

Abstract
RecG protein differs from other helicases analysed to atomic resolution in that it mediates strand separation via translocation on double-stranded (ds) rather than single-stranded (ss) DNA. We describe a highly conserved helical hairpin motif in RecG and show it to be important for helicase activity. It places two arginines (R609 and R630) in opposing positions within the component helices where they are stabilized by a network of hydrogen bonds involving a glutamate from helicase motif VI. We suggest that disruption of this feature, triggered by ATP hydrolysis, moves an adjacent loop structure in the dsDNA-binding channel and that a swinging arm motion of this loop drives translocation. Substitutions that reverse the charge at R609 or R630 reduce DNA unwinding and ATPase activities, and increase dsDNA binding, but do not affect branched DNA binding. Sequences forming the helical hairpin and loop structures are highly conserved in Mfd protein, a transcription-coupled DNA repair factor that also translocates on dsDNA. The possibility of type I restriction enzymes and chromatin-remodelling factors using similar structures to drive translocation on dsDNA is discussed.
Keywords: DNA replication/Holliday junctions/RecB/recombination/repair/RuvABC
Introduction
RecG helicase was first identified in Escherichia coli as a protein involved in DNA recombination and repair, a role supported by the subsequent discovery that it could catalyse branch migration of Holliday junctions (Lloyd and Sharples, 1993). RecG also dissociates D-loops, removes RNA from R-loops and reduces the copy number of plasmids that rely on R-loops for initiating DNA synthesis (Whitby et al., 1994; Vincent et al., 1996; Fukuoh et al., 1997; McGlynn et al., 1997; Whitby and Lloyd, 1998). More recent work revealed that RecG functions as a monomer and that it catalyses the formation of Holliday junctions from replication forks (McGlynn and Lloyd, 2000, 2001; McGlynn et al., 2000, 2001). The interconversion of forks and junctions facilitates interplay between DNA replication, recombination and repair. Such interplay underpins chromosome replication and is essential for faithful genome transmission (Chakraverty and Hickson, 1999; Cox et al., 2000; Marians, 2000; Kuzminov, 2001; Tercero and Diffley, 2001; McGlynn and Lloyd, 2002; Sogo et al., 2002). RecG has been linked specifically with mechanisms that promote direct rescue of replication forks stalled at lesions in or on the DNA, especially in the template for leading strand synthesis (McGlynn and Lloyd, 2000, 2002; Bolt and Lloyd, 2002; Gregg et al., 2002).
The formation of a Holliday junction from a replication fork requires the simultaneous unwinding of both leading and lagging strands, the subsequent annealing of these strands and re-annealing of the parental strands. Insight into how a single molecule of RecG is able to catalyse these reactions has come from the atomic structure of the Thermatoga maritima protein in a complex with a replication fork substrate (Singleton et al., 2001) (see Figure 2A). RecG has well-conserved helicase domains linked to a novel N-terminal ‘wedge’ domain that provides the specificity for binding a three-way branched DNA structure. It has been proposed that the helicase motor acts as a double-stranded (ds) DNA translocase, pulling the parental strands of a replication fork structure through two separate channels flanking the wedge domain, neither wide enough to accommodate dsDNA (Singleton et al., 2001). This has the effect of stripping off the nascent leading and lagging strands, and allowing the parental strands to re-anneal, as suggested by earlier biochemical studies (McGlynn and Lloyd, 2000, 2001). The unwound leading and lagging strands can then also anneal; this may be promoted by binding sites on the protein that hold the displaced strands in a position to facilitate this process. The end result would be the formation of a Holliday junction. As the protein continues to translocate along the rewound parental duplex, a ‘daughter duplex’ is spooled out in front of the wedge. This final stage is almost certainly equivalent to the Holliday junction branch migration reaction catalysed by RecG. The structure specificity of DNA binding and the mechanism of unwinding can also explain the ability of RecG to dissociate the invading strand from D- and R-loop structures.
