

Abstract
In eukaryotic cells, DNA mismatch repair is initiated by a conserved family of MutS (Msh) and MutL (Mlh) homolog proteins. Mlh1 is unique among Mlh proteins because it is required in mismatch repair and for wild-type levels of crossing over during meiosis. In this study, 60 new alleles of MLH1 were examined for defects in vegetative and meiotic mismatch repair as well as in meiotic crossing over. Four alleles predicted to disrupt the Mlh1p ATPase activity conferred defects in all functions assayed. Three mutations, mlh1-2, -29, and -31, caused defects in mismatch repair during vegetative growth but allowed nearly wild-type levels of meiotic crossing over and spore viability. Surprisingly, these mutants did not accumulate high levels of postmeiotic segregation at the ARG4 recombination hotspot. In biochemical assays, Pms1p failed to copurify with mlh1-2, and two-hybrid studies indicated that this allele did not interact with Pms1p and Mlh3p but maintained wild-type interactions with Exo1p and Sgs1p. mlh1-29 and mlh1-31 did not alter the ability of Mlh1p-Pms1p to form a ternary complex with a mismatch substrate and Msh2p-Msh6p, suggesting that the region mutated in these alleles could be responsible for signaling events that take place after ternary complex formation. These results indicate that mismatches formed during genetic recombination are processed differently than during replication and that, compared to mismatch repair functions, the meiotic crossing-over role of MLH1 appears to be more resistant to mutagenesis, perhaps indicating a structural role for Mlh1p during crossing over.
In eukaryotes, mismatch repair plays a critical role in mutation avoidance and is carried out by the MutSLH family of proteins (for reviews, see references 13, 36, and 40). During vegetative growth, these proteins recognize and bind DNA mispairs that result primarily from replication errors or DNA damage. In Escherichia coli, MutS binding to DNA mispairs results in the recruitment of MutL, a matchmaker protein that functions in postreplicative mismatch repair by interacting with both the MutH endonuclease and UvrD helicase (22, 23). These interactions coordinate mispair recognition with DNA strand-specific signals so that mispairs are removed via excision and resynthesis steps that occur on the newly replicated strand.
Eukaryotes contain multiple MutS (Msh) and MutL (Mlh) homologs, with six Msh and four Mlh homologs present in Saccharomyces cerevisiae (36). Genetic and biochemical studies have shown that the eukaryotic homologs display specialized functions with respect to the types of DNA substrates on which they act (10, 36, 40). In S. cerevisiae, the Mlh proteins form heterodimers (Mlh1p-Pms1p, Mlh1p-Mlh3p, and Mlh1p-Mlh2p) that display unique functions. Mlh1p is considered a central member of this group because heterodimers have not been identified among the other members (45, 70). The Mlh1p-Pms1p complex plays a major role in postreplicative mismatch repair, while the other two Mlh complexes appear to be redundant with Mlh1p-Pms1p and are required for the repair of only a limited set of DNA mispairs (18, 27).
Yeast mutants lacking Mlh1p or Pms1p display spontaneous mutation rates that are much higher than that of the wild type, and their mutations are epistatic to mutations deleting Msh2p, a central player in mispair recognition (36). Like MutL, Mlh1p-Pms1p has been shown to bind and hydrolyze ATP in a reaction that drives conformational changes thought to be important for mismatch repair (26, 50, 63, 64). Also like MutL, Mlh1p-Pms1p has been shown to bind DNA (25), though it is less clear how this activity functions in its matchmaking role.
While the role of E. coli MutL has been relatively well characterized, our understanding of the mechanistic steps employed by the Mlh proteins is still in the early stages. As hypothesized for a matchmaking protein, Mlh1p has been shown to physically interact with Exo1p, a 5′-3′ double-stranded DNA exonuclease that is thought to act in excision steps of mismatch repair (65). Consistent with a role in mismatch repair, high copy numbers of Exo1p suppress the mutator phenotype of specific mismatch repair mutants; however, exo1 mutants do not display a mismatch repair-like mutator phenotype (5, 6, 59, 62, 65). Physical interactions have also been reported between Mlh1p and BLM/Sgs1p, a DNA helicase that has been hypothesized to repair stalled replication forks (37, 48). Mammalian cells defective in BLM/Sgs1p display a chromosome instability phenotype but do not display defects in mismatch repair (37), suggesting that this helicase may not be required in mismatch repair but acts with Mlh proteins in other repair pathways.
In addition to mismatch repair, Msh and Mlh proteins have novel meiotic recombination functions. Genetic studies in yeast and mammalian cells have shown that the Msh4p-Msh5p and Mlh1p-Mlh3p complexes play important roles in meiotic crossing over (30, 32, 54, 70; reviewed in reference 10). Yeast mutants lacking any one of these factors display approximately half the number of meiotic crossover events; in these mutants, spore viability is reduced as a result of nondisjunction events in meiosis I (reviewed in reference 10). MLH1-deficient mice display a more severe crossover defect and are sterile (72). In addition to its role in crossing over, Msh4p is required for establishing crossover interference (43). Msh4p and Mlh1p interact physically and genetically (32, 56), suggesting that MutS and MutL homologs might function together to mediate crossing over in a mechanism that is still unclear.
Recent findings in yeast meiosis suggest that noncrossover (gene conversion) and crossover recombinants form through sequential and distinct pathways (4, 31). These studies proposed a model in which recombination is initiated through the formation of single-end invasion structures that later mature into double Holliday junction intermediates that can be resolved into crossovers. In this model, however, large portions of the single-ended invasions are processed to noncrossovers without ever forming stable Holliday junction intermediates. Msh4p has been hypothesized to bind Holliday junctions (30, 54); such a function could be important in stabilizing the Holliday junction intermediates proposed in such a model.
The role of MLH1 in mismatch repair has been studied primarily through the use of deletion and site-specific mutations; however, only the deletion mutation has been analyzed in meiotic crossing over. Furthermore, most site-specific mutations in MLH1 have been created in amino-terminal residues which have been suggested by crystallographic analysis to be important in ATP binding and/or hydrolysis (9, 26, 50, 63, 64). This approach has been limited by the fact that only the first 349 residues of MutL have been crystallized and that the approximately 400-amino-acid carboxy-terminal regions of Mlh proteins show modest sequence homology. Previous studies have focused on the mismatch repair aspect of MLH1 function because of its implication in hereditary nonpolyposis colorectal cancer (17, 44) but a comprehensive characterization of the meiotic functions of MLH1 has yet to be pursued.
In this study, we employed systematic mutagenesis of MLH1 with the goal of identifying regions required for mismatch repair and meiotic functions. This analysis identified both previously studied and uncharacterized regions of MLH1 that are required for mismatch repair and crossing over. In addition, we isolated separation-of-function alleles that conferred defects in postreplicative mismatch repair but did not disrupt meiotic crossing over. We also identified mutants that were functional for meiotic but not vegetative mismatch repair, suggesting that these repair processes are distinct.
MATERIALS AND METHODS
S. cerevisiae strains.
The SK1 and S288C strain backgrounds were used to analyze the effect of mlh1 mutations in mismatch repair and in meiotic crossing over (Table 1). Strains HTY1212 and HTY1213, congenic with SK1, were kindly provided by H. Tsubouchi and H. Ogawa (66). mlh1Δ (EAY841 and EAY844) and mlh1Δ mlh3Δ (EAY975 and EAY976) derivatives of the HTY strains were constructed via single-step gene replacement. The mlh1Δ::hisG (6) and mlh1Δ::KanMX4 alleles each contain only the amino-terminal 12 amino acids of the 769-amino-acid MLH1 coding region. The mlh3Δ::KanMX4 allele lacks all but 25 amino acids of the 715-amino-acid MLH3 coding region. The single-step mlh1Δ and mlh3Δ disruption plasmids and the oligonucleotide primers used to confirm the disruptions by PCR are available upon request. The remaining strains listed in Table 1 are derived from the MGD (S288C) background (52) and were generated by either single-step gene replacement or genetic cross.
TABLE 1.
Diploid strains used in the meiotic analysis of mlh1 mutantsa
| Strain | Genotype | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||||
|
|
|
|
||||||||||||
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|||||||||
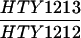

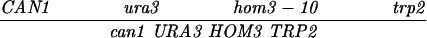
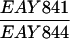

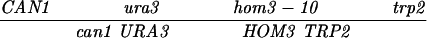


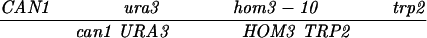

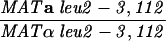
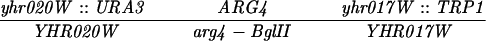
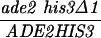
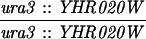
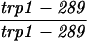
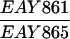
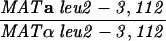
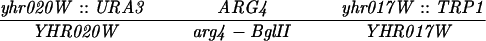
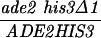
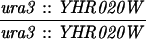
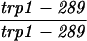
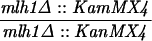
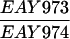
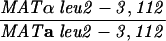
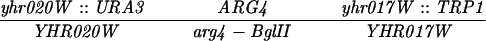
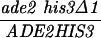
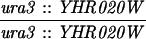
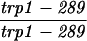
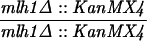
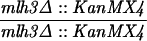
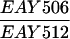
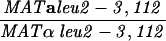
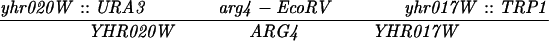
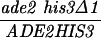
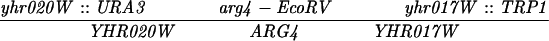
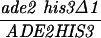
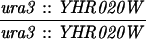
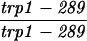
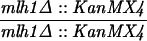
The diploid strains used in tetrad analysis were created by mating the indicated MATa and MATα strains. Markers grouped above and under a single line are located on the same chromosome and were used in the crossing-over studies. HTY1212 and HTY1213 as well as derivatives EAY841, EAY844, EAY975, and EAY976 are congenic with SK1 (66). EAY492, EAY502, EAY506, and EAY512 as well as derivatives EAY861, EAY865, EAY869, EAY874, EAY973, and EAY974 are derived from the MGD (S288C) strain background (6, 52). The arg4-BglII marker contains a 4-bp insertion at position 1274 in ARG4 relative to the initiating ATG, and the arg4-EcoRV marker contains a 2-bp deletion at 260.
