

Abstract
Recent studies suggested that the protection of cell apoptosis by AKT involves phosphorylation and inhibition of FKHR and related FOXO forkhead transcription factors and that androgens provide an AKT-independent cell survival signal in prostate cancer cells. Here, we report receptor-dependent repression of FKHR function by androgens in prostate cancer cells. Transcriptional analysis demonstrated that activation of the androgen receptor caused an inhibition of both wild-type FKHR and a mutant in which all three known AKT sites were mutated to alanines, showing that the repression is AKT independent. In vivo and in vitro coprecipitation studies demonstrated that the repression is mediated through protein-protein interaction between FKHR and the androgen receptor. Mapping analysis localized the interacting domains to the carboxyl terminus between amino acids 350 and 655 of FKHR and to the amino-terminal A/B region and the ligand binding domain of the receptor. Further analysis demonstrated that the activated androgen receptor blocked FKHR's DNA binding activity and impaired its ability to induce Fas ligand expression and prostate cancer cell apoptosis and cell cycle arrest. These studies identify a new mechanism for androgen-mediated prostate cancer cell survival that appears to be independent of the activity of the receptor on androgen response element-mediated transcription and establish FKHR and related FOXO forkhead proteins as important nuclear targets for both AKT-dependent and -independent survival signals in prostate cancer cells.
Androgens are the male steroid hormone responsible for the development, maintenance, and regulation of the male phenotype and reproductive physiology. The activity of androgens is mediated through the androgen receptor (7, 12, 31, 49), which belongs to the steroid/thyroid nuclear receptor superfamily, a group of ligand-regulated, zinc finger-containing transcription factors (11, 50).
Similar to other steroid receptors, the androgen receptor, both structurally and functionally, is modular in nature and composed of an N-terminal A/B region (NT) containing the major activation function (AF-1), a DNA binding domain containing two zinc fingers, a short hinge region, and a C-terminal ligand binding domain which contains a weaker activation function (AF-2) (21, 22). A third activation function, AF-5, has also been described (21).
Although modular in nature, the different domains of androgen receptor act in a highly coordinated fashion during the receptor activation. In the absence of androgens, the ligand binding domain represses the activation functions until androgens bind to it and relieve the repression by inducing the formation of a conformation suitable for interaction with a complex of transcriptional coactivators, including common coactivators (1, 34), steroid receptor coactivators (3, 16, 18, 40), as well as possible androgen receptor-selective coactivators (51). The coactivators often are histone acetyltransferases themselves or associated with the activity. They convey the effect of androgen receptor on androgen response element-mediated transcription by directly remodeling the chromatin structure through the acetylation of core histones.
In addition to androgen response element-mediated gene regulation, androgens have also been reported to regulate the expression of many genes in which no well-defined androgen response elements have been identified. Kallio et al. (23) have shown that, in a proper context, androgen receptor is capable of eliciting both trans-repression and trans-activation through protein-protein interactions with other transcription factors without interacting directly with specific DNA elements.
Besides their established physiological functions, androgens are implicated in multiple pathological processes, including benign prostatic hyperplasia and prostate cancer. Physiological levels of androgens are chronically required for the normal growth and development of the prostate gland. In addition to the stimulation of cell proliferation, inhibition of prostatic cell death plays an important role in the maintenance of the total prostatic cell number by androgens in adulthood (19). Similar to normal prostatic glandular epithelial cells, most prostate cancer cells are androgen receptor positive in situ and remain androgen sensitive in vivo. Due to this retained androgen sensitivity, androgen ablation therapy is an effective first-line therapy for metastatic prostate cancer. However, prostate cancer patients frequently relapse due to conversion of the tumor cells to an androgen-independent status. To overcome the resistance of prostate cancer cells to androgen ablation, it is essential to understand the mechanisms underlying the androgen protection of prostate cancer cells from death.
While the ability of androgens to stimulate cell proliferation is relatively well explained by the effects of androgens on cell cycle regulators such as CDK2, CDK4, and p16 CDK inhibitor (33), the mechanism by which androgens protect prostatic cells against apoptosis remains largely unknown. Previous studies have shown that androgens protect prostate cancer cells from death induced by PTEN tumor suppressor (30) and phosphoinositide 3-kinase inhibitors (5, 30), which both suppress the activity of AKT, by activating a survival pathway that is independent of the AKT signaling pathway or by activating the same survival pathway at steps downstream from AKT.
AKT is a serine/threonine protein kinase that is activated by membrane targeting through interaction with phosphatidylinositol (3,4,5)-triphosphate, which is generated by active phosphoinositide 3-kinase (6). Active AKT is known to phosphorylate and suppress the activity of many pro-apoptotic proteins, including FKHR (forkhead in rhabdomyosarcoma) (14, 47) and related FOXO transcription factors such as FKHRL1 (4) and AFX (35), which represent the mammalian homologues of Caenorhabditis elegans Daf-16 forkhead protein (39). AKT phosphorylation results in the translocation of these FOXO forkhead transcription factors to the cytoplasm, leading to the suppression of the transcription of proapoptotic genes such as Fas ligand (9), Bim (10), and Bcl-6 (48).
PTEN tumor suppressor is a lipid phosphatase that removes the D3 phosphate from the inositol ring of phosphatidylinositol (3,4,5)-triphosphate, resulting in inactivation of AKT (42, 45). Through its negative effect on AKT, PTEN regulates FKHR activity positively and induces Fas ligand expression (9). The opposing effects of PTEN and AKT on Fas ligand expression are believed to contribute to their pro- and antiapoptotic effects, respectively. Since apoptosis induced by androgen ablation was reported to be impaired in Fas-null mice (lpr−/−) (46), it is conceivable that androgens may also target FOXO forkhead factors to protect prostate cancer cells against apoptosis.
In the following pages, we will provide experimental evidence that the androgen receptor forms a complex with FKHR and that the active androgen receptor blocks the binding of FKHR to its DNA response elements, leading to suppression of FKHR's transcriptional activity and its ability to regulate Fas ligand expression and to induce apoptosis and cell cycle arrest of prostate cancer cells. These findings suggest that FOXO forkhead transcription factors are important nuclear targets for both AKT-dependent and -independent survival signals in prostate cancer cells.
MATERIALS AND METHODS
Plasmids.
pcDNA3-FLAG-FKHR:WT (13, 47), pcDNA3-FLAG-FKHR:TSS (47), pAlter-FKHR (14), 3×insulin response sequence-Luc (14, 47), pCMVβ (30), HA-FKHRL1-WT (4), HA-FKHRL1-MT (4), FHRE-Luc (4), AREe1bLuc (30), pCMVhAR (30), pLENβgal (28, 29), pLENhERα (28, 29), EREe1bLuc (28, 29), pSG5L-HA-PTEN:WT (30, 42), pSG5L-HA-PTEN:G129R (30, 42), CBP (32), SRC1 (40), AIB1 (2), and GST-AR-DBD (41) have already been described.
GST-AR-NT was constructed by inserting the ClaI/Klenow- and HindIII-digested 1.7-kb androgen receptor NT fragment from pAR3 (21) into pRSET-GST-SRC(782-7346) (44) digested with NcoI/Klenow/HindIII. GST-AR-LBD was constructed by inserting the NarI/BamHI/Klenow-treated 0.9-kb androgen receptor ligand binding domain fragment from pAR28.1 (20) into the pRSET-GST vector, which is derived from pRSET-GST-SRC(782-1139) digested with BamHI/HindIII/Klenow.
To construct Gal4-FKHR(1-150), FKHR cDNA fragment coding the first 150 amino acids were amplified by PCR with primers 5′-CGGGGGTCACCGGATCCATGGCCGAGGC3′ and 5′-GCTGCTCTTGCGTCTAGACTAGGCTGCCCCGCG3′, which generates BamHI and XbaI sites at 5′ and 3′ ends of the DNA fragment, respectively. The amplified FKHR(1-150) fragment was cloned into the BamHI and XbaI sites of pFA-CMV vector (Stratagene, La Jolla, Calif.). Gal4-FKHR(211-655) and Gal4-FKHR(350-655) were constructed similarly by generating a BamHI site at the 5′ end and a XbaI site at the 3′ end of the corresponding FKHR cDNA fragments. The primers for the 5′ end were 5′-ATTATGACGAATGGATCCCTTCCAGCCCGCCGAG-3′ for Gal-FKHR(211-655) and 5′-GGCCATCTTTGCGGATCCTGGCGGGTACACCATAG-3′ for Gal-FKHR(350-655). The 3′-end primer was 5′-CTCAGTTCCTGCTTCTAGACAATCTGAAGTAC-3′. pLENhERβ was constructed by replacing the BamHI fragment containing the β-galactosidase cDNA of pLENβgal with the full-length human estrogen receptor beta (ERβ) cDNA released from pKCRE.ERβ (36) with BamHI.
Transfections and reporter assays.
