence of levodopa was completely restored by inclusion of NSD in the medium (Fig. 4), suggesting a role for an NSD-inhibited enzyme in mediation of the effects of levodopa on insulin-stimulated glucose transport.
Figure 3.
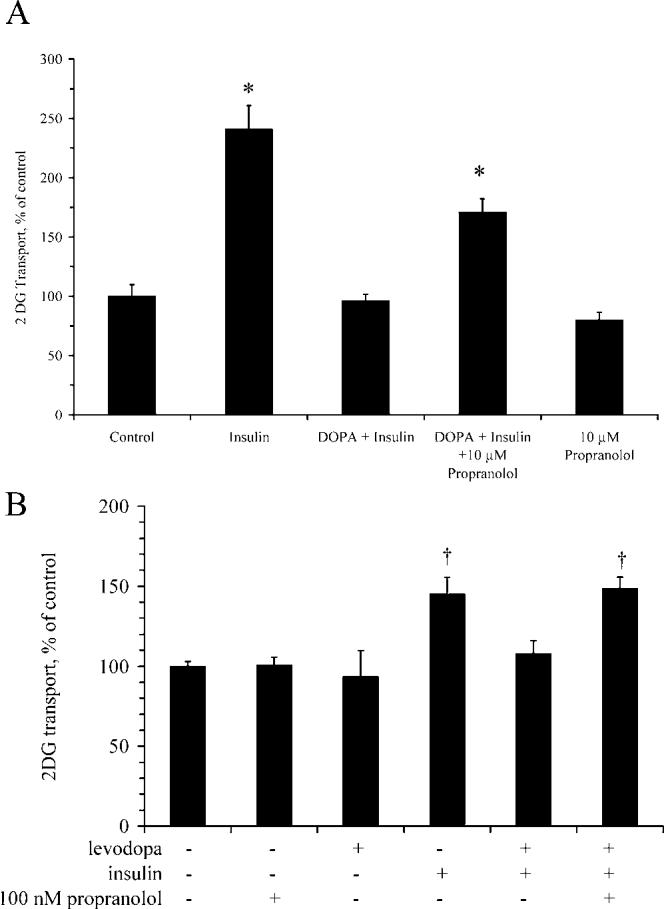
Levodopa inhibits insulin-stimulated glucose transport in a propranolol-sensitive manner. Myotubes were assayed for 2DG transport in the presence or absence of 15 μM levodopa, propranolol, and 100 nM insulin, as described in the Materials and Methods section. Propranolol is a β-adrenergic antagonist. (A) Transport experiments with 10 μM propranolol. (B) Transport experiments with 100 nM propranolol. * Significantly different from control, levodopa + insulin, and propranolol (P < 0.001; n = 6 per group); †significantly different from all other groups not marked with †(P < 0.005; n = 3–6 per group). Bars denote means ± SE.
Figure 4.
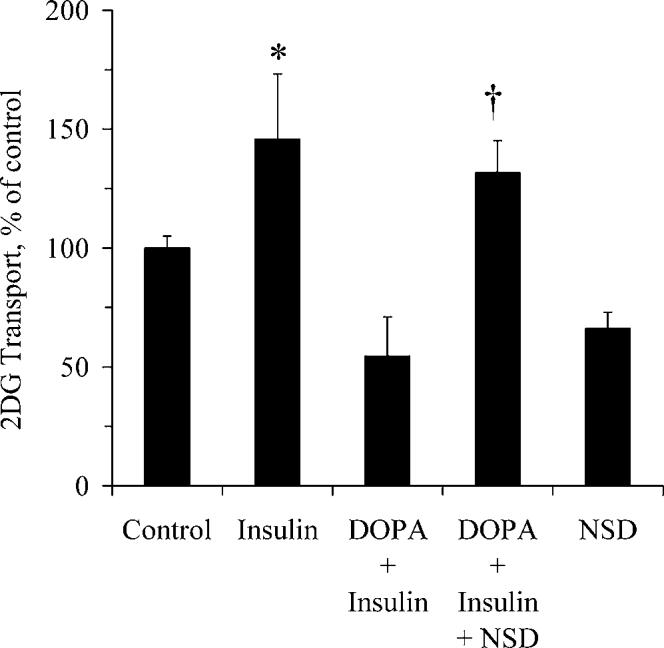
The decrease in insulin-stimulated glucose transport caused by levodopa is prevented by NSD-1015. Myotubes were assayed for glucose transport in the presence or absence of 15 μM levodopa, 250 μM NSD-1015, and 100 nM insulin, as described in the Materials and Methods section. NSD is an inhibitor of DDC, the enzyme that converts levodopa to dopamine, and monoamine oxidase, the enzyme that catalyzes the conversion of levodopa to dopaquinone. *Significantly different from control, levodopa + insulin, and NSD; †significantly different from levodopa + insulin and from NSD (P < 0.05; n = 6 per group). Bars denote means ± SE.