The mlh1Δ strains shown in Table 1 were transformed with either pRS415 (LEU2, ARSH4, and CEN6) (14), pEAA109 (MLH1, LEU2, ARSH4, and CEN6) (6), or pEAA109 derivatives containing the site-specific mlh1 mutations. pEAA109 contains a genomic MLH1 clone derived from the S288C background. In control studies, we found that wild-type strains and mlh1Δ strains transformed with pEAI109 were indistinguishable with respect to mismatch repair and meiotic phenotypes for both the S288C and SK1 strain backgrounds (data not shown).
Yeast strains were grown in either yeast extract-peptone-dextrose (YPD) or minimal selective medium (53). When required, canavanine (Sigma) was included in minimal selective medium at 60 mg/liter, and geneticin (Gibco) was included in YPD at 200 mg/liter (69). 5-Fluoroorotic acid (U.S. Biologicals) plates were prepared as described previously (53), and sporulation plates and procedures were as described previously (15).
Mutagenesis.
pEAA109 was mutagenized with the QuickChange kit (Stratagene) to create the 60 mlh1 alleles listed in Table 2. Oligonucleotides were purchased from MWG Biotech. Approximately 500 bp of DNA surrounding each cluster mutation was sequenced. The entire open reading frame, including approximately 200 bp of DNA upstream and downstream, was sequenced for the 16 mlh1 alleles which conferred the strongest defects in either the mismatch repair or spore viability assay. DNA sequencing was performed by the Cornell Bioresource Facility.
TABLE 2.
mlh1 substitutions and phenotypic classification
| MLH1 allele | Residues mutated to alaninea | Structural featureb | Summary of phenotypec
|
|
|---|---|---|---|---|
| Vegetative DNA mismatch repair | Meiotic crossing over | |||
| MLH1 | + | + | ||
| mlh1Δ | − | − | ||
| mlh1-4 | R4, K6 | Amino-terminal tail (S) | ± | + |
| mlh1-5 | K30, E31 | ATP I, hydrolysis (B) | − | − |
| mlh1-6 | K49, E50 | S | + | + |
| mlh1-7 | K67, D69 | Near ATP II (S) | − | ± |
| mlh1-8 | E75, R76 | Amino-heterodimerization (S) | ± | ± |
| mlh1-9 | K84, E86, D87 | S | ± | − |
| mlh1-10 | R97, E99 | ATP III (B) | − | − |
| mlh1-11 | K117, E118, D119, R120 | S | + | + |
| mlh1-12 | E129, K131 | S | + | + |
| mlh1-13 | K142, D143 | Near ATP IV (S) | + | + |
| mlh1-14 | E150, D151 | Near ATP IV (S) | ± | − |
| mlh1-15 | D168, E169 | Near ATP binding groove (B) | ± | ± |
| mlh1-16 | K185, D186 | S | + | + |
| mlh1-17 | K192, K193 | B | ± | + |
| mlh1-19 | K232, E234, D235 | S | + | + |
| mlh1-20 | D242, K244 | 242 (S), 244 domain interface (B) | + | + |
| mlh1-1 | K253, K254 | Near DNA binding groove (S) | − | − |
| mlh1-21 | R273, R274 | DNA binding groove (S) | − | − |
| mlh1-22 | K311, R312, E313 | Near ATP and domain interface (B) | − | − |
| mlh1-23 | D320, E321 | S | + | + |
| mlh1-24 | E324, K325 | B | + | + |
| mlh1-25 | K352, E354 | + | + | |
| mlh1-26 | E364, D366, R367 | + | + | |
| mlh1-27 | R369, K370 | + | + | |
| mlh1-28 | R390, K391 | ± | + | |
| mlh1-29d | K393, R394 | − | + | |
| mlh1-30 | E396, K398 | + | + | |
| mlh1-31 | R401, D403 | − | + | |
| mlh1-32 | K427, R428 | + | + | |
| mlh1-33 | E432, K434 | + | + | |
| mlh1-34 | E443, E445, K446 | + | + | |
| mlh1-35 | E451, E453 | + | + | |
| mlh1-36 | R456, D457 | + | + | |
| mlh1-37 | D463, D465 | + | + | |
| mlh1-38 | K467, D468 | + | + | |
| mlh1-39 | K471, K472, K473 | + | + | |
| mlh1-40 | D478, K480 | + | + | |
| mlh1-41 | D486, D487, E488, K489 | + | + | |
| mlh1-42 | K496, D497 | + | + | |
| mlh1-43 | K504, E505, R506 | ± | + | |
| mlh1-44 | K515, K516 | ± | + | |
| mlh1-45 | R518, E519, K520 | ± | + | |
| mlh1-46 | D522, D523 | ± | − | |
| mlh1-47 | R527, E528 | + | + | |
| mlh1-2 | D543, E544, E545, R546, R547 | − | ± | |
| mlh1-48 | D554, K556 | ± | + | |
| mlh1-49 | D593, D594 | ± | + | |
| mlh1-50 | E603, D605, E606 | ± | + | |
| mlh1-51 | D609, D610 | + | + | |
| mlh1-52 | K613, E614, K615 | + | + | |
| mlh1-54 | K648, K650 | ± | + | |
| mlh1-55 | K675, E676 | + | + | |
| mlh1-56 | E680, D681, E682 | − | − | |
| mlh1-57 | R691, E692 | + | + | |
| mlh1-58 | K704, D706 | + | + | |
| mlh1-3 | E714, D715, E716, K717 | + | + | |
| mlh1-59 | R723, K724, E725 | + | + | |
| mlh1-60 | K740, R741, R742 | − | ± | |
| mlh1-61 | K751, D752 | + | + | |
| mlh1-62 | E767, R768 | − | ± | |
Residues in Mlh1p that were changed to alanine. Underlined residues are those conserved in the crystal structure of E. coli MutL's LN40 fragment.
Putative structural features based on analogy to LN40 structure. B indicates residues buried inside the structure, and S indicates residues that are found on the structure's surface. ATP I to IV indicate the four conserved ATP binding motifs common among MutL, NgyrB, and Hsp90 (8). Domain interface refers to the interface between the amino and carboxy domains of the LN40 structure.
The vegetative mismatch repair phenotype classification was based on results from the canavanine patch assay: +, alleles that displayed up to threefold more papillations than the wild type; ±, alleles that conferred between 3- and 10-fold more papillations; −, alleles that conferred more than 10 fold more papillations. The meiotic crossing-over phenotype classification was a composite of spore viability and crossing over measured from a small number of tetrads. Spore viability: +, higher than 85%; ±, between 85% and 77%, −, less than 77%.
Bold indicates separation-of-function alleles that were characterized in detail.
Mutator assays.
For the semiquantitative canavanine resistance assay (Fig. 1), mlh1Δ strains containing pRS415 (mutant control), pEAA109 (wild-type control), or pEAA109 (mlh1 site-specific mutations) were streaked to leucine dropout plates to obtain single colonies. Eleven independent colonies were individually patched to minimal plates containing canavanine and incubated for 3 days at 30oC. The number of canavanine-resistant papillations in each patch was counted, and the median number of the 11 patches was recorded. Median numbers obtained from at least four repetitions were averaged to obtain the data presented for each mutant (Fig. 1). The rates of hom3-10 reversion, forward mutation to canavanine resistance, and dinucleotide repeat tract instability were calculated from the median mutation frequency by the method of Lea and Coulson (38). Reversion of hom3-10 to Thr+ was tested in EAY841, the forward mutation rate to canavanine resistance was measured in EAY841 and EAY861, and the rate of repeat tract instability (28) was determined in EAY774 (MATa mlh1Δ::hisG lys2-BglII leu2Δ-1 trp1Δ63 ura3-52 his3Δ; S288C background).
FIG. 1.

Functional organization of Mlh1p (A) and Pms1p (B). The vertical lines indicate the amino acid positions of the MLH1 alanine scan mutations, with the height of each line corresponding to the mutation frequency relative to the wild-type residue as measured in a canavanine resistance patch assay (Materials and Methods). The numbers directly above the vertical lines identify the MLH1 mutations specifically discussed in the text. The arrows indicate the mutation frequency for EAY841 containing pEAA109 (wild-type, 1×) and pRS415 (mlh1Δ, 16×). The amino acid substitutions indicated below Mlh1p and above Pms1p indicate the polymorphisms that exist between the S288C (first letter) and SK1 (second letter) strain backgrounds. The 447CEGT polymorphism corresponds to a four-residue insertion between residues 447 and 448 in the S288C sequence of Pms1p. The white bars indicate conserved ATP binding domain motifs (8), and the hatched areas indicate the Mlh1p-Pms1p interaction domain (45). The asterisks in MLH1 and PMS1 correspond to a residue in MutL which has been implicated in DNA binding (9).
Yeast two-hybrid analysis.
Target EXO1, SGS1, and PMS1 plasmids used in the two-hybrid analysis were generously provided by the Liskay, Kleckner, and Stagljar laboratories. The mlh1 alleles were subcloned into the bait LexA-Mlh1 vector pBTM-yMLH1 (45). The target MLH3 plasmid (pEAM98) contains a fusion between the Gal4p activation domain in pGAD10 and residues 481 to 715 of Mlh3p (70). The L40 strain used for two-hybrid analysis (68) was first transformed with plasmids carrying GAL4 activation domain fusions to PMS1, EXO1, SGS1, or MLH3, followed by transformation with pBTM-yMLH1 or mlh1 derivatives. Expression of the lacZ reporter gene was determined by color filter assays as described before (45).