DU145 and PC3 cells were plated in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at 2 × 105 cells/well in six-well plates, and LNCaP cells were plated in RPMI 1640 at 4 × 105 cells/well. One day after plating, cells were transfected by Lipofectamine Plus following the protocol from Gibco/BRL. Transfected cells were treated with synthetic androgens in medium containing 1% charcoal-stripped fetal bovine serum and washed with phosphate-buffered saline. Cell lysates were prepared by directly adding lysis buffer (25 mM Tris-phosphate [pH 7.8], 2 mM dithiothreitol, 2 mM 1,2-diaminocyclohexane-N′,N′,N′,N′-tetraacetic acid, 10% glycerol, 0.2% Triton X-100) to the cells on ice. Luciferase activity was determined with luciferase assay systems from Promega Corporation (Madison, Wis.) following the company's protocol. β-Galactosidase activity was determined as previously described (28, 29, 30).
Cell survival and apoptosis assays.
Determination of the survival and apoptotic index of transfected prostate cancer cells has been described (30). In brief, transfected cells were washed with phosphate-buffered saline and fixed in 3.7% formaldehyde. The viability of transfected cells in each well was determined by counting the total number of green cells in each well under a fluorescence microscope. For the demonstration of apoptotic cells, fixed cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI), and cells with green and blue fluorescence were observed with a Leitz Orthoplan 2 microscope. Representative micrographs were captured by a charge-coupled device camera with the Smart Capture Program (Vysis, Downers Grove, Ill.). The apoptotic index of green fluorescent protein (GFP)-positive cells was determined by scoring 300 GFP-positive cells for chromatin condensation and apoptotic body formation.
Immunological assays.
For immunoprecipitation with anti-Gal4 or M2 anti-Flag antibodies, DU145 cells were transfected and treated as described above for transcriptional assays. Whole-cell extracts were prepared in a buffer containing 0.5% NP-40, 20 mM Tris-HCl (pH 7.5), 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 10 μg of aprotinin per ml, 10 μg of leupeptin per ml, 10 mM benzamidine, and I mM phenylmethylsulfonyl fluoride. Cellular extracts were incubated with 4 μg of antibody for 4 h at 4°C and subsequently with protein G-agarose beads for an additional 4 h. Beads were washed four times, and the immunoprecipitates were heated in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer.
For immunoblotting, cell extracts or immunoprecipitates were separated on SDS-polyacrylamide gels, transferred to a nitrocellulose membrane, probed with anti-androgen receptor, anti-Gal4, or M2 anti-Flag antibodies, and visualized with enhanced chemiluminescence as described (28, 29).
Nuclear extract preparation and electrophoretic mobility shift assay.
Nuclear extracts were prepared as described (28). In brief, transfected DU145 cells were scraped in hypotonic buffer containing 20 mM HEPES (pH 7.9), 1 mM EDTA, 1 mM EGTA, 20 mM NaF, 1 mM Na3VO4, 1 mM Na2P2O7, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1% NP-40, and protease cocktail. Nuclei were separated from the cytosol by centrifugation at 4°C for 20 min. After centrifugation, pellets containing nuclei were resuspended in the same buffer but now containing 420 mM KCl and 20% glucose.
Electrophoretic mobility shift assay was performed in binding buffer containing 50 mM Tris (pH 7.5), 13 mM MgCl2, 1 mM EDTA, 5% glycerol, 1 mM dithiothreitol, 0.5 μg of poly(dI-dC), 0.5 μg of poly(dA-dT), 0.5 ng of insulin response sequence oligonucleotide probe (8, 14, 48) labeled with the 32P RTS T4 kinase labeling system (Life Technologies, Rockville, Md.) and nuclear extracts containing 10 μg of protein. After incubation at 22°C for 20 min, samples were subjected to analysis on a 5% polyacrylamide gel containing 0.5× Tris-borate-EDTA (45 mM Tris-borate, 1 mM EDTA).
GST pulldown assays.
FKHR protein was produced in vitro from pAlter-FKHR in the presence of [35S]methionine with T7 polymerase with the coupled transcription-translation kit (Promega, Madison, Wis.). Constructs in which GST was fused to the ligand binding domain, the DNA binding domain, and the NT region of the androgen receptor were transformed into Escherichia coli BL21(DE3) strain. Transformed bacteria were cultured at 37°C until the optical density at 600 nm reached 0.8, and the culturing was continued overnight at 30°C in the presence of 0.5 mM isopropylthiogalactopyranoside (IPTG). Then, the bacterial cultures were harvested and sonicated in buffer containing 50 mM Tris, 10 mM NaCl, 1 mM EDTA, 6 mM MgCl2, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride. GST fusion proteins were purified by binding to glutathione-Sepharose 4B beads (Amersham Pharmacia, Piscataway, N.J.). Then 50 μg of bead-bound GST proteins was incubated overnight at 4°C with 10 μl of 35S-labeled FKHR or androgen receptor in binding buffer with or without androgens. The beads were then washed with binding buffer containing 0.5% NP-40 and heated in SDS-PAGE sample buffer. After a brief centrifugation, the supernatant was resolved by SDS-8% PAGE, and the labeled protein was revealed by autoradiography.
Determination of Fas ligand expression on the cell surface of transfected cells by flow cytometry.
To quantify Fas ligand expression on the cell surface of transfected cells, DU145 and LNCaP cells grown in 100-mm dishes were cotransfected with CD20 with or without FKHR and androgen receptor and treated with R1881 or ethanol as a vehicle control. Then, cells were collected in phosphate-buffered saline containing 2.5 mM EDTA, washed twice with phosphate-buffered saline, and incubated at 4°C for 60 min with fluorescein isothiocyanate-conjugated mouse anti-CD20 antibody (20 μl/106 cells). After staining with CD20 antibody, the cells were fixed on ice for 30 min in phosphate-buffered saline-paraformaldehyde (1.0%, wt/vol). Fixed cells were incubated at 4°C for 60 min with a rabbit anti-Fas ligand antibody (2 μg/ml), which was followed by incubation with indocarbocyanine-conjugated anti-rabbit IgG (30 min at 4°C). Cell sorting and flow cytometry analysis were performed on a FACScan (Becton Dickinson, Mountain View, Calif.).
Cell cycle analysis of transfected DU145 cells.
DU145 cells were cotransfected with CD20 and treated as above for Fas ligand assays. After treatment, the cells were harvested, incubated with fluorescein isothiocyanate-conjugated anti-CD20 antibody, stained with propidium iodide, and analyzed by two-color fluorescence-activated cell sorting (Beckton-Dickinson).
RESULTS
Androgens repress FKHR activity in prostate cancer cells in an androgen receptor- and dose-dependent manner.
To investigate the possibility that androgen-mediated prostate cancer cell survival involves the suppression of FKHR function, PTEN-intact and androgen receptor-negative DU145 cells were transfected with wild-type and mutant FKHR(AAA), in which all three AKT phosphorylation sites were mutated to alanines (14, 47). FKHR transcriptional activity was determined by cotransfection with a reporter gene containing copies of the IGFBP-1 insulin response sequence immediately upstream of the minimal thymidine kinase promoter (14, 47).
As shown in Fig. 1A, IGF-1 inhibits transcriptional activity of the wild-type FKHR but not the AAA mutant, indicating that phosphorylation of these sites is critical for the ability of growth factors to suppress transactivation by FKHR, consistent with previous reports (37, 39). In cells transfected with the androgen receptor, treatment with R1881, a synthetic androgen receptor agonist, represses the activity of both the wild-type FKHR and the AAA mutant (Fig. 1B), indicating that repression of transactivation of FKHR by androgens occurs independently of FKHR phosphorylation by AKT and that androgens are more powerful repressors of FKHR than growth factors. R1881 does not affect the FKHR activity in the absence of the androgen receptor, and the expression of the androgen receptor does not have an effect on the FKHR activity in the absence of R1881, demonstrating that the FKHR inhibition is androgen receptor dependent and only occurs if androgen receptor is activated. Activated androgen receptor also decreases the reporter activity in the absence of ectopic FKHR (Fig. 1B), suggesting that, when activated, androgen receptor may also inhibit the activity of endogenous forkhead proteins in DU145 cells. Overall, the activity in Fig. 1A is lower than in Fig. 1B, presumably because the vehicle is ethanol for R1881 and medium for IGF-1.
FIG. 1.