NSD and Propranolol Prevent the Increase in cAMP Concentrations Caused by Levodopa. Figure 5 shows that myotube cAMP concentrations were increased more than 2-fold above baseline in the presence of levodopa (P < 0.001). Propranolol completely abolished the increase in cAMP concentrations brought about by levodopa (P < 0.001), suggesting that the levodopa-stimulated increase in [cAMP] was a β-adrenergic effect. Prevention of the levodopa-related increase in [cAMP] by NSD suggests that an NSD-inhibited enzyme mediates the effects of levodopa on [cAMP].
Figure 5.
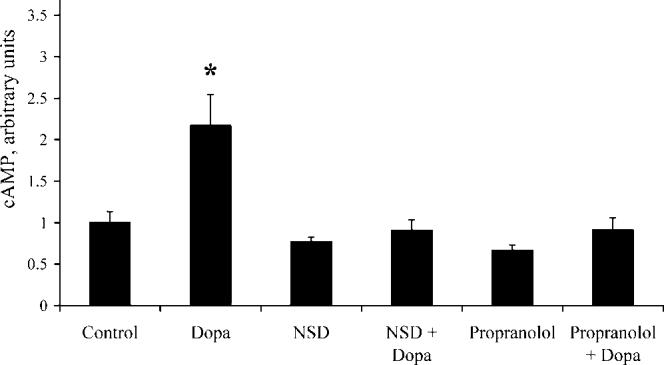
Levodopa stimulates increased cAMP concentrations. cAMP content of myotubes was assayed after incubation in the absence or presence of 15 μM levodopa, 10 μM propranolol, and 250 μM NSD, as described in the Materials and Methods section. * Significantly different from all groups (P < 0.001; n = 5 per group). Bars denote means ± SE.
Effects of Levodopa Are Catecholamine Independent. As previously described, NSD prevented both the levodopa-related inhibition of insulin action (Fig. 4) and the levodopa-stimulated increase in [cAMP] (Fig. 5). NSD is an inhibitor of DDC, the enzyme that catalyzes the conversion of levodopa to dopamine. However, we detected neither catecholamines in medium that had been incubated with cells (either in the absence or presence of levodopa) nor DDC protein in the myotubes (Fig. 6), so we do not believe that DDC mediates the effects of levodopa on insulin action or cAMP concentrations. NSD also inhibits monoamine oxidase (29, 30), an enzyme that catalyzes the conversion of levodopa to dopaquinone, suggesting that dopaquinone or one of its metabolites mediates the catecholamine-independent effects of levodopa on insulin action and cAMP concentrations.
Figure 6.

C2C12 myotubes do not contain DDC. Mouse kidney served as a positive control for Western blot analysis of DDC content of myotubes. DDC catalyzes the conversion of levodopa to dopamine.