Ternary complex formation assay.
Msh2p-Msh6p-DNA complexes were examined in gel retardation assays in the presence and absence of Mlh1p-Pms1p and ATP. Mlh1p-Pms1p and Msh2p-Msh6p were purified as described previously (1, 24). All protein concentrations were determined with the Bradford reagent (Bio-Rad) and bovine serum albumin (Sigma) as the standard. Binding reactions were performed at room temperature for 5 min in 20-μl reaction mixes containing 100 nM Msh2p-Msh6, 0 to 64 nM Mlh1p-Pms1p, 100 nM 32P-labeled +1 substrate (48 bp) (12), 1 mM ATP (Pharmacia), 20 mM HEPES (pH 7.6), 40 μg of bovine serum albumin per ml, 7% sucrose, and 25 mM NaCl. Samples were electrophoresed at 130 V for 1 h at room temperature in 4% (wt/vol) nondenaturing polyacrylamide gels containing 0.5× TBE (Tris-borate-EDTA) buffer. Gels were dried and then visualized with the Phosphor Imaging system and analyzed with Imagequant (Molecular Dynamics).
Meiotic analysis.
The diploid strains outlined in Table 1 were sporulated with the zero growth mating protocol as previously described (6, 51) with the exception that each mlh1Δ haploid parent was transformed with the appropriate control or pEAA109 allele plasmid prior to mating. Tetrads were dissected on YPD or minimal complete plates after zymolyase treatment. After 3 or 4 days of growth at 30°C, spore clones were replica plated onto relevant selective plates and incubated at 30°C. Aberrant segregations were scored 1 day after replica plating, and sectored colonies were confirmed by microscopic examination. In the case of MGD-derived strains, tetrads with aberrant segregations at ADE2 or HIS3 were discarded to eliminate possible false tetrads.
Genetic map distances were determined by the formula of Perkins (49). The expected number of nonparental ditype (NPD) tetrads was calculated for each interval with the equation of Papazian (46), and the ratio of observed and expected NPDs was examined as described before. Only four-spore viable asci that displayed Mendelian segregation for relevant markers were included in the calculation. Mapping and NPD ratios were evaluated with software available at the Stahl Laboratory Online Tools website (http://groik.com/stahl/). NPD ratios and aberrant segregation data were evaluated with a chi-squared test, and P values lower than 0.05 were considered significant.
RESULTS
Rationale for mutagenesis and phenotypic analysis.
We mutagenized MLH1 by substituting clusters of charged residues with alanines (Table 2, Fig. 1). This approach allowed mutagenesis of a large number of residues (142 out of 769) with the expectation that protein-protein interactions would involve solvent-exposed residues. This idea was tested by projecting the N-terminal Mlh1p domain onto the MutL LN40 crystal structure (8). About half of the 21 N-terminal mlh1 alleles contained substitutions in residues that were identical in Mlh1p and MutLp. With structure-viewing software, we found that residues in 15 of the 21 alleles aligned to surface-exposed residues in MutL (http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml).
The mismatch repair and meiotic functions conferred by each mlh1 allele were assessed in SK1 strains (Table 1) with a semiquantitative mutator assay and tetrad analysis, respectively. This allowed us to tentatively classify each allele as functional, intermediate, null, or separation of function. To investigate the meiotic phenotypes conferred by the alanine scan mutations, at least 60 tetrads were dissected from each mutant strain. While this small number of tetrads was insufficient to accurately calculate map distances, the spore viability data gave a reliable estimate of MLH1 meiotic function that was later confirmed in detailed tetrad analysis (Table 3; see Fig. 4).
TABLE 3.
Genetic map distances and crossing-over interference in wild-type and mlh1 SK1 and S288C strainsa
| Genetic interval | Relevant genotype | Distance (centimorgans) | No. of tetrads
|
No. of NPD expected | NPD ratio | P | ||
|---|---|---|---|---|---|---|---|---|
| PD | TT | NPD | ||||||
| CAN1-URA3 (SK1) | MLH1 | 35.8 ± 1.3 | 434 | 686 | 22 | 106 | 0.21 ± 0.05 | ≪0.001 |
| mlh1Δ | 23.6 ± 1.1 | 732 | 488 | 16 | 34 | 0.47 ± 0.12 | 0.002 | |
| mlh3Δ | 26.0 ± 1.4 | 516 | 376 | 16 | 28 | 0.57 ± 0.14 | 0.02 | |
| mlh1-2 | 32.0 ± 1.9 | 221 | 273 | 8 | 33 | 0.24 ± 0.08 | ≪0.001 | |
| mlh1-2 mlh3Δ | 19.3 ± 2.0 | 89 | 56 | 0 | 4 | 0.0 | 0.04 | |
| mlh1-29 | 36.3 ± 2.0 | 186 | 300 | 10 | 47 | 0.21 ± 0.07 | ≪0.001 | |
| mlh1-31 | 32.8 ± 1.9 | 187 | 247 | 7 | 32 | 0.22 ± 0.08 | ≪0.001 | |
| URA3-HOM3 (SK1) | MLH1 | 36.3 ± 1.6 | 500 | 596 | 38 | 68 | 0.56 ± 0.09 | ≪0.001 |
| mlh1Δ | 25.9 ± 1.2 | 693 | 515 | 20 | 40 | 0.51 ± 0.12 | 0.002 | |
| mlh3Δ | 26.7 ± 1.5 | 516 | 368 | 19 | 27 | 0.70 ± 0.17 | 0.12 | |
| mlh1-2 | 31.8 ± 2.0 | 234 | 261 | 10 | 29 | 0.35 ± 0.11 | ≪0.001 | |
| mlh1-2 mlh3Δ | 25.9 ± 3.3 | 80 | 63 | 2 | 5 | 0.39 ± 0.28 | 0.17 | |
| mlh1-29 | 34.4 ± 1.8 | 196 | 295 | 8 | 44 | 0.18 ± 0.07 | ≪0.001 | |
| mlh1-31 | 37.7 ± 2.7 | 188 | 235 | 16 | 28 | 0.58 ± 0.15 | 0.02 | |
| HOM3-TRP2 (SK1) | MLH1 | 13.3 ± 0.8 | 847 | 269 | 5 | 10 | 0.51 ± 0.23 | 0.11 |
| mlh1Δ | 11.3 ± 0.7 | 964 | 258 | 3 | 8 | 0.38 ± 0.21 | 0.08 | |
| mlh3Δ | 12.8 ± 1.0 | 690 | 199 | 5 | 7 | 0.76 ± 0.34 | 0.45 | |
| mlh1-2 | 15.2 ± 1.0 | 348 | 152 | 0 | 7 | 0.0 | 0.008 | |
| mlh1-2 mlh3Δ | 13.4 ± 1.8 | 106 | 39 | 0 | 2 | 0.0 | 0.16 | |
| mlh1-29 | 13.8 ± 1.3 | 368 | 124 | 2 | 5 | 0.42 ± 0.30 | 0.18 | |
| mlh1-31 | 17.7 ± 1.8 | 307 | 124 | 5 | 6 | 0.90 ± 0.41 | 0.68 | |
| ADE2-HIS3 (S288C) | MLH1 | 36.2 ± 1.3 | 453 | 733 | 24 | 116 | 0.21 ± 0.04 | ≪0.001 |
| mlh1Δ | 22.5 ± 1.0 | 638 | 443 | 8 | 32 | 0.25 ± 0.08 | ≪0.001 | |
| mlh1-2 | 34.5 ± 1.0 | 725 | 1,016 | 35 | 138 | 0.25 ± 0.04 | ≪0.001 | |
| mlh1-2 mlh3Δ | 24.7 ± 3.3 | 85 | 61 | 2 | 5 | 0.44 ± 0.31 | 0.18 | |
| mlh1-29 | 35.4 ± 1.9 | 158 | 275 | 6 | 48 | 0.12 ± 0.05 | ≪0.001 | |
| mlh1-31 | 35.7 ± 1.0 | 668 | 1,079 | 32 | 171 | 0.19 ± 0.03 | ≪0.001 | |
| URA3-TRP1 (S288C) | MLH1 | 7.8 ± 0.7 | 511 | 94 | 0 | 2 | 0.0 | 0.16 |
| mlh1Δ | 4.3 ± 0.6 | 440 | 41 | 0 | 0 | 0.0 | 1 | |
| mlh1-2 | 6.3 ± 0.6 | 688 | 100 | 0 | 2 | 0.0 | 0.16 | |
| mlh1-2 mlh3Δ | 4.6 ± 1.2 | 127 | 13 | 0 | 0 | 0.0 | 1 | |
| mlh1-29 | 8.7 ± 0.9 | 345 | 73 | 0 | 2 | 0.0 | 0.16 | |
| mlh1-31 | 7.7 ± 0.7 | 662 | 114 | 1 | 2 | 0.43 ± 0.43 | 0.48 | |
Map distances were calculated according to the formula of Perkins (49), and the number of expected NPDs was calculated with the formula of Papazian (46). Standard errors for map distances and NPD ratios were calculated with software available at the Stahl Laboratory Online Tools website (http://groik.com/stahl/). P values derived from χ2 analysis indicate the probability that the difference between the observed and expected number of NPDs was due to chance. PD, parental ditype tetrads; TT, tetratype tetrads; NPD, nonparental ditype tetrads.
FIG. 4.

Distribution of viable spores in SK1 tetrads. The distribution of classes containing 0 to 4 spores per tetrad is presented. Above each graph is the overall spore viability (spo. viab.) and the total number of tetrads dissected from each genotype.