Inhibition of FKHR reporter activity by activated androgen receptor. (A) Inhibition of wild-type FKHR by IGF-1. DU145 cells were transfected with 0.5 μg of 3×IRS-Luc, 0.2 μg of pCMVβ, 0.1 μg of FKHR:WT (WT) or FKHR:TSS (AAA). Transfected cells were treated for 24 h or not with 50 ng of IGF-1 per ml as indicated. Luciferase activity was determined and normalized to cognate β-galactosidase activity. FKHR activity was expressed as relative luciferase units (RLU) after normalization. Duplicate samples were analyzed for each data point, and the data have been reproduced three times. (B) Androgen receptor (AR)-dependent inhibition of wild-type and AAA mutant FKHR by androgens. DU145 cells were transfected with 0.2 μg of pCMVhAR, 0.2 μg of pCMVβ, 0.5 μg of 3×IRS-Luc, and 0.1 μg of FKHR:WT (WT) or FKHR:TSS (AAA). Transfected cells were treated for 24 h with 10−8 M R1881 or ethanol (EOH) as vehicle controls. FKHR activity was determined and expressed as for panel A. (C) Dosage-dependent inhibition of wild-type FKHR by activated androgen receptor (AR). DU145 cells were transfected with 0.5 μg of 3×IRS-Luc, 0.2 μg of pCMVβ, 0.1 μg of FKHR:WT, and the indicated amounts of pCMVhAR. Transfected cells were treated and FKHR activity was determined as for panel B. (D) Dosage-dependent inhibition of the AAA mutant by activated androgen receptor (AR). DU145 cells were transfected as in panel C except that 0.1 μg of FKHR:TSS was used in place of FKHR:WT. Transfected cells were treated and FKHR activity was determined as for panel B. (E) No decrease at the level of FKHR protein after androgen treatment. DU145 cells were transfected and treated as for panels C and D. Androgen receptor (AR) and FKHR proteins were detected by Western blotting with PG-21 anti-androgen receptor (Upstate) and M2 anti-Flag (Sigma) antibodies, respectively. Equal amounts of protein were loaded. W, wild-type FKHR; M, AAA mutant.
To directly demonstrate the dosage dependency, the activities of wild-type FKHR (Fig. 1C) and the AAA mutant (Fig. 1D) were determined in DU145 cells transfected with various amounts of androgen receptor vector. Although in the absence of androgens, the androgen receptor expressed at different levels has little effect on FKHR activity, in the presence of R1881, it decreases the activities of both wild-type FKHR and the AAA mutant in a dosage-dependent manner.
To determine whether the decrease in FKHR activity is due to a decrease in the level of FKHR protein, the level of FKHR and androgen receptor proteins was examined by immunoblotting under the same conditions that the reporter activity was determined. As shown in Fig. 1E, R1881 treatment of DU145 cells expressing different amounts of androgen receptor protein does not decrease the level of FKHR protein, suggesting that activated androgen receptor decreases the specific activity of FKHR.
To make sure that the decreased reporter activity truly reflects inhibition of FKHR, we next tested the effect of activated androgen receptor on the FKHR reporter in LNCaP cells without FKHR cotransfection. LNCaP cells contain no functional PTEN (42). AKT is constitutively active in these cells, which would phosphorylate endogenous FKHR, resulting in its cytoplasmic localization. As a consequence of the AKT phosphorylation, LNCaP cells contain little transcriptionally active FKHR in the nucleus. As shown in Fig. 2A, activated androgen receptor does not inhibit the basal activity of the FKHR reporter in LNCaP cells, demonstrating that the decreased reporter activity in DU145 cells without FKHR transfection must be due to the repression of endogenous activity by activated androgen receptor.
FIG. 2.

Androgen inhibition of FKHR activity mediated through endogenous androgen receptor. (A) Lack of an androgen effect on basal FKHR reporter activity in LNCaP cells. Cells were transfected with 0.1 μg of pCMVhAR, 0.2 μg of pCMVβ and 0.5 μg of 3×IRS-Luc. Transfected cells were treated and FKHR activity was determined as in Fig. 1B. (B) Androgen inhibition of FKHR activity mediated through endogenous androgen receptor (AR). LNCaP cells were transfected as in A but with 0.1 μg of FKHR:WT (WT) or FKHR:TSS (AAA). Transfected cells were treated and FKHR activity was determined as for Fig. 1B. (C) No decrease at the level of FKHR protein in LNCaP cells after androgen treatment. LNCaP cells were transfected and treated as for panel B. DU145 cells transfected with or without Flag-tagged FKHR were included as controls. Note that there is a background band recognized by the M2 antibody which moves slightly faster than Flag-FKHR in the gel. Androgen receptor (AR), FKHR, and β-actin proteins were detected by Western blotting with the PG-21, M2, and anti-β-actin monoclonal (Sigma) antibodies, respectively. Equal amounts of protein were loaded.
Wild-type FKHR introduced into LNCaP cells contains little activity (Fig. 2B), which is expected because the activity of wild-type FKHR, similar to the endogenous FKHR activity, should be suppressed by the AKT phosphorylation. Different from wild-type FKHR, the AAA mutant is active in the cells and its activity is inhibited by R1881 through endogenous androgen receptor (Fig. 2B), showing that FKHR inhibition is not limited to ectopic androgen receptor introduced into androgen receptor-negative cells. The transfection of additional androgen receptor enhances the androgen inhibition, confirming the data from DU145 cells that the degree of inhibition depends on the amount of androgen receptor in the cells. Similar to the data in Fig. 1, androgen treatment does not decrease the level of FKHR protein expression (Fig. 2C), demonstrating that R1881, acting through the endogenous androgen receptor, decreased the specific activity of the AAA mutant.
To determine whether androgens also repress the activity of other mammalian homologues of Daf-16, the effect of activated androgen receptor on FKHRL1 was determined in DU145 cells. As shown in Fig. 3A, activated androgen receptor inhibits the activity of FKHRL1 on the reporter constructed with the promoter of the Fas ligand gene (4), albeit to a lesser degree than FKHR (Fig. 1B). To test whether FKHRL1 activity is also inhibited by androgens through endogenous androgen receptor, the activity of a mutant FKHRL1, similar to the AAA of FKHR, was analyzed in LNCaP cells treated or not with R1881, As shown in Fig. 3B, the mutant FKHRL1 is again inhibited by R1881 treatment. The data suggest that androgen inhibition is not limited to FKHR and that androgens might be general suppressors of FOXO forkhead factors.
FIG. 3.

Inhibition of FKHRL1 activity by activated androgen receptor. (A) Inhibition of FKHRL1 activity by androgens in DU145 cells. Cells were transfected with 0.2 μg of pCMVhAR, 0.2 μg of pCMVβ, 0.5 μg of FHRE-Luc, and 0.1 μg of HA-FKHRL1-WT. Transfected cells were treated and FKHRL1 activity was determined as for Fig. 1B. (B) Inhibition of FKHRL1 activity by androgens through endogenous androgen receptor (AR) in LNCaP cells. Cells were transfected as for panel A except that HA-FKHRL1-MT was used in the place of HA-FKHRL1-WT and no pCMVhAR was included in the transfection. Transfected cells were treated and FKHRL1 activity was determined as for Fig. 1B.
PTEN reexpression in PTEN-null prostate cancer cells increases FKHR activity that is suppressed by activated androgen receptor.
As shown in our earlier studies (30), PTEN represses the transcriptional activity of the androgen receptor but is unable to override the protective effect of androgens on PTEN-induced apoptosis. It is thus possible that androgens protect prostate cancer cells from PTEN-induced death through inhibition of FKHR. If so, androgen repression of FKHR should occur despite the PTEN repression of androgen receptor transcriptional activity. Thus, we assayed the androgen repression of FKHR in PTEN-null PC3 cells transfected with wild-type PTEN.
As shown in Fig. 4, FKHR activity in PC3 cells is increased by wild-type PTEN in comparison to an inactive PTEN mutant, and the increase is repressed by R1881. As expected, PTEN and androgen receptor vectors express the corresponding proteins in the cells (Fig. 4B). The data suggest that the repression of FKHR by activated androgen receptor may contribute to the androgen receptor-mediated androgen protection of prostate cancer cells from PTEN-induced apoptosis.
FIG. 4.

Activated androgen receptor repressed PTEN-induced FKHR activity. (A) Inhibition of PTEN-induced FKHR activity by activated androgen receptor (AR). PC3 cells were transfected with 0.2 μg of pCMVhAR, 0.2 μg of pCMVβ, 0.5 μg of 3×IRS-Luc, 0.1 μg of pSG5L-HA-PTEN:WT (WT) or pSG5L-HA-PTEN:G129R (MT), and 0.1 μg of FKHR:WT (WT). Transfected cells were treated and FKHR activity was determined as for Fig. 1B. (B) Expression of PTEN and androgen receptor (AR) in PC3 cells. Cells were transfected and treated as for panel A. PTEN and androgen receptor proteins were detected by Western blotting with anti-PTEN (Upstate), PG-21 anti-androgen receptor, and anti-β-actin antibodies.
Androgen repression of FKHR is not due to competition for transcriptional coactivators.