Effects of Levodopa on Insulin Signaling. As shown in Figure 7, levodopa did not alter the insulin-stimulated phosphorylation of Akt. Thus, levodopa appears to act in a fashion similar to caffeine, which increases [cAMP] and inhibits insulin-stimulated glucose transport without blunting the activation of Akt by insulin (31).
Figure 7.
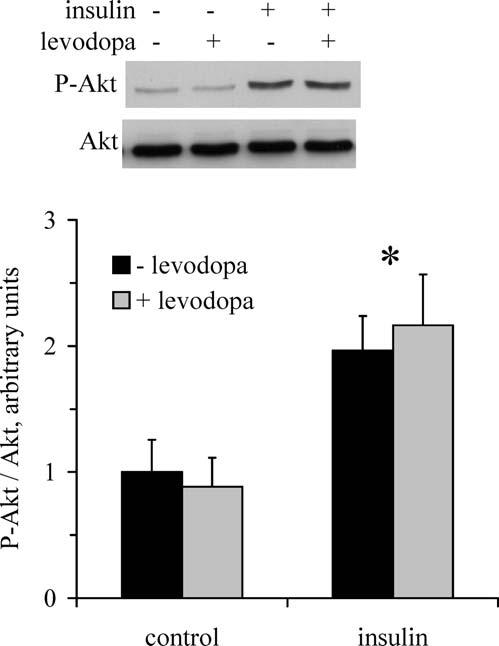
Effects of levodopa on insulin-stimulated Akt phosphorylation. Western blots were performed for myotubes incubated in the absence and presence of levodopa and the absence and presence of insulin to assess T308 phosphorylation of Akt. * Main effect of insulin (P < 0.05; n = 3 per group).
Discussion
The new information provided by this study is that endogenous or exogenous levodopa (or a noncatecholamine levodopa metabolite) appears to have hormone-like actions in muscle cells, impinging on glucose metabolism through a β-adrenergic mechanism. Inhibition of insulin action by levodopa has previously been reported for skeletal muscle (5), which contains peripheral nerves (potential sites for levodopa-related catecholamine synthesis) in addition to muscle fibers. The current data extends previous findings by demonstrating the possibility of direct DOPAergic action in C2C12 myotubes, independent of catecholamine synthesis.
Accumulating evidence suggests that levodopa has hormone-like or neurotransmitter-like properties and may function through direct interaction with β-adrenergic receptors without prior conversion to catecholamines (32, 33). For instance, exogenously applied levodopa stimulates the β-adrenoceptor–dependent release of norepinephrine and dopamine from rat hypothalamic slices (9). These actions were observed in the presence of a DDC inhibitor, so they did not appear to be secondary to the conversion of levodopa to dopamine and/or norepinephrine. In addition, for neurons in several regions of the central nervous system, levodopa itself has been reported to produce presynaptic and postsynaptic responses (8, 7, 34). Although selective β1- and β2-adrenergic antagonists both reportedly interfere with DOPAergic signaling (35), levodopa does not displace bound 3H-dihydroalprenol (a β-adrenoceptor ligand; Ref. 35). In addition, although propranolol prevents actions of levodopa, it does so in a noncompetitive manner (36). These previous findings suggest that either there is a separate levodopa receptor (i.e., a receptor that is not an adrenoceptor) that is inhibited by β-adrenergic antagonists or that it is a levodopa metabolite (not levodopa per se) that binds β-adrenoceptors. Efforts to identify specific receptors that mediate the hormone-like effects of levodopa are reviewed by Slominski and Paus (37). The current findings of DOPAergic action in C2C12 myotubes suggest that signaling properties of levodopa are not limited to neural tissue.