Structure-function analysis of MLH1.
The effects of the mlh1 mutations on mismatch repair were determined with the canavanine patch assay (Fig. 1). The 60 alleles were distributed into three classes, with 33 functional, 15 intermediate, and 12 defective for mismatch repair. This analysis matched well with previously characterized functional regions. First, all four of the alleles (mlh1-5, -7, -10, and -14) located very near or within conserved ATP binding/hydrolysis domain motifs conferred severe mismatch repair defects (9, 17, 44, 64). Tetrad analysis of these alleles showed that the Mlh1p ATPase was also required for meiotic functions (Table 2); such studies had not been done previously.
In addition, four mlh1 mutations (mlh1 E31K, D60N, G95D, and G98S) that corresponded to dominant negative alleles of mutL (7) and mapped to the Mlh family ATP binding domain, also conferred null phenotypes in all genetic assays (data not shown). These observations are consistent with the Mlh1p ATP binding/hydrolysis domain's playing a role in both mismatch repair and meiotic crossing over (8, 26, 64). Second, 11 alleles localizing to the C-terminal Pms1p interaction domain conferred mutator phenotypes. Third, mlh1-62, which maps beyond the Pms1p interaction domain, conferred a strong mutator phenotype that was similar to that conferred by a small deletion in this region (45).
Our analysis also supported important roles for two less well characterized regions of Mlh1p and identified a third region that appeared to be insensitive to mutagenesis. First, the region between residues 253 and 313 contains three consecutive alleles (mlh1-1, -21, and -22), each of which conferred a mutator phenotype. The corresponding region in MutL contains residues which have been implicated in DNA binding (9). Projection of the residues that were substituted in the mlh1-21 allele onto the MutL LN40 structure suggested that they map within the putative DNA binding groove (9). Recent studies have also identified mutations in this region that were functionally important in mismatch repair (5, 6, 17, 58). Second, a novel functional region was detected between residues 390 and 403 (mlh1-28, -29, and -31), which contains three alleles that conferred a strong mutator phenotype. Sia and colleagues (58), in a screen for new mismatch repair factors, identified mlh1-I409N, which maps very close to our alleles. Interestingly, none of the alleles in this region were found to affect the meiotic functions of Mlh1p (see below). Third, no mutations between amino acids 427 and 497 conferred a mutator phenotype despite the relatively high number of targeted mutation sites. Secondary-structure prediction analysis indicated that residues 350 to 500 form a random coil (33); this suggests that deletion mutations might be required to determine the role of this region.
As shown in Fig. 4, mlh1 alleles functional for crossing over conferred high spore viability, while the mlh1Δ mutation conferred lower viability and displayed a meiosis I chromosome nondisjunction pattern in which 0, 2, and 4 viable spore tetrads predominated (54). Only two alleles, mlh1-22 and -46, conferred a low spore viability that was comparable to that of the mlh1Δ strain. In all other cases, the alleles conferred a more severe mutator phenotype than the meiotic defect, and alleles displaying a more severe meiotic phenotype relative to the mutator phenotype were not observed. Therefore, mismatch repair functions were more easily disrupted then meiotic functions. This suggests that the catalytic role of Mlh1p may be more important during mismatch repair, whereas during crossing over, Mlh1p is playing an important structural role, perhaps in the context of a larger protein complex (see the Discussion).
Characterization of separation-of-function mutations in mutator assays.
Three mismatch repair-negative, meiosis-positive separation-of-function alleles, mlh1-2, -29, and -31, were chosen for detailed analysis. While the mlh1-2 allele belongs to a group of intermediate separation-of-function alleles (-2, -7, -60, and -62), it was chosen for this analysis because it contains mutations that localize to the Pms1p interaction domain (45). Strains bearing these alleles were analyzed for forward mutation to canavanine resistance, dinucleotide repeat tract instability, and reversion of the hom3-10 (+1 frameshift) Thr− phenotype. As shown in Fig. 2, the mlh1-2 and mlh1-31 alleles conferred null phenotypes in all mutator assays. Surprisingly, the mlh1-29 allele, mutagenized from an S288C clone, conferred a null phenotype in mutator assays performed in the SK1 strain background but conferred a nearly wild-type phenotype in the S288C strain background.
FIG. 2.

Relative mutation rates of mlh1 alleles. Forward mutation to canavanine resistance (A) and hom3-10 reversion (B and C) are presented for the SK1-derived strain (EAY841), and forward mutation to canavanine resistance (D, EAY874) and repeat tract instability (E, EAY774) are presented for the S288C strains. Mutation rates were determined by the method of the median (at least seven cultures per experiment) (38). Presented are the averages and standard deviations of three independent repetitions relative to the wild-type control (mlh1Δ transformed with pEAA109). The rates for the wild-type controls were as follows: 3.2 × 10−6 (A), 7.2 × 10−7 (B), 2.3 × 10−6 (D), and 9.8 × 10−6 (E). In panel C, all strains contained both the MLH1 experimental plasmid and an ARS-CEN-URA3 plasmid bearing the PMS1 gene from S288C (pJH480). In this experiment, the rate of hom3-10 reversion is presented relative to that of EAY841 containing pEAA109 and pJH480 (2.5 × 10−7).
We investigated the strain-specific mutator phenotype of the mlh1-29 allele by conducting segregation analysis with hybrid diploids formed by mating SK1 and S288C strains bearing the mlh1-29 allele. None of 100 tetrads dissected produced four viable spores, suggesting chromosome segregation incompatibilities between the two strain backgrounds. However, random spore clones displayed an approximately 1:1 (27:24) ratio between mutators and nonmutators in the canavanine patch assay, suggesting that a single gene in SK1 was responsible for the mlh1-29 mutator phenotype (J. Wanat, unpublished observations). It is important to note that the hybrid diploid strain did not display a mutator phenotype, indicating that the SK1 locus responsible for the mlh1-29 mutator phenotype was recessive (data not shown).
We then tested whether known mismatch repair components such as PMS1 or MSH2 could suppress the mlh1-29 SK1 mutator phenotype. As shown in Fig. 2C, a single copy of the S288C-derived PMS1 gene dramatically reduced the hom3-10 reversion rate in the mlh1-29 SK1 strain to nearly wild-type levels (12-fold above the wild-type level, compared to 263-fold above the wild-type level for mlh1Δ), while MSH2 did not influence the phenotype of mlh1-29 (data not shown). This observation encouraged us to sequence the MLH1 and PMS1 genes from the SK1-derived strain HTY1212 (Fig. 1). We found six polymorphisms in Mlh1p and eight in Pms1p, including a four-residue insertion. These polymorphisms were scattered across the two open reading frames, with some mapping near previously identified domains. Two other alleles, mlh1-56 and mlh1-60, conferred an SK1-specific mutator phenotype which could also be suppressed by a single copy of the S288C PMS1 gene (J. Heck, unpublished information). Together, this information suggests that the amino acid differences between the SK1 and S288C mismatch repair components could exacerbate the mutant phenotype of the mlh1 alleles. An attractive possibility is that the SK1 and S288C substitutions are functionally compensatory.
Two-hybrid and biochemical characterization of separation-of-function alleles.
A two-hybrid analysis was performed to test whether the separation-of-function phenotypes conferred by the mlh1-2, -29, and -31 mutations resulted from impaired protein-protein interactions between Mlh1p and known interactors (Pms1p, Mlh3p, Sgs1p, and Exo1p). As shown in Fig. 3A, mlh1-29 and mlh1-31 strains were functional for all interactions; however, mlh1-2 mutant strains displayed defects in both Mlh1p-Pms1p and Mlh1p-Mlh3p interactions, while Mlh1p-Sgs1p and Mlh1p-Exo1p interactions were unaffected.
FIG. 3.

Two-hybrid and biochemical analysis of MLH1 mutations. (A) Two-hybrid interactions between lexA-mlh1 and GAL4-PMS1, -MLH3, -EXO1, and -SGS1 fusion constructs. Plates overlaid with paper filters were incubated at 30°C, and expression of the lacZ reporter gene was determined (Materials and Methods). (B) Purification of Msh2p-Msh6p and mutant Mlh1p-Pms1p complexes. Approximately 4 pmol of Msh2p-Msh6p, Mlh1p, and the indicated Mlh1p-Pms1p complexes were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (8% gel). Lane M, size standards (in kilodaltons). The gels were visualized with Coomassie blue. (C) Ternary complex formation involving Msh2p-Msh6p, Mlh1p-Pms1p, and +1 mismatch substrate. Binding reactions and gel retardation assays were performed as described in Materials and Methods.
We tested the ability of the mutant Mlh1 proteins to form complexes with Pms1p. An intein-chitin tag was fused to Mlh1p, and the Mlh1p-Pms1p complex was purified on a chitin affinity column as previously described (24). As shown in Fig. 3B, the mlh1-29 and mlh1-31 mutations did not affect the stability of Mlh1p or its ability to interact with Pms1p. In contrast, the mlh1-2 mutation did not affect Mlh1p stability but disrupted Mlh1-2p-Pms1p complex formation. Western blot analysis confirmed that Pms1p was absent from the purified Mlh1-2p fraction, yet was expressed at a similar level in mlh1-2 and wild-type Mlh1p whole-cell extracts (data not shown).
Recently, Shcherbakova et al. (57) observed that Mlh1p overexpression in wild-type strains conferred a dominant negative phenotype. They hypothesized that this phenotype was due to Mlh1p homodimers interfering with the assembly of a functional mismatch repair complex. One possibility is that the mlh1-2 mutation did not disrupt Pms1p interactions, as suggested above, but instead promoted Mlh1-2p homodimerization. In such a model, Pms1p overexpression in mlh1-2 strains would be expected to drive complex formation towards a functional heterodimeric complex and thus attenuate the mutator phenotype. We found, however, that the mlh1-2 mutator phenotype was not rescued by 2μm overexpression of PMS1 through either the PMS1 or GAL10 promoter (data not shown). This observation, together with the finding that the high levels of meiotic crossing over observed in mlh1-2 strains required MLH3 (see below), argues against a preferential self-dimerization model.