Competition for the limited amounts of transcriptional coactivators in cells has been held accountable for the inhibition between AP-1 and steroid receptors (24) as well as among steroid receptors (40). To test whether the FKHR repression by activated androgen receptor is due to competition for coactivators, DU145 cells were transfected with various amounts of coactivators, and the repression of FKHR by activated androgen receptor was determined. As shown in Fig. 5A, expression of the coactivators at different dosages does not relieve the FKHR repression. As controls, the transcriptional activity of ERα (Fig. 5B) is increased by the increased dosages of the coactivators. In addition, different amounts of activated ERβ do not affect FKHR activity (Fig. 5C). Since steroid receptors share coactivators, activated ERβ would also be expected to repress FKHR activity if the repression were due to the competition for coactivators. Overall, the data in Fig. 5 indicate that the FKHR repression by androgens is not due to competition between the activated androgen receptor and FKHR for coactivators.
FIG. 5.

Lack of effect of coactivator expression on inhibition of FKHR by androgens and differential effect of activated ERα and ERβ on FKHR activity. (A) Lack of an effect of coactivator expression on the inhibition of FKHR by activated androgen receptor (AR). DU145 cells were transfected with 0.2 μg of pCMVhAR or control vector, 0.2 μg of pCMVβ, 0.5 μg of 3×IRS-Luc, 0.1 μg of FKHR:WT (WT), and the indicated amounts of coactivators. Transfected cells were treated and FKHR activity was determined as for Fig. 1B. (B) Enhancement of ERα activity by coactivators. DU145 cells were transfected with 0.1 μg of pLENhERα, 0.5 μg of pLENβgal, 0.5 μg of EREe1bLuc, and the indicated amounts of CBP, SRC1, or AIB1. Transfected cells were treated for 24 h with 10−8 M 17β-estradiol (E2) or ethanol (EOH) as vehicle controls. ERα activity was determined and expressed as for Fig. 1B. (C) Lack of an effect on FKHR activity of activated ERβ. DU145 cells were transfected with 0.5 μg of pLENβgal, 0.5 μg of 3×IRS-Luc, 0.1 μg FKHR:WT (WT) or FKHR:TSS (AAA), and 0.1 or 0.5 μg of pLENhERβ. Transfected cells were treated with 17β-estradiol or ethanol as for panel B, and FKHR activity was determined as for Fig. 1B. (D) Inhibition of FKHR activity by activated ERα. DU145 cells were transfected as for panel C except that 0.1 μg of pLENhERα was used in place of pLENhERβ. Transfected cells were treated with 17β-estradiol or ethanol, and FKHR activity was determined as for panel C. Co-A, coactivator.
Different from ERβ but similar to androgen receptor, ERα represses FKHR in the presence but not in the absence of 17β-estradiol (Fig. 5D), suggesting that FKHR repression is not limited to the androgen receptor. The data are consistent with two recent studies that identified FKHR as an ERα-interacting protein in yeast two-hybrid screening and in cells (43, 52).
Androgen receptor forms a complex with FKHR.
Since the repression of FKHR by androgens does not appear to be due to competition for coactivators, we next tested the possibility that activated androgen receptor represses FKHR through protein-protein interaction. DU145 cells were transfected with Flag-FKHR and androgen receptor vectors, and complex formation was determined by coimmunoprecipitation with M2 anti-Flag antibody. As shown in Fig. 6A, the androgen receptor is coimmunoprecipitated with FKHR only in cells transfected with both FKHR and the androgen receptor, and the coprecipitation is enhanced by R1881 (Fig. 6A, lanes 1 and 2). Without Flag-FKHR, the M2 anti-Flag antibody does not bring down androgen receptor in either the presence (Fig. 6A, lane 3) or the absence of R1881 (Fig. 6A, lane 4), demonstrating that there is no cross-reactivity between androgen receptor and M2 antibody.
FIG. 6.

Complex formation between FKHR and the androgen receptor in prostate cancer cells and in vitro. (A) Coimmunoprecipitation (IP) of FKHR and the androgen receptor (AR). DU145 cells were transfected with 0.5 μg of pCMVhAR, 0.5 μg of FKHR:WT (WT), and 0.2 μg of pCMVβ. Transfected cells were treated as for Fig. 1B. Immunoprecipitates with M2 anti-Flag antibody were probed with M2 anti-Flag (top panel) or PG-21 anti-androgen receptor (middle panel) antibodies. Androgen receptor (AR) protein expression in cell lysates was detected with the PG-21 anti-androgen receptor antibody (bottom panel). IB, immunoblot. (B) Interaction between FKHR and two separate regions of the androgen receptor (AR). 35S-labeled FKHR synthesized in in vitro transcription-translation reactions was incubated with GST-androgen receptor fusion proteins, precipitated with glutathione beads, and visualized by autoradiography (top panel). The amount of GST proteins used in the pulldown assays was visualized by Coomassie blue staining after SDS-PAGE (bottom panel). Note that the amount of reticulocyte lysate used for precipitations was 10 times the input amount. EOH, ethanol. (C) A diagram depicting the FKHR-interacting regions of the androgen receptor. aa, amino acids; DBD, DNA binding domain; LBD, ligand binding domain.
In cells transfected only with Flag-FKHR, no androgen receptor is detected in the precipitates (Fig. 6A, lanes 5 and 6), showing that the band detected in lane 1 is indeed androgen receptor. Since androgens are known to increase the stability of androgen receptor protein, which is confirmed under our conditions for R1881 (Fig. 6A, lanes 1 and 2 of the bottom panel), the increased amount of androgen receptor protein in the immunoprecipitates may be due simply to the higher amount of total androgen receptor protein in R1881-treated cells. Based on our estimation of the enhanced chemiluminescence signals on the immunoblots, the amount of the androgen receptor in the immunoprecipitates, after normalization to the total androgen receptor protein, is induced by R1881 about fourfold. This experiment shows that the androgen receptor and FKHR coexist in a protein complex in prostate cancer cells and that complex formation is increased by R1881.
To determine whether the androgen receptor and FKHR interact in vitro, GST fusions with the androgen receptor amino-terminal A/B region (GST-AR-NT), the DNA binding domain (GST-AR-DBD), or the ligand binding domain (GST-AR-LBD) were produced in and purified from bacteria. These fusion proteins were used to precipitate 35S-labeled FKHR protein produced by in vitro transcription-translation reactions. In these so-called GST pulldown assays, FKHR coprecipitates with GST-AR-NT (Fig. 6B, lane 2) but not with GST-DBD (Fig. 6B, lane 5). In addition, the coprecipitation could be detected with GST-AR-LBD in the presence (Fig, 6B, lane 4) but not in the absence of R1881 (Fig. 6B, lane 3). This demonstrates that androgen receptor interacts with FKHR through both the NT region and the ligand binding domain (Fig. 6C). No FKHR was detected in GST precipitates (Fig. 6B, lane 6), showing the specificity of FKHR coprecipitation with the androgen receptor fragments.
Compared to the input (Fig. 6B, lane 1), the NT region showed a much stronger interaction with FKHR than the liganded ligand binding domain. Coomassie blue staining shows that the amounts of GST and GST-AR-DBD used in the pulldown experiment are not lower than the amounts of the GST-AR-NT or GST-AR-LBD constructs and that the amount of GST-AR-LBD used for the pulldown assays performed in the presence or absence of R1881 is comparable (Fig. 6B, lower panel), eliminating the possibility that the difference in the FKHR signal in the pulldown assays is due to different amounts of GST protein used for the precipitations. Multiple bands smaller than the predicted size of the fusion protein are present in the GST-AR-NT and GST-AR-LBD lanes. Presumably, these are the products of partial degradation of the full-length GST fusion proteins. It is known that androgen receptor expressed in bacteria as GST fusion proteins is not stable, and so far nobody has succeeded in generating a GST fusion protein with a full-length androgen receptor.
To determine which region of FKHR interacts with the androgen receptor, we transfected DU145 cells with androgen receptor and FKHR fragments fused to the Gal4 DNA binding domain and determined the binding in coimmunoprecipitation assays with an anti-Gal4 antibody. As shown in Fig. 7, no interaction with the androgen receptor is detected with Gal4-FKHR(1-150) in either the absence or the presence of R1881, whereas interaction with the androgen receptor is detected with both Gal4-FKHR(211-655) and Gal4-FKHR(350-655) in the presence but not the absence of R1881. As controls, Gal4-FKHR proteins are detected in the immunoprecipitates (Fig. 7, middle panel), demonstrating that the lack of androgen receptor interaction with Gal4-FKHR fusions in the absence of R1881 as well as with Gal4-FKHR(1-150) in the presence of R1881 is not due to insufficient amounts of Gal4-FKHR protein expression.
FIG. 7.

Mapping the androgen receptor-interacting domain of FKHR by coimmunoprecipitation. DU145 cells were transfected with 0.5 μg of pCMVhAR, 0.5 μg of Gal4-FKHR, and 0.2 μg of pCMVβ and treated with R1881 or ethanol (EOH). Immunoprecipitates (IP) of anti-Gal4 antibody were probed with PG-21 anti-androgen receptor (AR) antibody (upper panel) or the same anti-Gal4 antibody (lower panel). Note that Gal4-FKHR(1-150), Gal4-FKHR(350-655), and IgG light chain run close to one another in the gel due to their similar molecular weights. After quantifying the intensity of the corresponding bands, the ratio of androgen receptor to Gal4-FKHR in the immunoprecipitates was plotted (bottom panel). IB, immunoblot; H-chain, heavy chain.