In addition to inhibiting DDC, NSD also inhibits monoamine oxidase, an enzyme that is present in skeletal muscle (38) and catalyzes the conversion of levodopa to dopaquinone. In the absence of any countervailing effects on levodopa action attributable to inhibition of DDC by NSD, the prevention of the effects of levodopa by NSD suggests that the monoamine oxidase product dopaquinone or one of its metabolites mediates DOPAergic action in myotubes. Intriguingly, the dopaquinone synthetic pathway is dysregulated in the Agouti mouse, a model of obesity and insulin resistance (39). Several dominant mutations of agouti, a secreted paracrine factor that regulates adipocyte transcription factors, lead to increased body-fat content, insulin resistance, impaired glucose tolerance, and hyperglycemia (40). Agouti regulates lipid metabolism and adipogenesis, and altered expression levels of agouti disrupt the control of body weight in mice (39, 41). Thus, there are now two separate lines of evidence (i.e., the current data and previous reports regarding agouti) that suggest potential roles for levodopa-related pathways in metabolic regulation.
Skeletal muscle is the primary site of glucose disposal (13, 14) and, consequently, glucose metabolism in muscle plays a significant role in whole-body glucose homeostasis. Emerging evidence suggests that β-adrenergic action or cAMP, a second-messenger mediator of β-adrenergic signaling, may interfere with insulin signaling and/or stimulation of glucose transport by insulin (42-44). For example, epinephrine has been reported to prevent stimulation of muscle glucose transport by a physiologic concentration of insulin (12). In addition, for transgenic mice with reduced expression of Gsα protein (a key element in signal transduction for G protein–coupled receptors such as β-adrenergic receptors), responsiveness of skeletal glucose transport to insulin was increased compared with insulin responsiveness in muscle from wild-type littermates (42). The current data suggest that C2C12 myotubes might possess an autocrine DOPAergic/β-adrenergic system that could impinge on insulin action.
In conclusion, the data illustrate β-adrenergic inhibition of insulin action by levodopa in cells that cannot synthesize catecholamines, suggesting that levodopa or one of its noncatecholamine metabolites has hormone-like effects in C2C12 myotubes. Furthermore, prevention of endogenous levodopa production potentiated insulin-stimulated glucose transport, suggesting a novel means for the improvement of insulin action.
Footnotes
JSF was supported by the National Institute for Diabetes and Diseases of the Kidney (K01 DK066330). Funding for materials was provided by the Saint Louis University Beaumont Faculty Development Fund. JRL was a member of the Pfizer-Solutia STARS research program that provided additional funding for supplies and is administered by Kenneth Mares (University of Missouri-St. Louis, MO).
References
- 1.Boyd AE, III, Lebovitz HE, Feldman JM. Endocrine function and glucose metabolism in patients with Parkinson's disease and their alteration by L-Dopa. J Clin Endocrinol Metab. 1971;33:829–837. doi: 10.1210/jcem-33-5-829. [DOI] [PubMed] [Google Scholar]
- 2.Hirai K, Gondo K. Über Dopa-Hyperglykämie. Biochem Z. 1927;189:92–100. [Google Scholar]