We also tested the ability of these mutations to affect the formation of a ternary complex in native acrylamide gels containing +1 mismatch substrate, Msh2p-Msh6p, and Mlh1p-Pms1p (Materials and Methods). Ternary complex formation is thought to reflect an early step in the mismatch repair process (11, 21). As shown in Fig. 3C, Mlh1-29p-Pms1p and Mlh1-31p-Pms1p complexes were completely functional in the ternary complex assay. This result was expected for Mlh1-29p, considering that all proteins used were derived from S288C strains and mlh1-29 strains are not defective in mismatch repair in this background. The result with Mlh1-31p is more intriguing because this mutation conferred a strong mutator phenotype. Taking these results together with the two-hybrid data, we favor a model in which mlh1-31 causes defects in signaling steps that occur after ternary complex formation (see the Discussion).
Characterization of separation-of-function mutations in spore viability and meiotic crossing-over assays.
Diploid strains bearing the mlh1-2, -29, and -31 alleles were analyzed in depth for meiotic defects. Figure 4 shows the spore viability data for the mlh1 alleles in the SK1 strain background. Spore viability results in the S288C background were similar with the exception of the mlh1-29 strain, which displayed a completely wild-type phenotype (data not shown). As expected, the wild-type strain displayed high spore viability (92%), while the mlh1Δ (68%) and mlh3Δ (77%) strains displayed patterns favoring tetrads with four, two, and zero viable spores; this bias reflects meiosis I nondisjunction events due to reduced crossing over (70). In contrast, strains bearing each of the three mlh1 alleles displayed high spore viability (83 to 88%) and did not display the mlh1Δ spore viability distribution.
Previous studies have indicated that defects in mismatch repair have a negative influence on spore viability, presumably due to the accumulation of recessive mutations in diploids (2, 51). For example, the msh2Δ and pms1Δ mutations, which confer mismatch repair defects but do not appear to affect meiotic crossing over, typically cause a reduction in overall spore viability to about 80%, but this reduction is not associated with a bias in spore viability distribution (2). We hypothesize that the mild reduction in spore viability seen in mlh1-2, mlh1-29, and mlh1-31 strains results from their mutator phenotypes during vegetative growth.
Meiotic crossing over was measured in SK1 strains at the CAN1-URA3, URA3-HOM3, and HOM3-TRP2 intervals and in the S288C strains at the ADE2-HIS3 and URA3-TRP1 intervals (Table 3). Statistical analysis of the mapping data was performed with the Stahl Laboratory online tools website (http://groik.com/stahl/). Map distances were considered significant when at least one of two criteria were met: the absolute value of the difference between the two map distances was greater than twice the standard error (1), or there was no overlap between map distance plus/minus standard error between two genotypes (2). Unless noted, both criteria were met.
As shown in Table 3, significant differences in map distance were observed between the wild-type and mlh1Δ and mlh3Δ strains for all intervals with the exception of HOM3-TRP2, which was shown previously to be insensitive to other mutations that affect meiotic crossing over (43, 66). For these intervals, the mlh1Δ and mlh3Δ mutations conferred an approximately 40% reduction in map distance. All three mlh1 strains, however, were competent in meiotic crossing over. For all intervals, mlh1-29 and mlh1-31 strains displayed map distances that were statistically indistinguishable from that of the wild-type. mlh1-2 strains displayed a minor crossover defect. Map distances in mlh1-2 strains were not significantly different from those in MLH1 strains when the first statistical test was applied. However, for the CAN1-URA3, URA3-HOM3, and URA3-TRP1 intervals, the map distance values for the mlh1-2 and MLH1 strains did not overlap within their standard errors but were separated by less than 1 centimorgan (second statistical test). Compared to mlh1Δ strains, mlh1-2 strains displayed map distances that were significantly different for all relevant intervals.
Because mlh1-2 strains displayed nearly wild-type levels of crossing over but defective Mlh1p-Mlh3p two-hybrid interactions, we tested whether the high levels of crossing over in mlh1-2 strains occurred through an MLH3-independent mechanism. As shown in Table 3 and Fig. 4, mlh1-2 mlh3Δ double mutants displayed spore viability and crossing-over patterns that were indistinguishable from those of mlh1Δ or mlh3Δ mutants, indicating that the meiotic viability observed in mlh1-2 strains was dependent on MLH3 function despite the apparent defect in Mlh1p-Mlh3p interactions observed in the two-hybrid assay.
Separation-of-function mutants are functional for mismatch repair during genetic recombination.
Previously, MLH1 was shown to be required to repair DNA mispairs that form during meiotic recombination at the HIS4 and ARG4 loci (6, 32). The ARG4 locus has often been used to monitor mismatch repair during meiosis because it displays high levels of meiotic gene conversion that are thought to result primarily from the repair of DNA mismatches in heteroduplex DNA (3, 42, 61, 71). The persistence of DNA mispairs is inferred from a sectored colony phenotype seen in spore clones derived from strains heterozygous for mutations in auxotrophic markers. These events are referred to as postmeiotic segregations.
Recombination at ARG4 displays a gene conversion gradient in which high frequencies of non-Mendelian segregation (aberrant events) are observed for genetic markers located near a meiotically induced double-strand break site located within the ARG4 promoter, while lower levels are observed for markers located further away (16, 20, 39, 42, 55,). In strains defective in the MSH2, MLH1, and PMS1 genes, approximately half of aberrant segregation events at loci such as ARG4 and HIS4 are detected as postmeiotic segregations (2, 6, 32, 67).
Table 4 shows the results from tetrad analysis of wild-type and mlh1 mutants performed in the S288C background. Strains were constructed so that they were heterozygous for ARG4 markers located at the high (arg4-EcoRV) or low (arg4-BglII) end of the ARG4 conversion gradient (Table 1) (42). Postmeiotic segregation events (sum of events at arg4-EcoRV and arg4-BglII) were rare in the wild type (3.8% of aberrant events) but frequent in mlh1Δ strains (52.5%). Postmeiotic segregation events were also rare in mlh1-31 strains (7.9%, χ2 = 0.88, P ≫ 0.05, indistinguishable from the wild-type value), while mlh1-2 strains displayed an intermediate level of postmeiotic segregation (21.1%) that was distinct from that of both MLH1 (χ2 = 8.2, P < 0.01) and mlh1Δ (χ2 = 21.5, P < 0.01) strains. These results indicate that the mlh1-31 and mlh1-2 strains were at least partially competent for mismatch repair in meiosis. It is important to note that the mlh1-29 strains displayed a postmeiotic segregation phenotype that was indistinguishable from that of the wild type; this result was expected because the mlh1-29 allele appears to be fully functional in the S288C background that was used in this experiment.
TABLE 4.
Aberrant segregations at the ARG4 locus in wild-type and mlh1 S288C diploidsa
| ARG4 marker | Genotype | Total no. of tetrads | % Aberrant | No. of tetrads in each class of aberrant segregation
|
% Postmeiotic segregations (total no. of aberrants) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 6:2 | 2:6 | 5:3 | 3:5 | Ab4:4 | 0:8 | |||||
| arg4-EcoRV | MLH1 | 575 | 6.8 | 29 | 9 | 0 | 1 | 0 | 0 | 2.5 (39) |
| mlh1Δ | 558 | 10.4 | 21 | 4 | 24 | 8 | 1 | 0 | 56.9 (58) | |
| mlh1-2 | 942 | 6.3 | 27 | 21 | 6 | 5 | 0 | 0 | 18.6 (59) | |
| mlh1-31 | 965 | 4.4 | 16 | 22 | 3 | 1 | 0 | 0 | 9.5 (42) | |
| arg4-BglII | MLH1 | 635 | 2.2 | 8 | 5 | 1 | 0 | 0 | 0 | 7.1 (14) |
| mlh1Δ | 531 | 7.7 | 17 | 4 | 15 | 2 | 2 | 1 | 46.3 (41) | |
| mlh1-2 | 834 | 5.4 | 21 | 12 | 10 | 1 | 0 | 1 | 24.4 (45) | |
| mlh1-29 | 439 | 1.4 | 3 | 3 | 0 | 0 | 0 | 0 | 0 (6) | |
| mlh1-31 | 818 | 2.6 | 10 | 10 | 0 | 1 | 0 | 0 | 4.7 (21) | |
Aberrant events include all tetrads that could not be classified as 2 Arg+:2 Arg− (19).
In addition to displaying a postmeiotic segregation phenotype, mismatch repair mutations alter the conversion gradient at ARG4 so that the frequency of aberrant events is raised to a greater extent at the low end of the gradient compared to the high end (2, 6, 16). Several models have been proposed to explain this effect. In one model, the gradient forms as the result of repairing mispairs near the double-strand break to gene conversions and repairing mispairs far from the double-strand break to restorations (16). Other models have invoked the formation of conversion gradients through heteroduplex DNA rejection (2, 29, 42). As shown in Table 4, both MLH1 and mlh1Δ strains displayed ARG4 aberrant events at frequencies similar to those found in previous studies (MLH1, 6.8% at EcoRV versus 2.2% at BglII, P < 0.001; mlh1Δ, 10.4% at EcoRV versus 7.7% at BglII, P > 0.05) (6). mlh1-31 strains displayed an aberrant segregation frequency at the BglII marker that was similar to that of the wild type (2.6% for mlh1-31 versus 2.2% for MLH1, P > 0.05); the frequency at the EcoRV marker was only marginally different (4.4% for mlh1-31 versus 6.8% for MLH1, P = 0.04). These data, combined with wild-type levels of postmeiotic segregations and crossing over in mlh1-31 strains, suggest that this mutation does not affect MLH1 functions in meiosis.