Based on the estimation of the enhanced chemiluminescence signals on the immunoblots, the amount of the androgen receptor in the immunoprecipitates, after normalization to the amount of Gal4-FKHR fusion proteins, is increased by R1881 more than 10-fold (Fig. 7, bottom panel). Although the quantitative data shown in Fig. 7, bottom panel, suggest an androgen induction of complex formation between the androgen receptor and FKHR, the data do not exclude the possibility that the increased complex formation detected in the presence of androgens is the result of androgen-induced accumulation of both androgen receptor and FKHR to a higher level in the cells.
Activated androgen receptor inhibits the ability of FKHR to bind DNA.
Since the androgen receptor and FKHR exist in the same protein complex and activated androgen receptor represses FKHR activity, we performed electrophoretic mobility shift assays to test whether activated androgen receptor inhibits the ability of FKHR to bind DNA. A synthetic oligonucleotide probe (8, 14, 47) containing an insulin response sequence from the IGFBP-1 promoter was used in the electrophoretic mobility shift assay. As shown in Fig. 8A, neither androgen receptor expression nor t treatment with R1881 decreased the level of FKHR expression, consistent with data presented earlier (Fig. 1E and 2C). Compared to the control (Fig. 8B, lane 2), androgen receptor coexpression does not decrease FKHR DNA binding in the absence of R1881 (Fig. 8B, lane 3, and 8C, lane 9), whereas, after R1881 treatment, the androgen receptor blocks the formation of FKHR-DNA complex (Fig. 8B, lane 4, and 8C, lane 10).
FIG. 8.

Androgen receptor-dependent inhibition of FKHR DNA binding by androgens. (A) Detection of transfected Flag-FKHR by immunoblotting. DU145 cells in 100-mm dishes were transfected with 2.5 μg of Flag-tagged FKHR or empty vector with or without 2.5 μg of pCMVhAR and treated with R1881 or ethanol (EOH). Then 50 μg of nuclear extracts of transfected cells was subjected to immunoblotting analysis with M2 anti-Flag antibody. Lane 1, transfected with control vectors; lane 2, transfected with Flag-FKHR; lane 3, transfected with both Flag-FKHR and androgen receptor; lane 4, transfected with Flag-FKHR and androgen receptor and treated with 10−8 M R1881. (B) Inhibition of FKHR DNA binding by activated androgen receptor. We used 10 μg of nuclear extracts for the electrophoretic mobility shift assay with radiolabeled insulin response sequence (IRS) probe as described in the text. The arrow points to the DNA complex containing Flag-FKHR protein. Lanes 1 to 4, as in panel A; lane 2b, as in lane 2, but the nuclear extract was incubated with M2 antibody. (C) Characterization of the FKHR-DNA complexes. DU145 cells were transfected as for panel A, and electrophoretic mobility shift assays performed as for panel B. Cotreatment with antibodies and an excess amount of nonradiolabeled wild-type (WT) and mutant (MT) insulin response sequence oligonucleotides was performed as indicated.
Interestingly, the unliganded androgen receptor appears to enhance the FKHR binding (Fig. 8B, lane 3, and 8C, lane 9), for which the mechanism is unclear. The specificity of the FKHR DNA complex is demonstrated by the inhibition of complex formation with the M2 anti-Flag antibody (Fig. 8B, lane 2b, and 8C, lane 2), but not with 12CA5 antihemagglutinin (anti-HA) antibody (Fig. 8C, lane 1). Further analysis shows that excess amounts of nonradiolabeled wild-type insulin response sequence (Fig. 8C, lanes 5 and 6), but not that of a mutant insulin response sequence (Fig. 8C, lanes 7 and 8), inhibit the complex formation, confirming the specificity of the FKHR complex. Although the activity of endogenous FKHR is detected in DU145 cells (Fig. 1B), no specific DNA binding is observed with nuclear extracts from DU145 cells transfected with control vector (Fig. 8B, lane 1, and Fig. 8C, lane 3), presumably due to the limited sensitivity of electrophoretic mobility shift assays.
Activated androgen receptor suppresses FKHR-induced Fas ligand expression.
Since activated androgen receptor inhibited the transcriptional activity of FKHR as well as its ability to bind DNA, it is reasonable to expect that the activated androgen receptor would suppress the expression of FKHR target genes such as Fas ligand. To test this possibility, DU145 cells were transfected with the mutant FKHR and an expression vector for a cell surface marker, CD20, with or without the androgen receptor. CD20-positive cells were separated from CD20-negative cells by flow cytometry-based sorting, and Fas ligand expression on the surface of the CD20-positive cells was analyzed. As shown in Fig. 9A and B, the expression of AAA in DU145 cells increases Fas ligand expression on the cell surface about fourfold, and the increase is suppressed by R1881 in the presence of androgen receptor. In the absence of AAA expression, androgens and androgen receptor, alone or in combination, have little effect on the basal level of Fas ligand on the cell surface. Similar analysis in LNCaP cells shows that the endogenous androgen receptor is sufficient to mediate the androgen inhibition of FKHR-induced Fas ligand expression (Fig. 9C).
FIG. 9.

Inhibition of FKHR-induced cell surface Fas ligand expression by activated androgen receptor. (A) Determination of cell surface Fas ligand expression by flow cytometry. DU145 cells were transfected with 3.0 μg of pCMVCD20 and 2.5 μg of FKHR:TSS (AAA) with or without 2.5 μg of pCMVhAR. Transfected cells were treated with 10−8 M R1881 or vehicle for 36 h. CD20-positive cells were separated from CD20-negative cells, and Fas ligand (FasL) expression on the surface of CD20-positive cells was determined. Similar data were obtained in multiple experiments, and a representative experiment is shown. Cy3, indocarbocyanine. (B) A bar graph showing the inhibition of FKHR-induced Fas ligand expression by activated androgen receptor (AR). (C) Inhibition of Fas ligand expression by androgens mediated through the endogenous androgen receptor in LNCaP cells. LNCaP cells were transfected with CD20 with or without AAA. Transfected cells were treated with R1881 or ethanol (EOH) as for panel A. The data from two independent experiments are presented as in panel B.
Activated androgen receptor impairs the ability of FKHR to induce prostate cancer cell apoptosis and cell cycle arrest.
Up to this point, our investigation has provided strong evidence that activated androgen receptor binds and inhibits FKHR transcriptional activity in prostate cancer cells as well as the ability of FKHR to bind DNA and induce target gene expression. To determine the effect of the inhibition on the biological activity of FKHR in prostate cancer cells, FKHR-induced cell apoptosis was analyzed in the presence and absence of R1881. LNCaP cells were transfected with wild-type FKHR or the AAA mutant together with a GFP expression vector, and the survival of transfected cells was examined. As shown in Fig. 10A, expression of the AAA mutant decreases LNCaP cell survival by fourfold, whereas the expression of wild-type FKHR has little effect. The decrease in cell survival induced by the AAA mutant is prevented by R1881, showing that the endogenous androgen receptor is sufficient to mediate the androgen protection.
FIG. 10.

Androgen protection of prostate cancer cells from FKHR-induced cell death. (A) Androgen effect on the viability of FKHR-transfected LNCaP cells. Cells were transfected with 0.5 μg of pLNCE and 0.1 μg of FKHR:WT (WT) or FKHR:TSS (AAA) and treated with R1881 or ethanol (EOH). The viability of transfected cells in each well was determined by counting the total number of green cells. Triplicate samples were analyzed for each data point, and the data were reproduced three times. (B) Representative micrographs of LNCaP cells transfected with FKHR:TSS. Cells were transfected with FKHR:TSS and GFP vectors as for panel A, fixed, and stained with DAPI. Representative micrographs were captured by a charge-coupled device camera attached to the fluorescence microscope. (C) Androgen effect on FKHR-induced increase in apoptotic index of LNCaP cells. Cells were transfected as for panel A and processed as for panel B. Apoptotic index of GFP-positive cells was determined by scoring 300 GFP-positive cells for chromatin condensation and nuclear fragmentation. Triplicate samples were analyzed per data point, and the graph represents three independent experiments. (D) Androgen effect on the viability of FKHR-transfected DU145 cells. Cells were transfected as for panel A but with or without 0.2 μg of pCMVhAR. Transfected cells were treated and determined for cell viability as for panel A. (E) Dosage-dependent protection of DU145 cells from FKHR-induced cell death by activated androgen receptor (AR). Cells were transfected as for panel D except that different amount of pCMVhAR were used, as indicated. Transfected cells were treated and cell viability was determined as for panel A.