- 3.Sirtori CR, Bolme P, Azarnoff DL. Metabolic responses to acute and chronic L-dopa administration in patients with Parkinsonism. New Engl J Med. 1972;287:729–733. doi: 10.1056/NEJM197210122871501. [DOI] [PubMed] [Google Scholar]
- 4.Hakanson R, Lundquist I, Rerup C. On the hyperglycaemic effect of DOPA and dopamine. Eur J Pharmacol. 1967;1:114–119. doi: 10.1016/0014-2999(67)90047-7. [DOI] [PubMed] [Google Scholar]
- 5.Smith JL, Ju JS, Saha BM, Racette BA, Fisher JS. Levodopa with carbidopa diminishes glycogen concentration, glycogen synthase activity, and insulin-stimulated glucose transport in rat skeletal muscle. J Appl Physiol. 2004;97:2339–2346. doi: 10.1152/japplphysiol.01219.2003. [DOI] [PubMed] [Google Scholar]
- 6.Schubert D, Tarikas H, LaCorbiere M. Neurotransmitter regulation of adenosine 3′,5′-monophosphate in clonal nerve, glia, and muscle cell lines. Science. 1976;192:471–473. doi: 10.1126/science.176728. [DOI] [PubMed] [Google Scholar]
- 7.Misu Y, Goshima Y, Ueda H, Okamura H. Neurobiology of L-DOPAergic systems. Prog Neurobiol. 1996;49:415–454. doi: 10.1016/0301-0082(96)00025-1. [DOI] [PubMed] [Google Scholar]
- 8.Misu Y, Yue JL, Goshima Y. L-DOPA systems for blood pressure regulation in the lower brainstem. Neurosci Res. 1995;23:147–158. doi: 10.1016/0168-0102(95)00939-q. [DOI] [PubMed] [Google Scholar]
- 9.Goshima Y, Kubo T, Misu Y. Biphasic actions of L-DOPA on the release of endogenous noradrenaline and dopamine from rat hypothalamic slices. Br J Pharmacol. 1986;89:229–234. doi: 10.1111/j.1476-5381.1986.tb11139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang CH. Beta-adrenergic blockade attenuates insulin resistance induced by tumor necrosis factor. Am J Physiol. 1993;264:R984–R991. doi: 10.1152/ajpregu.1993.264.5.R984. [DOI] [PubMed] [Google Scholar]
- 11.Zierath JR, Handberg A, Tally M, Wallberg-Henriksson H. C-peptide stimulates glucose transport in isolated human skeletal muscle independent of insulin receptor and tyrosine kinase activation. Diabetologia. 1996;39:306–313. doi: 10.1007/BF00418346. [DOI] [PubMed] [Google Scholar]
- 12.Hunt DG, Ding Z, Ivy JL. Clenbuterol prevents epinephrine from antagonizing insulin-stimulated muscle glucose uptake. J Appl Physiol. 2001;92:1285–1292. doi: 10.1152/japplphysiol.01009.2001. [DOI] [PubMed] [Google Scholar]
- 13.Jue T, Rothman DL, Shulman GI, Tavitian BA, DeFronzo RA, Shulman RG. Direct observation of glycogen synthesis in human muscle with 13C NMR. Proc Natl Acad Sci USA. 1989;86:4489–4491. doi: 10.1073/pnas.86.12.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 15.Somwar R, Kim DY, Sweeney G, Huang C, Niu W, Lador C, Ramlal T, Klip A. GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem J. 2001;359:639–649. doi: 10.1042/0264-6021:3590639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith JL, Patil PB, Fisher JS. AICAR and hyperosmotic stress increase insulin-stimulated glucose transport. J Appl Physiol. 2005;99:878–883. doi: 10.1152/japplphysiol.01297.2004. [DOI] [PubMed] [Google Scholar]
- 17.Tortorella LL, Pilch PF. C2C12 myocytes lack an insulin-responsive vesicular compartment despite dexamethasone-induced GLUT4 expression. Am J Physiol Endocrinol Metab. 2002;283:E514–E524. doi: 10.1152/ajpendo.00092.2002. [DOI] [PubMed] [Google Scholar]