In mlh1-2 strains, roughly the same aberrant segregation frequency was observed at both markers (6.3% at EcoRV, 5.4% at BglII, P > 0.05). Compared to MLH1, mlh1-2 strains displayed a higher frequency of aberrant events only at the low end of the gradient (mlh1-2 versus MLH1 at BglII, P < 0.01). Compared to mlh1Δ, these strains displayed a lower frequency of aberrant events only at the high end of the gradient (mlh1-2 versus mlh1Δ at EcoRV, P < 0.01). Together, these results suggested that mlh1-2 disrupted the ARG4 conversion gradient to a greater extent than mlh1Δ (see Discussion).
mlh1Δ strains display crossover interference at three genetic intervals.
In S. cerevisiae, biochemical and genetic analyses have implicated Mlh1p-Mlh3p and Msh4p-Msh5p complexes in meiotic crossing over (30, 32, 54, 70). In addition, analysis of msh4 mlh1 double mutants suggested that these proteins all act in the same crossover pathway (32). In wild-type S. cerevisiae undergoing meiosis, two crossovers rarely occur within the same genetic interval; this phenomenon is known as crossover interference. Interference is calculated by the difference between the number of double crossovers predicted from the frequency of single crossovers compared to the number of double crossovers observed (NPDs). Recently, MSH4 has been shown to be important for crossover interference in S. cerevisiae (43). In msh4Δ diploids, the number of NPD tetrads was often close to the number expected in the absence of interference, producing an NPD observed/expected ratio of approximately 1.0 at five intervals, including the CAN1-URA3, URA3-HOM3, and HOM3-TRP2 intervals that we examined (43).
The tetrad analysis presented in Table 3 allowed us to examine whether MLH1 plays a role similar to MSH4 in establishing crossover interference. Interference was examined in both SK1 and S288C strains. The SK1 strains contain three of the same genetic intervals and are isogenic to the ones used in the MSH4 study (43). mlh1Δ diploids displayed appreciable levels of crossover interference; mlh3Δ diploids showed a similar trend, but the data were less conclusive because fewer tetrads and intervals were examined, and statistical significance was only observed at CAN1-URA3 (P = 0.02).
As shown in Table 3, mlh1Δ strains produced NPD ratios that were significantly smaller than 1.0 at three genetic intervals (CAN1-URA3, URA3-HOM3, and ADE2-HIS3) and were similar to wild-type ratios at two of them (URA3-HOM3 and ADE2-HIS3). A higher NPD ratio was observed in the mlh1Δ strain at the CAN1-URA3 interval, but the ratio was significantly below 1.0. Accurate measurements of interference could not be made at HOM3-TRP2 and URA3-TRP1 because NPD tetrads were observed at low frequency in these short intervals. It is important to note that mlh1Δ and mlh3Δ strains displayed a 1:2:1 ratio of single crossovers involving two, three, and four chromatids at the CAN1-URA3-HOM3 interval, indicating an absence of chromatid interference like that observed in wild-type yeast strains (Table 5) (41). Together, these studies suggest that Mlh1p may stimulate crossing over in a different way than Msh4p. A detailed analysis of crossover interference in mlh3 strains has been carried out by N. Hunter and N. Kleckner (personal communication) with analogous conclusions.
TABLE 5.
Analysis of chromatid interference at the CAN1-URA3-HOM3 interval in wild-type and mlh1 SK1 strainsa
| Relevant genotype | No. of tetrads with:
|
P | ||
|---|---|---|---|---|
| Two strands | Three strands | Four strands | ||
| MLH1 | 84 | 179 | 95 | 0.71 |
| mlh1Δ | 49 | 89 | 50 | 0.76 |
| mlh3Δ | 33 | 77 | 45 | 0.26 |
| mlh1-2 | 38 | 70 | 26 | 0.30 |
| mlh1-2 mlh3Δ | 7 | 13 | 2 | 0.22 |
| mlh1-29 | 43 | 76 | 50 | 0.32 |
| mlh1-31 | 33 | 66 | 25 | 0.46 |
All tetrads containing tetratypes at both CAN1-URA3 and URA3-HOM3 are shown. P values derived from χ2 analysis indicate the probability that the number of tetrads with exchanges involving two, three, and four chromatids follows a 1:2:1 distribution.
DISCUSSION
The alanine scanning mutagenesis described in this paper provides a comprehensive view of the functional organization of MLH1. Mutations that disrupt known functions of MLH1 were identified, and separation-of-function mutations were found in which mismatch repair functions of MLH1 were compromised without disrupting meiotic crossing over. Mutations affecting mismatch repair were also mapped to previously uncharacterized regions. In addition, three alleles that conferred strain-specific phenotypes were identified, and a large region was found to be insensitive to mutagenesis. This analysis should provide a useful tool for the study of Mlh protein functions in higher eukaryotes. The identification of separation-of-function alleles in S. cerevisiae, for example, should make it easier to create fertile mice that are specifically defective in MLH1 mismatch repair functions. Similar separation-of-function mlh1 mutations have been identified independently (29a).
ATP binding-hydrolysis domain of Mlh1p is required for meiotic function, while a second domain appears insensitive to mutagenesis.
A large number of studies have analyzed the effect of ATP binding-hydrolysis domain mutations on the mismatch repair functions of Mlh family proteins (26, 50, 60, 63, 64). These studies suggest that ATP binding and/or hydrolysis is important for triggering conformational changes within Mlh proteins that are required in downstream mismatch repair steps. For example, a mutation that disrupted ATP binding by E. coli MutL (E32K) also prevented MutSL-dependent activation of the MutH endonuclease. This mutation, however, did not interfere with interactions between MutL and MutH, MutS, and UvrD (60). In S. cerevisiae, an mlh1 mutation predicted to disrupt ATP binding (G98A) conferred a strong mutator phenotype and also prevented conformational changes that promote interactions between the amino-terminal domains of Mlh1p and Pms1p (64).
In this study, we found that mlh1 ATP binding domain mutants are defective in meiotic crossing over. These observations suggest that Mlh1p may activate mismatch repair and crossing-over factors through a common mechanism that is not well understood. A potential candidate for activation by Mlh1p is Exo1p, a factor which also promotes meiotic crossing over and physically interacts with Mlh1p (34, 35, 65, 66). Additional genetic analyses will be required to determine whether Mlh1p and Exo1p function in a common meiotic pathway. Genetic studies have already shown that Exo1p and Msh4p function in distinct crossover pathways (34).
A large region between amino acids 350 and 500 in Mlh1p is predicted to form a random coil or unstructured domain (33). This region, which is not well conserved among Mlh proteins, was heavily targeted for mutagenesis because it contains a high concentration of charged residues. Because a large part of this interval (approximately residues 400 to 500) was insensitive to mutagenesis, we were unable to assign it a specific function. One attractive possibility that needs to be tested by deletion analysis is that it serves as a flexible linker between the carboxy-terminal heterodimerization domain and the amino-terminal ATPase and DNA binding domains. Such a role might allow the Mlh proteins to interact with different sets of DNA substrates and/or repair factors.
Analysis of mlh1-2, -29, and -31 separation-of-function alleles reveals distinct properties.
The mlh1-29 (K393A, R394A) and mlh1-31 (R401A, D403A) alleles map to the N terminus of the predicted unstructured domain. Interestingly, these alleles conferred distinct mismatch repair properties. The mlh1-29 mutation, derived from an S288C clone, conferred mismatch repair defects only in SK1 strains. These defects, however, were almost completely eliminated by transforming mlh1-29 SK1 strains with a single-copy vector containing PMS1 from S288C. This observation indicates that the SK1-S288C amino acid polymorphisms that exist in the MLH1 and PMS1 genes are silent but are capable of conferring detectable phenotypes under special circumstances. The phenotype of mlh1-29 mutants in SK1 strains is reminiscent of the synergistic mutator phenotype observed in strains containing multiple weak mutations in mismatch repair components such as MLH1, PMS1, and EXO1 (5, 65). The synergistic mutator phenotype is thought to reflect structural interactions between components important for stabilizing multiprotein mismatch repair complexes (see below) (5).
In contrast to mlh1-29, the mlh1-31 mutation conferred severe defects in vegetative mismatch repair in both the SK1 and S288C strain backgrounds. Interestingly, meiotic mismatch repair was only minimally affected (only the S288C background was tested). How can we explain this difference in mismatch repair function? Mismatch repair during vegetative growth is thought to occur immediately after mispair formation, possibly within the context of a replication fork (13, 40). In contrast, the meiotic mismatch repair assay that we used detects mispairs that result from strand invasion and branch migration steps catalyzed by recombination enzymes (reviewed in reference 47). These DNA mispairs form in heteroduplex DNA and are detected as Arg+/− postmeiotic segregation events.
Within this framework, at least three models can be developed to explain the unique mismatch repair phenotype in mlh1-31 strains. (i) The mlh1-31 mutation specifically disrupts Mlh1p interactions with DNA replication factors that are required only for vegetative mismatch repair without compromising meiotic mismatch repair. (ii) The mlh1-31 mutation weakens interactions that are common to both processes, but defects in vegetative mismatch repair become more apparent because vegetative mismatch repair is a time-dependent repair process that is tightly coupled to DNA replication. (iii) The mlh1-31 mutation disrupts interactions in both processes, but a meiosis-specific factor can compensate for the meiotic defect. At present, we are unable to distinguish between these hypotheses because few downstream components of mismatch repair have been identified. Furthermore, the mlh1-31 mutation did not disrupt two-hybrid interactions with known Mlh1 interaction proteins or affect ternary complex assembly.