To show that the decreased prostate cancer cell survival induced by FKHR is due to apoptosis, LNCaP cells transfected with the AAA mutant and GFP vectors were stained with DAPI and examined for features of cell apoptosis under a fluorescence microscope. As shown in representative micrographs in Fig. 10B, two of eight of the transfected cells are going through apoptosis, while none of the surrounding nontransfected cells are dying of apoptosis. After scoring 300 cells, the results suggest that the AAA mutant increases the apoptotic index of LNCaP cells by fourfold and that R1881 treatment restores the index to the basal level (Fig. 10C). In a parallel analysis, wild-type FKHR causes only a marginal increase in the apoptotic index, which is nevertheless also restored to the basal level by R1881. Consistent with the fact that PTEN is intact in DU145 cells, wild-type FKHR, like the AAA mutant, decreased the survival of DU145 cells (Fig. 10D). R1881 treatment restores the cell survival to the control level in cells cotransfected with the androgen receptor but showed no effect in the absence of androgen receptor, suggesting that the protection of prostate cancer cells from FKHR-induced death by androgens is androgen receptor dependent. By varying the amount of androgen receptor introduced into the cells, it is obvious that the ability of the androgen receptor to mediate the protection is dose dependent (Fig. 10E).
To extend the functional analysis beyond cell death, the effect of activated androgen receptor on the regulation of DU145 cell cycle progression by FKHR was determined with a strategy similar to what we have used for the Fas ligand study. DU145 cells were transfected with AAA, CD20, and androgen receptor, and cell cycle distribution of CD20-positive cells was analyzed by flow cytometry. As shown in Fig. 11, the expression of AAA in DU145 cells decreases the percentage of cells in S phase and increases the percentage in G1 phase about 15%. The effect of FKHR on both G1/G0 and S phases is suppressed by R1881 treatment. Similar analysis in LNCaP cells shows that the endogenous androgen receptor is sufficient to mediate the androgen suppression of FKHR-induced alteration in cell cycle progression, with the exceptions that FKHR induces the accumulation of LNCaP cells in G2/M instead of G1/G0 phase and that the effect of FKHR on LNCaP cell cycle progression is relatively minor (about 5%) (data not shown).
FIG. 11.
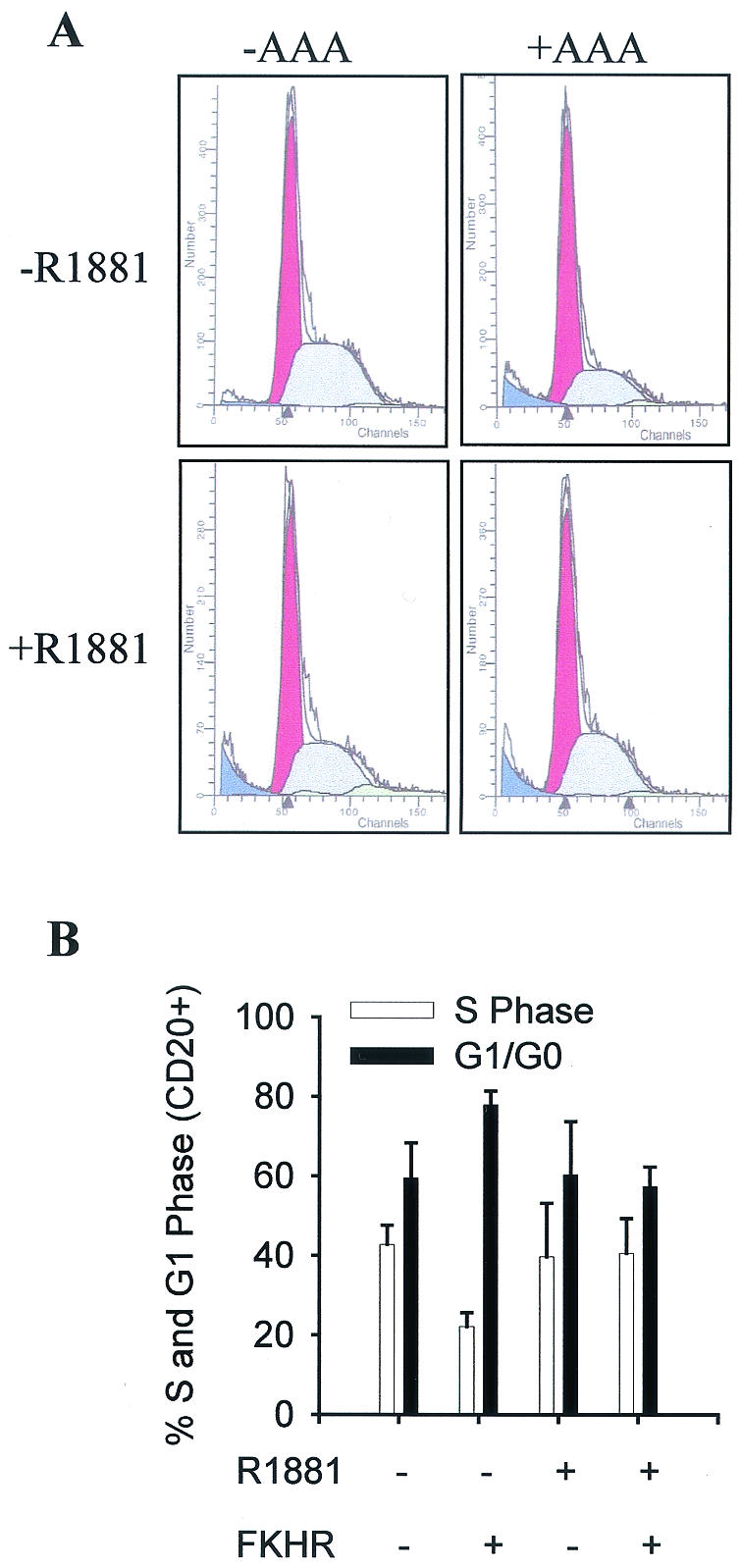
Inhibition of FKHR-induced cell cycle arrest by activated androgen receptor. (A) Flow cytometry profile of CD20-positive DU145 cells. DU145 cells were transfected with 3.0 μg of pCMVCD20, 2.5 μg of FKHR TSS (AAA), and 2.5 μg of pCMVhAR. Transfected cells were treated with 10−8 M R1881 for 36 h, fixed, and stained with propidium iodide. CD20-positive cells were separated from CD20-negative cells and subjected to flow cytometry analysis. Data from a representative experiment are shown. (B) A bar graph showing the inhibition of FKHR-induced cell cycle arrest by activated androgen receptor in DU145 cells. Each data point was analyzed in duplicate.
DISCUSSION
Due to the clinical relevance, the mechanism underlying prostate cancer cell death induced by androgen ablation is the continued focus of many investigations. A previous study has shown that the introduction of synthetic androgen response element oligonucleotides into LNCaP cells induces apoptosis (25), suggesting that androgen response element-mediated transcription may play an important role in prostate cancer cell survival. If androgen response element-mediated transcription were required for the androgen protection of prostate cancer cell death induced by PTEN, our previous data (30) that PTEN-induced prostate cancer cell apoptosis was blocked by activated androgen receptor while the androgen receptor transcriptional activity on androgen response element reporters was repressed by PTEN under the same conditions would appear paradoxical.
The ability of activated androgen receptor to suppress transactivation of FKHR through protein-protein interaction as demonstrated in the current study reveals a new mechanism for the androgen-mediated prostate cancer cell survival that appears to be independent of androgen response element-mediated transcription, which could explain why the effect of androgen receptor transcriptional activity on androgen response element-mediated reporters and its antiapoptotic function are separable (30). The facts that apoptosis induced by androgen ablation was reported to be impaired in Fas-null mice (lpr−/−) (46) and that FKHR-induced Fas ligand expression is blocked by activated androgen receptor, as shown in this study, suggest that the androgen response element-independent inhibition of FKHR through protein-protein interaction might be more relevant to the in vivo function of androgens in protecting prostate epithelial cells from apoptosis under physiological and pathological conditions.
The existence of androgen response element-dependent (25) and -independent mechanisms mediating androgen protection of prostate cancer cells from apoptosis suggests that prostate cancer cell survival in vivo may be the result of the cooperative actions of separate pathways that appear redundant in in vitro analyses. Alternatively, prostate cancer cells may have developed distinct pathways to mediate androgen protection from specific death stimuli. For example, androgen repression of FKHR activity may be specifically involved in the protection of prostate cancer cells from death induced by the PTEN tumor suppressor or other natural or synthetic inhibitors of the phosphoinositide 3-kinase/AKT signaling pathways. Although definitive proof that FKHR mediates PTEN-induced apoptosis in prostate cancer cells is still lacking, this possibility is supported by our data, which indicate that FKHR activity is induced by restored PTEN expression in PTEN-null cells (Fig. 4). It is also consistent with a recent report that correlated FKHR activity in prostate cancer cells with the functional status of the PTEN tumor suppressor (38).