- 18.Winder AJ. A stopped spectrophotometric assay for the dopa oxidase activity of tyrosinase. J Biochem Biophys Methods. 1994;28:173–183. doi: 10.1016/0165-022x(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 19.Klip A, Li G, Logan WJ. Induction of sugar uptake response to insulin by serum depletion in fusing L6 myoblasts. Am J Physiol. 1984;247:E291–E296. doi: 10.1152/ajpendo.1984.247.3.E291. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama A, Wiegn P, McKnite S, Lurie KG. Enzymatic fluorometric assay for tissue cAMP. J Clin Lab Anal. 1994;8:437–442. doi: 10.1002/jcla.1860080616. [DOI] [PubMed] [Google Scholar]
- 21.Ju JS, Smith JL, Oppelt PJ, Fisher JS. Creatine feeding increases GLUT4 expression in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E347–E352. doi: 10.1152/ajpendo.00238.2004. [DOI] [PubMed] [Google Scholar]
- 22.Arita DY, Di Marco GS, Schor N, Casarini DE. Purification and characterization of the active form of tyrosine hydroxylase from mesangial cells in culture. J Cell Biochem. 2002;87:58–64. doi: 10.1002/jcb.10277. [DOI] [PubMed] [Google Scholar]
- 23.Di Marco GS, Naffah-Mazzacoratti Md Mda G, Vio CP, Dos Santos OF, Schor N, Casarini DE. Mesangial cells are able to produce catecholamines in vitro. J Cell Biochem. 2003;89:144–151. doi: 10.1002/jcb.10485. [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto K, Hirshman MF, Aschenbach WG, Goodyear LJ. Contraction regulation of Akt in rat skeletal muscle. J Biol Chem. 2002;277:11910–11917. doi: 10.1074/jbc.M112410200. [DOI] [PubMed] [Google Scholar]
- 25.Li SW, Lin TS, Minteer S, Burke WJ. 3,4-dihydroxyphenylacetalde-hyde and hydrogen peroxide generate a hydroxyl radical: possible role in Parkinson's disease pathogenesis. Brain Res Mol Brain Res. 2001;93:1–7. doi: 10.1016/s0169-328x(01)00120-6. [DOI] [PubMed] [Google Scholar]
- 26.McCullough LA, Westfall TC. Neuropeptide Y inhibits depolarization-stimulated catecholamine synthesis in rat pheochromocytoma cells. Eur J Pharmacol. 1995;287:271–277. doi: 10.1016/0014-2999(95)00496-3. [DOI] [PubMed] [Google Scholar]
- 27.Tabata I, Schluter J, Gulve EA, Holloszy JO. Lithium increases susceptibility of muscle glucose transport to stimulation by various agents. Diabetes. 1994;43:903–907. doi: 10.2337/diab.43.7.903. [DOI] [PubMed] [Google Scholar]
- 28.Gulve EA, Cartee GD, Zierath JR, Corpus VM, Holloszy JO. Reversal of enhanced muscle glucose transport after exercise: roles of insulin and glucose. Am J Physiol. 1990;259:E685–E691. doi: 10.1152/ajpendo.1990.259.5.E685. [DOI] [PubMed] [Google Scholar]
- 29.Treseder SA, Rose S, Summo L, Jenner P. Commonly used L-amino acid decarboxylase inhibitors block monoamine oxidase activity in the rat. J Neural Transm. 2003;110:229–238. doi: 10.1007/s00702-002-0778-4. [DOI] [PubMed] [Google Scholar]
- 30.Hunter LW, Rorie DK, Tyce GM. Inhibition of aromatic L-amino acid decarboxylase under physiological conditions: optimization of 3-hydroxybenzylhydrazine concentration to prevent concurrent inhibition of monoamine oxidase. Biochem Pharmacol. 1993;45:1363–1366. doi: 10.1016/0006-2952(93)90292-5. [DOI] [PubMed] [Google Scholar]
- 31.Thong FS, Derave W, Kiens B, Graham TE, Urso B, Wojtaszewski JF, Hansen BF, Richter EA. Caffeine-induced impairment of insulin action but not insulin signaling in human skeletal muscle is reduced by exercise. Diabetes. 2002;51:583–590. doi: 10.2337/diabetes.51.3.583. [DOI] [PubMed] [Google Scholar]