Mlh1p is likely to play a structural role in meiosis.
Four observations suggest that Mlh1p is a component of a larger complex that specifically promotes meiotic crossing over. First, the vegetative mismatch repair functions of Mlh1p were more easily disrupted than the meiotic crossing-over functions, suggesting that factors exist in meiosis that compensate for defective Mlh1 proteins. Second, mlh1-2 strains were defective in Mlh1p-Mlh3p interaction yet were nearly wild type for meiotic crossing over. Crossing over in these strains was still MLH3 dependent, suggesting that additional meiotic factors interacting with Mlh1p allowed the Mlh1p-Mlh3p heterodimer to remain functional. Third, in a previous screen, we identified conditional mlh1 mutants (I296S and F228S) that were functional in meiotic crossing over but defective in meiotic mismatch repair (6). Protein and two-hybrid analyses indicated that these mutant proteins were unstable (data not shown) (6), suggesting that a meiotic crossover complex can stabilize these mutant proteins. Finally, we failed to identify any alleles that disrupted crossing over without affecting mismatch repair, and only in rare cases did we see a complete loss of the crossover function.
Three mutations, mlh1-9, -14, and -46, that caused a slightly greater defect in the spore viability assay (68 to 75%) than in the vegetative mismatch repair assay (six- to ninefold increased) were identified; however, these phenotypes were subtle, and these were not considered clean separation-of-function mutations. If Mlh1p functions as part of a large complex that provides a structural function in meiosis, it may be difficult to identify mlh1 alleles that disrupt crossing over without affecting mismatch repair. Together, these observations are consistent with Mlh1p's being an integral component of a multiprotein complex that promotes crossing over. In this scenario, the ATPase mutations dramatically disrupt the function of Mlh1p because they prevent the conformational changes that are required to coordinate key regulatory steps similar to those that occur in vegetative mismatch repair.
ARG4 conversion gradient is altered in mlh1-2 strains.
As shown in Table 4, mlh1-2 strains displayed a gene conversion gradient that differed from that of both the wild-type and mlh1Δ strains and was essentially flat. An attractive model for conversion gradient formation suggests that they form by repairing mispairs located near a double-strand break towards gene conversion, while markers further away from the double-strand break are randomly repaired (16). Other versions of this model suggest that markers distant to a double-strand break are repaired to restoration as the result of nicks created by Holliday junction cleavage (2).
In meiosis, unrepaired DNA mispairs remain in heteroduplex DNA until spore germination. Such a long time interval could allow a very inefficient mismatch repair system to act on a DNA mispair that would normally escape repair in a fast-growing culture. Because mlh1-2 strains displayed meiotic mismatch repair efficiencies that were approximately 60% of the wild-type value, we hypothesize that this mutation confers a weakly active allele that delays the repair process, resulting in a flat gradient in which repair is random at both marker sites. In this model, random repair occurs because nicks present in recombination intermediates that are thought to direct mismatch repair have already been sealed prior to the initiation of mismatch repair. A prediction of this model is that mutations that completely disrupt the enzymatic activity of Mlh1p should display an mlh1Δ phenotype in meiotic mismatch repair. Consistent with this, we found that mutations in the ATPase binding domain (E31K, G95G, and G98S) conferred postmeiotic segregation and aberrant segregation phenotypes at ARG4 that were indistinguishable from those of the null mutant (data not shown). A more rigorous test of this model will require the selective expression of mismatch repair components at times in meiosis when double-strand break repair is already complete.
Acknowledgments
We thank members of the Alani laboratory, Neil Hunter, Mike Liskay, and Wei Yang for helpful discussions and comments on the manuscript, Jennifer Wanat for analysis of SK1/S288C hybrid strains, and Rhona Borts for sharing unpublished data. We are grateful to M. Hall and T. Kunkel for providing the MLH1 and PMS1 overexpression vectors, H. Tsubouchi and H. Ogawa for providing the parental SK1 strains, and R. Lahue for anti-Pms1p antibody.
E.A. and A.W.K. were supported by National Institutes of Health grant GM53085. J.L.A. was supported by a CAPES fellowship awarded by the Brazilian government. J.H. was supported by a Department of Education training grant. M.W. and S.S. were funded by undergraduate research fellowships from the Howard Hughes Medical Institute awarded to Cornell University. M.W. was also supported by a Cornell Presidential Undergraduate Scholarship.
REFERENCES
- 1.Alani, E. 1996. The Saccharomyces cerevisiae Msh2 and Msh6 proteins form a complex that specifically binds to duplex oligonucleotides containing mismatched DNA base pairs. Mol. Cell. Biol. 16:5604-5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alani, E., R. A. G. Reenan, and R. D. Kolodner. 1994. Interaction between mismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics 137:19-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allers, T., and M. Lichten. 2001. Intermediates of yeast meiotic recombination contain heteroduplex DNA. Mol. Cell 8:225-231. [DOI] [PubMed] [Google Scholar]
- 4.Allers, T., and M. Lichten. 2001. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106:47-57. [DOI] [PubMed] [Google Scholar]
- 5.Amin, N. S., M. N. Nguyen, S. Oh, and R. D. Kolodner. 2001. exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol. Cell. Biol. 21:5142-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Argueso, J. L., D. Smith, J. Yi, M. Waase, S. Sarin, and E. Alani. 2002. Analysis of conditional mutations in the Saccharomyces cerevisiae MLH1 gene in mismatch repair and in meiotic crossing over. Genetics 160:909-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aronshtam, A., and M. G. Marinus. 1996. Dominant negative mutator mutations in the mutL gene of Escherichia coli. Nucleic Acids Res. 24:2498-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ban, C., and W. Yang. 1998. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell 95:541-552. [DOI] [PubMed] [Google Scholar]
- 9.Ban, C., M. Junop, and W. Yang. 1999. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell 97:85-97. [DOI] [PubMed] [Google Scholar]
- 10.Borts, R. H., S. R. Chambers, and M. F. Abdullah. 2000. The many faces of mismatch repair in meiosis. Mutat. Res. 451:129-150. [DOI] [PubMed] [Google Scholar]
- 11.Bowers, J., P. T. Tran, R. M. Liskay, and E. Alani. 2000. Analysis of yeast MSH2-MSH6 suggests that the initiation of mismatch repair can be separated into discrete steps. J. Mol. Biol. 302:327-338. [DOI] [PubMed] [Google Scholar]
- 12.Bowers, J., P. T. Tran, A. Joshi, R. M. Liskay, and E. Alani. 2001. MSH-MLH complexes formed at a DNA mismatch are disrupted by the PCNA sliding clamp. J. Mol. Biol. 306:957-968. [DOI] [PubMed] [Google Scholar]
- 13.Buermeyer, A. B., S. M. Deschenes, S. M. Baker, and R. M. Liskay. 1999. Mammalian DNA mismatch repair. Annu. Rev. Genet. 33:533-564. [DOI] [PubMed] [Google Scholar]
- 14.Christianson, T. W., R. S. Sikorski, M. Dante, J. H. Shero, and P. Hieter. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene 110:119-122. [DOI] [PubMed] [Google Scholar]
- 15.Detloff, P., J. Sieber, and T. D. Petes. 1991. Repair of specific base pair mismatches formed during meiotic recombination in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 11:737-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Detloff, P., M. A. White, and T. D. Petes. 1992. Analysis of a gene conversion gradient at the HIS4 locus in Saccharomyces cereviae. Genetics 132:113-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellison, A. R., J. Lofing, and G. A. Bitter. 2001. Functional analysis of human MLH1 and MSH2 missense variants and hybrid human-yeast MLH1 proteins in Saccharomyces cerevisiae. Hum. Mol. Genet. 10:1889-1900. [DOI] [PubMed] [Google Scholar]
- 18.Flores-Rozas, H., and R. D. Kolodner. 1998. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. USA 95:12404-12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fogel, S., R. Mortimer, K. Lusnak, and F. Tavares. 1978. Meiotic gene conversion: a signal of the basic recombination event in yeast. Cold Spring Harbor Symp. Quant. Biol. 43:1325-1341. [DOI] [PubMed] [Google Scholar]
- 20.Fogel, S., R. K. Mortimer, and K. Lusnak. 1981. Mechanisms of meiotic gene conversion, or “wanderings on a foreign strand,” p. 289-339. In J. N. Strathern, E. W. Jones, and J. R. Broach (ed.), The molecular biology of the yeast Saccharomyces. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 21.Habraken, Y., P. Sung, L. Prakash, and S. Prakash. 1998. ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2-MSH6 and MLH1-PMS1 protein complexes. J. Biol. Chem. 273:9837-9841. [DOI] [PubMed] [Google Scholar]
- 22.Hall, M. C., J. R. Jordan, and S. W. Matson. 1998. Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. EMBO J. 17:1535-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall, M. C., and S. W. Matson. 1999. The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J. Biol. Chem. 274:1306-1312. [DOI] [PubMed] [Google Scholar]
- 24.Hall, M. C., and T. A. Kunkel. 2001. Purification of eukaryotic MutL homologs from Saccharomyces cerevisiae with self-affinity technology. Protein Expr. Purif. 21:333-342. [DOI] [PubMed] [Google Scholar]
- 25.Hall, M. C., H. Wang, D. A. Erie, and T. A. Kunkel. 2001. High affinity cooperative DNA binding by the yeast Mlh1-Pms1 heterodimer. J. Mol. Biol. 312:637-647. [DOI] [PubMed] [Google Scholar]