Schuur et al. (43) recently reported that FKHR interacted specifically with the ligand binding domain of ERα but did not detect an interaction with androgen receptor under their conditions. Here, we demonstrate that the androgen receptor and FKHR exist in the same protein in prostate cancer cells and in vitro, which involves a possible androgen-regulated binding of FKHR with the ligand binding domain and a constitutive interaction with the NT region of the androgen receptor. Compared to the interaction with the NT region, the interaction with the androgen receptor ligand binding domain is weak, which may explain why previous studies failed to detect such an interaction (43). Since the activity of the NT region in full-length androgen receptor is also regulated by androgens, the interaction between FKHR and the NT region in the full-length receptor is likely to be regulated by androgens in a way similar to interaction with the ligand binding domain. This is consistent with our finding that the interaction between FKHR and full-length androgen receptor in coimmunoprecipitation assays is regulated by androgens (Fig. 6 and 7). The fact that, in addition to the androgen receptor, the activated ERα also inhibits FKHR activity is consistent with recent publications in which FKHR was identified as an ER-interacting protein in yeast two-hybrid screening and in cells (43, 52). Different from our findings that FKHR interacts strongly with androgen receptor NT region, the ER studies showed that it binds with the ER ligand binding domain. Since recent studies suggested that ERα and ERβ may have different roles in prostate tumorigenesis and that the ratio between them changes during prostate cancer development (17, 27), it is interesting that the suppressive effect of estrogens on FKHR is selectively mediated through the ERα but not the ERβ (Fig. 5).
Our data suggest that androgen-induced complex formation with androgen receptor prevents FKHR from binding to DNA response elements. Interestingly, the unliganded androgen receptor appears to be enhancing the binding of FKHR to the insulin response sequence (Fig. 8B and 8C), the reason for which is unclear. Since our data only showed an androgen enhancement of the interaction and did not rule out the binding of androgen receptor to FKHR in the absence of the ligand, it is possible that the mechanism underlying the inhibition of FKHR DNA binding by activated androgen receptor is more complicated than a quantitative change in protein-protein interaction. Androgens may induce a qualitative change in the interaction that switches stimulation to inhibition.
Besides the interaction with the carboxyl terminus of FKHR, we also detected a relatively weak interaction between the androgen receptor and the forkhead domain of FKHR that appears to be enhanced by androgens (data not shown). In addition, FKHR interacts strongly with the androgen receptor NT region but weakly with the androgen receptor ligand binding domain, although the inhibition of FKHR DNA binding by the androgen receptor is only detected in the presence of androgens (Fig. 8B and 8C). Although this is speculative, the carboxyl terminus of FKHR may interact with the NT region of the androgen receptor in the absence of the hormone, providing a docking site that stabilizes the weak interaction between the forkhead domain and the liganded androgen receptor ligand binding domain. Overall, the interaction detected in the in vitro assays is generally weak, leaving open the possibility that the interaction between androgen receptor and FKHR may be indirectly mediated through a “bridging” factor that also happens to be present in the rabbit reticulocyte lysates.
Since both the androgen receptor NT region and the ligand binding domain participate in the FKHR interaction, the androgen-induced conformational change in the androgen receptor three-dimensional structure, such as the interaction between amino and carboxyl terminuses (26), may play an important role in the inhibition of FKHR DNA binding. In addition to a transcriptional activation domain, the carboxyl-terminal region of FKHR contains a nuclear receptor interaction motif, LxxLL, frequently present in receptor coactivators, which may mediate the androgen receptor interaction. However, studies by He et al. (15) showed that the liganded androgen receptor ligand binding domain preferentially binds the FxxLF motifs versus the LxxLL motifs in the androgen receptor NT region and that the interaction between the amino and the carboxyl termini inhibits the binding of the androgen receptor ligand binding domain to LxxLL motifs of coactivators, raising the possibility that it may not play a major role in the FKHR interaction with the androgen receptor.
Overall, our study points to FOXO forkhead transcriptional factors as being the common targets for both AKT- and androgen-mediated prostate cancer cell survival. Furthermore, the fact that PTEN-intact DU145 cells do not die despite the lack of both active AKT and androgen receptor suggests the existence of an alternative mechanism in prostate cancer cells that opposes the negative effect of FOXO forkhead factors on cell growth and survival, which is obviously independent of androgens and AKT. Overall, our studies demonstrate that multiple survival signals converge on FOXO forkhead factors in prostate cancer cells, raising the possibility that augmenting the function of FKHR may provide an effective approach to overcome the resistance of prostate cancer cells to apoptosis induced by androgen ablation, tumor suppressors, and other natural or synthetic proapoptosis drugs used in prostate cancer therapy.
Acknowledgments
We thank Betty A. Bing for technical help, F. G. Barr for permission to use FKHR in the studies, K. L. Guan for the Flag-tagged wild-type and mutant FKHR plasmids, M. E. Greenberg for the FKHRL1 and FHRE-Luc plasmids, B. W. O'Malley and P. S. Meltzer for receptor coactivator plasmids, S. Mosselman for plasmid pKCRE.ERβ, and Paul Byvoet for reading the manuscript. Fas ligand expression and cell cycle were analyzed by the Flow Cytometry Core Facility at the H. Lee Moffitt Cancer Center and Research Institute.
The work was supported by Public Health Service grants R29 CA79530 (W.B.) and R01 CA93666 (W.B.) and DOD Prostate Cancer Idea Grant DAMD17-02-1-0140 (W.B.).
REFERENCES
- 1.Aarnisalo, P., J. J. Palvimo, and O. A. Janne. 1998. CREB-binding protein in androgen receptor-mediated signaling. Proc. Natl. Acad. Sci. USA 95:2122-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzick, S. L., J. Kononen, R. L. Walker, D. O. Azorsa, M. M. Tanner, X. Y. Guan, G. Sauter, O. P. Kallioniemi, J. M. Trent, and P. S. Meltzer. 1997. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277:965-968. [DOI] [PubMed] [Google Scholar]
- 3.Bevan, C. L., S. Hoare, F. Claessens, D. M. Heery, and M. G. Parker. 1999. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol. Cell. Biol. 19:8383-8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunet, A., A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson, K. C. Arden, J. Blenis, and M. E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857-868. [DOI] [PubMed] [Google Scholar]
- 5.Carson, J. P., G. Kulik, and M. J. Weber. 1999. Antiapoptotic signaling in LNCaP prostate cancer cells: a survival signaling pathway independent of phosphatidylinositol 3′-kinase and Akt/protein kinase B. Cancer Res. 59:1449-1453. [PubMed] [Google Scholar]
- 6.Chan, T. O., S. E. Rittenhouse, and P. N. Tsichlis. 1999. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68:965-1014. [DOI] [PubMed] [Google Scholar]
- 7.Chang, C., J. Kokontis, and S. Liao. 1988. Structural analysis of cDNA and amino acid sequences of human and rat androgen receptors. Proc. Natl. Acad. Sci. USA 85:7211-7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cichy, S. B., S. Uddin, A. Danilkovich, S. Guo, A. Klippel, and T. G. Unterman. 1998. Protein kinase b/akt mediates effects of insulin on hepatic insulin-like growth factor-binding Protein-1 gene expression through a conserved insulin response sequence. J. Biol. Chem. 273:6482-6487. [DOI] [PubMed] [Google Scholar]
- 9.Di Cristofano, A., P. Kotsi, Y. F. Peng, C. Cordon-Cardo, K. B. Elkon, and P. P. Pandolfi. 1999. Impaired Fas response and autoimmunity in Pten+/− mice. Science 285:2122-2125. [DOI] [PubMed] [Google Scholar]
- 10.Dijkers, P. F., R. H. Medema, J. W. Lammers, L. Koenderman, and P. J. Coffer. 2000. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 10:1201-1204. [DOI] [PubMed] [Google Scholar]
- 11.Evans, R. M. 1988. The steroid and thyroid hormone receptor superfamily. Science 240:889-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faber, P. W., G. G. Kuiper, H. C. van Rooij, J. A. van der Korput, A. O. Brinkmann, and J. Trapman. 1989. The N-terminal domain of the human androgen receptor is encoded by one large exon. Mol. Cell. Endocrinol. 61:257-262. [DOI] [PubMed] [Google Scholar]