- 32.Goshima Y, Nakamura S, Misu Y. L-dopa facilitates the release of endogenous norepinephrine and dopamine via presynaptic beta 1- and beta 2-adrenoceptors under essentially complete inhibition of L-aromatic amino acid decarboxylase in rat hypothalamic slices. Jpn J Pharmacol. 1990;53:47–56. doi: 10.1254/jjp.53.47. [DOI] [PubMed] [Google Scholar]
- 33.Tedroff JM. The neuroregulatory properties of L-DOPA: a review of the evidence and potential role in the treatment of Parkinson's disease. Rev Neurosci. 1997;8:195–204. doi: 10.1515/revneuro.1997.8.3-4.195. [DOI] [PubMed] [Google Scholar]
- 34.Nishihama M, Miyamae T, Goshima Y, Okumura F, Misu Y. An L-dopaergic relay from the posterior hypothalamic nucleus to the rostral ventrolateral medulla and its cardiovascular function in anesthetized rats. Neuroscience. 1999;92:123–135. doi: 10.1016/s0306-4522(98)00720-9. [DOI] [PubMed] [Google Scholar]
- 35.Goshima Y, Nakamura S, Misu Y. L-DOPA facilitates the release of endogenous norepinephrine and dopamine via presynaptic β1- and β2-adrenoceptors under essentially complete inhibition of L-aromatic amino acid decarboxylase in rat hypothalamic slices. Jpn J Pharmacol. 1990;53:47–56. doi: 10.1254/jjp.53.47. [DOI] [PubMed] [Google Scholar]
- 36.Goshima Y, Nakamura S, Misu Y. L-dihydroxyphenylalanine methyl ester is a potent competitive antagonist of the L-dihydroxyphenylala-nine-induced facilitation of the evoked release of endogenous norepinephrine from rat hypothalamic slices. J Pharmacol Exp Ther. 1991;258:466–471. [PubMed] [Google Scholar]
- 37.Slominski A, Paus R. Towards defining receptors for L-tyrosine and Ldopa. Mol Cell Endocrinol. 1994;99:C7–C11. doi: 10.1016/0303-7207(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 38.Koenig H, Goldstone A, Lu CY. Androgens regulate mitochondrial cytochrome c oxidase and lysosomal hydrolases in mouse skeletal muscle. Biochem J. 1980;192:349–353. doi: 10.1042/bj1920349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mynatt RL, Stephens JM. Agouti regulates adipocyte transcription factors. Am J Physiol Cell Physiol. 2001;280:C954–C961. doi: 10.1152/ajpcell.2001.280.4.C954. [DOI] [PubMed] [Google Scholar]
- 40.Yen TT, Gill AM, Frigeri LG, Barsh GS, Wolff GL. Obesity, diabetes, and neoplasia in yellow A(vy)/- mice: ectopic expression of the agouti gene. FASEB J. 1994;8:479–488. doi: 10.1096/fasebj.8.8.8181666. [DOI] [PubMed] [Google Scholar]
- 41.Smith SR, Gawronska-Kozak B, Janderova L, Nguyen T, Murrell A, Stephens JM, Mynatt RL. Agouti expression in human adipose tissue: functional consequences and increased expression in type 2 diabetes. Diabetes. 2003;52:2914–2922. doi: 10.2337/diabetes.52.12.2914. [DOI] [PubMed] [Google Scholar]
- 42.Yu S, Castle A, Chen M, Lee R, Takeda K, Weinstein LS. Increased insulin sensitivity in Gsa knockout mice. J Biol Chem. 2001;276:19994–19998. doi: 10.1074/jbc.M010313200. [DOI] [PubMed] [Google Scholar]
- 43.Kim S, Jee K, Kim D, Koh H, Chung J. Cyclic AMP inhibits Akt activity by blocking the membrane localization of PDK1. J Biol Chem. 2003;276:12864–12870. doi: 10.1074/jbc.M001492200. [DOI] [PubMed] [Google Scholar]
- 44.Lee AD, Hansen PA, Schluter J, Gulve EA, Gao J, Holloszy JO. Effects of epinephrine on insulin-stimulated glucose uptake and GLUT-4 phosphorylation in muscle. Am J Physiol. 1997;273:C1082–C1087. doi: 10.1152/ajpcell.1997.273.3.C1082. [DOI] [PubMed] [Google Scholar]