- 26.Hall, M. C., P. V. Shcherbakova, and T. A. Kunkel. 2002. Differential ATP binding and intrinsic ATP hydrolysis by amino-terminal domains of the yeast Mlh1 and Pms1 proteins. J. Biol. Chem. 277:3673-3679. [DOI] [PubMed] [Google Scholar]
- 27.Harfe, B. D., B. K. Minesinger, and S. Jinks-Robertson. 2000. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr. Biol. 10:145-148. [DOI] [PubMed] [Google Scholar]
- 28.Henderson, S. T., and T. D. Petes. 1992. Instability of simple sequence DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 12:2749-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hillers, K. J., and F. W. Stahl. 1999. The conversion gradient at HIS4 of Saccharomyces cerevisiae. I. Heteroduplex rejection and restoration of Mendelian segregation. Genetics 153:555-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a.Hoffmann, E. R., P. V. Shcherbakova, T. A. Kunkel, and R. H. Borts. MLH1 mutations differentially affect meiotic functions in Saccharomyces cerevisiae. Genetics, in press. [DOI] [PMC free article] [PubMed]
- 30.Hollingsworth, N. M., L. Ponte, and C. Halsey. 1995. MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes Dev. 9:1728-1739. [DOI] [PubMed] [Google Scholar]
- 31.Hunter, N., and N. Kleckner. 2001. The single-end invasion: an asymmetric intermediate at the double-strand break to double-Holliday junction transition of meiotic recombination. Cell 106:59-70. [DOI] [PubMed] [Google Scholar]
- 32.Hunter, N., and R. H. Borts. 1997. Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev. 11:1573-1582. [DOI] [PubMed] [Google Scholar]
- 33.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195-202. [DOI] [PubMed] [Google Scholar]
- 34.Khazanehdari, K. A., and R. H. Borts. 2000. EXO1 and MSH4 differentially affect crossing-over and segregation. Chromosoma 109:94-102. [DOI] [PubMed] [Google Scholar]
- 35.Kirkpatrick, D. T., J. R. Ferguson, T. D. Petes, and L. S. Symington. 2000. Decreased meiotic intergenic recombination and increased meiosis I nondisjunction in exo1 mutants of Saccharomyces cerevisiae. Genetics 156:1549-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolodner, R. D., and G. T. Marsischky. 1999. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9:89-96. [DOI] [PubMed] [Google Scholar]
- 37.Langland, G., J. Kordich, J. Creaney, K. Heppner Goss, K. Lillard-Wetherell, K. Bebenek, T. A. Kunkel, and J. Groden. 2001. The BLM helicase interacts with hMLH1 but is not required for DNA mismatch repair. J. Biol. Chem. 276:30031-30035. [DOI] [PubMed] [Google Scholar]
- 38.Lea, D. E., and C. A. Coulson. 1949. The distribution of the numbers of mutants in bacterial populations. J. Genet. 49:264-285. [DOI] [PubMed] [Google Scholar]
- 39.Lissouba, P., J. Mouseau, G. Rizet, and J. L. Rossignol. 1962. Fine structure of genes in the ascomycete Ascobolus immersus. Adv. Genet. 11:343-380. [Google Scholar]
- 40.Marti, T. M., C. Kunz, and O. Fleck. 2002. DNA mismatch repair and mutation avoidance pathways. J. Cell Physiol. 191:28-41. [DOI] [PubMed] [Google Scholar]
- 41.Mortimer, R., and S. Fogel. 1974. Genetical interference and gene conversion, p. 263-275 In R. Grell (ed.), Mechanisms in recombination. Plenum Press, New York, N.Y.
- 42.Nicolas, A., D. Treco, N. P. Schultes, and J. W. Szostak. 1989. An initiation site for meiotic gene conversion in the yeast Saccharomyces cerevisiae. Nature 338:35-39. [DOI] [PubMed] [Google Scholar]
- 43.Novak, J. E., P. B. Ross-Macdonald, and G. S. Roeder. 2001. The budding yeast Msh4 protein functions in chromosome synapsis and the regulation of crossover distribution. Genetics 158:1013-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nystrom-Lahti, M., C. Perrera, M. Raschle, E. Panyushkina-Seiler, G. Marra, A. Curci, B. Quaresima, F. Costanzo, M. D'Urso, S. Venuta, and J. Jiricny. 2002. Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes Chromosomes Cancer 33:160-167. [PubMed] [Google Scholar]
- 45.Pang, Q., T. A. Prolla, and R. M. Liskay. 1997. Functional domains of the Saccharomyces cerevisiae Mlh1p and Pms1p DNA mismatch repair proteins and their relevance to human hereditary nonpolyposis colorectal cancer-associated mutations. Mol. Cell. Biol. 17:4465-4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papazian, H. P. 1952. The analysis of tetrad data. Genetics 37:175-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paques, F., and J. E. Haber. 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63:349-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedrazzi, G., C. Perrera, H. Blaser, P. Kuster, G. Marra, S. L. Davies, G. H. Ryu, R. Freire, I. D. Hickson, J. Jiricny, and I. Stagljar. 2001. Direct association of Bloom's syndrome gene product with the human mismatch repair protein MLH1. Nucleic Acids Res. 29:4378-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perkins, D. D. 1949. Biochemical mutants in the smut fungus Ustilago maydis. Genetics 34:607-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raschle, M., P. Dufner, G. Marra, and J. Jiricny. 2002. Mutations within the hMLH1 and hPMS2 subunits of the human MutLa mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSa. J. Biol. Chem. 277:21810-21820. [DOI] [PubMed] [Google Scholar]
- 51.Reenan, R. A., and R. D. Kolodner. 1992. Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics 132:975-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rocco, V., B. de Massy, and A. Nicolas. 1992. The Saccharomyces cerevisiae ARG4 initiator of meiotic gene conversion and its associated double-strand DNA breaks can be inhibited by transcriptional interference. Proc. Natl. Acad. Sci. USA 89:12068-12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rose, M. D., F. Winston, and P. Hieter. 1990. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 54.Ross-Macdonald, P., and G. S. Roeder. 1994. Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell 79:1069-1080. [DOI] [PubMed] [Google Scholar]
- 55.Rossignol, J. L., A. Nicolas, H. Hamza, and T. Langin. 1984. Origins of gene conversion and reciprocal exchange in Ascobolus. Cold Spring Harbor Symp. Quant. Biol. 49:13-21. [DOI] [PubMed] [Google Scholar]
- 56.Santucci-Darmanin, S., D. Walpita, F. Lespinasse, C. Desnuelle, T. Ashley, and V. Paquis-Flucklinger. 2000. MSH4 acts in conjunction with MLH1 during mammalian meiosis. FASEB J. 14:1539-1547. [DOI] [PubMed] [Google Scholar]
- 57.Shcherbakova, P. V., M. C. Hall, M. S. Lewis, S. E. Bennett, K. J. Martin, P. R. Bushel, C. A. Afshari, and T. A. Kunkel. 2001. Inactivation of DNA mismatch repair by increased expression of yeast MLH1. Mol. Cell. Biol. 21:940-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sia, E. A., M. Dominska, L. Stefanovic, and T. D. Petes. 2001. Isolation and characterization of point mutations in mismatch repair genes that destabilize microsatellites in yeast. Mol. Cell. Biol. 21:8157-8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sokolsky, T., and E. Alani. 2000. EXO1 and MSH6 are high-copy suppressors of conditional mutations in the MSH2 mismatch repair gene of Saccharomyces cerevisiae. Genetics 155:589-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spampinato, C., and P. Modrich. 2000. The MutL ATPase is required for mismatch repair. J. Biol. Chem. 275:9863-9869. [DOI] [PubMed] [Google Scholar]
- 61.Sun, H., D. Treco, N. P. Schultes, and J. W. Szostak. 1989. Double-strand breaks at an initiation site for meiotic gene conversion. Nature 338:87-90. [DOI] [PubMed] [Google Scholar]
- 62.Tishkoff, D. X., A. L. Boerger, P. Bertrand, N. Filosi, G. M. Gaida, M. F. Kane, and R. D. Kolodner. 1997. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci. USA 94:7487-7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomer, G., A. B. Buermeyer, M. M. Nguyen, and R. M. Liskay. 2002. Contribution of human Mlh1 and Pms2 ATPase activities to DNA mismatch repair. J. Biol. Chem. 277:21801-21809. [DOI] [PubMed] [Google Scholar]
- 64.Tran, P. T., and R. M. Liskay. 2000. Functional studies on the candidate ATPase domains of Saccharomyces cerevisiae MutLa. Mol. Cell. Biol. 20:6390-6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tran, P. T., J. A. Simon, and R. M. Liskay. 2001. Interactions of Exo1p with components of MutLa in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 98:9760-9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsubouchi, H., and H. Ogawa. 2000. Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol. Biol. Cell 11:2221-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vedel, M., and A. Nicolas. 1999. CYS3, a hot spot of meiotic recombination in Saccharomyces cerevisiae: effects of heterozygosity and mismatch repair functions on gene conversion and recombination intermediates. Genetics 151:1245-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vojtek, A. B., S. M. Hollenberg, and J. A. Cooper. 1993. Mammalian ras interacts directly with the serine/threonine kinase Raf. Cell 74:205-214. [DOI] [PubMed] [Google Scholar]
- 69.Wach, A., A. Brachat, R. Pohlmann, and P. Philippsen. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793-1808. [DOI] [PubMed] [Google Scholar]
- 70.Wang, T. F., N. Kleckner, and N. Hunter. 1999. Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc. Natl. Acad. Sci. USA 96:13914-13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White, J. H., K. Lusnak, and S. Fogel. 1985. Mismatch-specific postmeiotic segregation frequency in yeast suggests a heteroduplex recombination intermediate. Nature 315:350-352. [DOI] [PubMed] [Google Scholar]
- 72.Woods, L. M., C. A. Hodges, E. Baart, S. M. Baker, R. M. Liskay, and P. A. Hunt. 1999. Chromosomal influence on meiotic spindle assembly: abnormal meiosis I in female Mlh1 mutant mice. J. Cell Biol. 145:1395-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]