- 13.Galili, N., R. J. Davis, W. J. Fredericks, S. Mukhopadhyay, F. J. Rauscher 3rd, B. S. Emanuel, G. Rovera, and F. G. Barr. 1993. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet.. 5:230-235. [DOI] [PubMed] [Google Scholar]
- 14.Guo, S., G. Rena, S. Cichy, X. He, P. Cohen, and T. G. Unterman. 1999. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274:17184-17192. [DOI] [PubMed] [Google Scholar]
- 15.He, B., J. T. Minges, L. W. Lee, and E. M. Wilson. 2002. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J. Biol. Chem. 277:10226-10235. [DOI] [PubMed] [Google Scholar]
- 16.Hong, H., K. Kohli, M. J. Garabedian, and M. R. Stallcup. 1997. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol. Cell. Biol. 17:2735-2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horvath, L. G., S. M. Henshall, C. S. Lee, D. R. Head, D. I. Quinn, S. Makela, W. Delprado, D. Golovsky, P. C. Brenner, G. O'Neill, R. Kooner, P. D. Stricker, J. J. Grygiel, J. A. Gustafsson, and R. L. Sutherl. 2001. Frequent loss of estrogen receptor-beta expression in prostate cancer. Cancer Res.. 61:5331-5335. [PubMed] [Google Scholar]
- 18.Irvine, R. A., H. Ma, M. C. Yu, R. K. Ross, M. R. Stallcup, and G. A. Coetzee. 2000. Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Hum. Mol. Genet. 9:267-274. [DOI] [PubMed] [Google Scholar]
- 19.Isaacs, J. T. 1994. Role of androgens in prostatic cancer. Vitamins Hormones 40:433-503. [DOI] [PubMed] [Google Scholar]
- 20.Jenster, G., J. Trapman, and A. O. Brinkmann. 1993. Nuclear import of the human androgen receptor. Biochem. J. 293:761-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenster, G., H. A. van der Korput, J. Trapman, and A. O. Brinkmann. 1995. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J. Biol. Chem. 270:7341-7346. [DOI] [PubMed] [Google Scholar]
- 22.Jenster, G., H. A. van der Korput, C. van Vroonhoven, T. H. van der Kwast, J. Trapman, and A. O. Brinkmann. 1991. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol. Endocrinol. 5:1396-1404. [DOI] [PubMed] [Google Scholar]
- 23.Kallio, P. J., H. Poukka, A. Moilanen, O. A. Janne, and J. J. Palvimo. 1995. Androgen receptor-mediated transcriptional regulation in the absence of direct interaction with a specific DNA element. Mol. Endocrinol. 9:1017-1028. [DOI] [PubMed] [Google Scholar]
- 24.Kamei, Y., L. Xu, T. Heinzel, J. Torchia, R. Kurokawa, B. Gloss, S. C. Lin, R. A. Heyman, D. W. Rose, C. K. Glass, and M. G. Rosenfeld. 1996. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85:403-414. [DOI] [PubMed] [Google Scholar]
- 25.Kuratsukuri, K., K. Sugimura, K. Harimoto, H. Kawashima, and T. Kishimoto. 1999. “Decoy” of androgen-responsive element induces apoptosis in LNCaP cells. Prostate 41:121-126. [DOI] [PubMed] [Google Scholar]
- 26.Langley, E., J. A. Kemppainen, and E. M. Wilson. 1998. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J. Biol. Chem. 273:92-101. [DOI] [PubMed] [Google Scholar]
- 27.Lau, K. M., M. LaSpina, J. Long, and S. M. Ho. 2000. Expression of estrogen receptor (ER)-alpha and ER-beta in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer Res. 60:3175-3182. [PubMed] [Google Scholar]
- 28.Lee, H., and W. Bai. 2002. Inhibition of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol. Cell. Biol. 22:5835-5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, H., F. Jiang, Q. Wang, S. V. Nicosia, J. Yang, B. Su, and W. Bai. 2000. MEKK1 activation of human estrogen receptor, α., and stimulation of the agonistic activity of 4-hydroxytamoxifen in endometrial and ovarian cancer cells. Mol. Endocrinol. 14:1882-1896. [DOI] [PubMed] [Google Scholar]
- 30.Li, P., S. V. Nicosia, and W. Bai. 2001. Antagonism between PTEN/Mmac1/Tep-1 and androgen receptor in growth and apoptosis of prostate cancer cells. J. Biol. Chem. 276:20444-20450. [DOI] [PubMed] [Google Scholar]
- 31.Lubahn, D. B., D. R. Joseph, M. Sar, J. Tan, H. N. Higgs, R. E. Larson, F. S. French, and E. N. M. Wilson. 1988. The human androgen receptor: complementary deoxyribonucleic acid cloning, sequence analysis and gene expression in prostate. Mol. Endocrinol. 2:1265-1275. [DOI] [PubMed] [Google Scholar]
- 32.Lundblad, J. R., R. P. Kwok, M. E. Laurance, M. L. Harter, and R. H. Goodman. 1995. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional coactivator CBP. Nature 374:85-88. [DOI] [PubMed] [Google Scholar]
- 33.Lu, S., S. Y. Tsai, and M. J. Tsai. 1997. Regulation of androgen-dependent prostatic cancer cell growth: androgen regulation of CDK2, CDK4, and CKI p16 genes. Cancer Res. 57:4511-4516. [PubMed] [Google Scholar]
- 34.McEwan, I. J., and J. Gustafsson. 1997. Interaction of the human androgen receptor transactivation function with the general transcription factor TFIIF. Proc. Natl. Acad. Sci. USA 94:8485-8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medema, R. H., G. J. Kops, J. L. Bos, and B. M. Burgering. 2000. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404:782-787. [DOI] [PubMed] [Google Scholar]
- 36.Mosselman, S., J. Polman, and R. Dijkema. 1996. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. 392:49-53. [DOI] [PubMed] [Google Scholar]
- 37.Nakae, J., V. Barr, and D. Accili. 2000.. Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 19:989-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura, N., S. Ramaswamy, F. Vazquez, S. Signoretti, M. Loda, and W. R. Sellers. 2000. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell. Biol. 20:8969-8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogg, S., S. Paradis, S. Gottlieb, G. L. Patterson, L. Lee, H. A. Tissenbaum, and G. Ruvkun. 1997. The fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389:994-999. [DOI] [PubMed] [Google Scholar]
- 40.Onate, S. A., S. Y. Tsai, M. J. Tsai, and B. W. O'Malley. 1995. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270:1354-1357. [DOI] [PubMed] [Google Scholar]
- 41.Phan, D., X. Sui, D. T. Chen, S. M. Najjar, G. Jenster, and S. H. Lin. 2001. Androgen regulation of the cell-cell adhesion molecule-1 (Ceacam1) gene. Mol. Cell. Endocrinol. 184:115-123. [DOI] [PubMed] [Google Scholar]
- 42.Ramaswamy, S., N. Nakamura, F. Vazquez, D. B. Batt, S. Perera, T. M. Roberts, and W. R. Sellers. 1999. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA 96:2110-2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuur, E. R., A. V. Loktev, M. Sharma, Z. Sun, R. A. Roth, and R. J. Weigel. 2001. Ligand-dependent interaction of estrogen receptor-alpha with members of the forkhead transcription factor family. J. Biol. Chem. 276:33554-33560. [DOI] [PubMed] [Google Scholar]
- 44.Spencer, T. E., G. Jenster, M. M. Burcin, C. D. Allis, J. Zhou, C. A. Mizzen, N. J. McKenna, S. A. Onate, S. Y. Tsai, M. J. Tsai, and B. W. O'Malley. 1997. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 389:194-198. [DOI] [PubMed] [Google Scholar]
- 45.Stambolic, V., A. Suzuki, J. L. de la Pompa, G. M. Brothers, C. Mirtsos, T. Sasaki, J. Ruland, J. M. Penninger, D. P. Siderovski, and T. W. Mak. 1998. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29-39. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki, A., A. Matsuzawa, and T. Iguchi. 1996. Down regulation of Bcl-2 is the first step on Fas-mediated apoptosis of male reproductive tract. Oncogene 13:31-37. [PubMed] [Google Scholar]
- 47.Tang, E. D., G. Nunez, F. G. Barr, and K. L. Guan. 1999. Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274:16741-16746. [DOI] [PubMed] [Google Scholar]
- 48.Tang, T. T., D. Dowbenko, A. Jackson, L. Toney, D. A. Lewin, A. L. Dent, and L. A. Lasky. 2002.. The forkhead transcription factor AFX activates apoptosis by induction of the BCL-6 transcriptional repressor. J. Biol. Chem. 277:14255-14265. [DOI] [PubMed] [Google Scholar]
- 49.Tilley, W. D., M. Marcelli, J. D. Wilson, and M. J. McPhaul. 1989. Characterization and expression of a cDNA encoding the human androgen receptor. Proc. Natl. Acad. Sci. USA 86:327-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai, M. J., and B. W. O'Malley. 1994. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 63:451-486. [DOI] [PubMed] [Google Scholar]
- 51.Yeh, S., and C. Chang. 1996. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc. Natl. Acad. Sci. USA 93:5517-5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao, H. H., R. E. Herrera, E. Coronado-Heinsohn, M. C. Yang, J. H. Ludes-Meyers, K. J. Seybold-Tilson, Z. Nawaz, D. Yee, F. G. Barr, S. G. Diab, P. H. Brown, S. A. Fuqua, and C. K. Osbo. 2001. Forkhead homologue in rhabdomyosarcoma functions as a bifunctional nuclear receptor-interacting protein with both coactivator and corepressor functions. J. Biol. Chem. 276:27907-27912. [DOI] [PubMed] [Google Scholar]