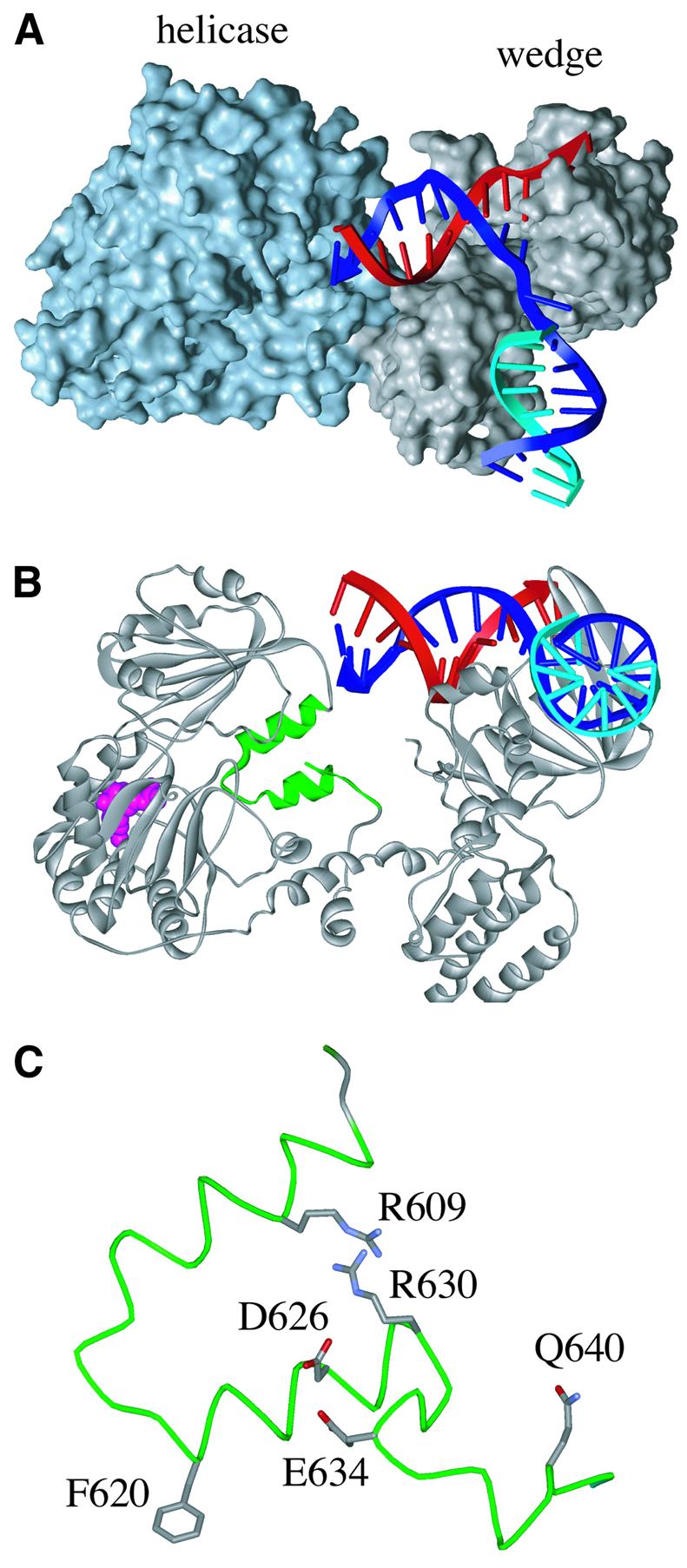
Fig. 2. Structure of RecG protein and its TRG motif. (A) Domain structure of RecG from Thermatoga maritima in a complex with a partial replication fork structure lacking a leading strand. (B) Ribbon representation of the RecG–DNA complex viewed from the end of the lagging strand arm of the fork, showing in green the two α-helices of the helical hairpin and loop structures of the TRG motif. The bound ADP is shown in pink. (C) Detailed structure of the TRG motif from the E.coli RecG structure modelled on the structure of the T.maritima protein, showing the locations of the two opposed arginines and other conserved residues investigated.
Sequence analysis identifies RecG as a superfamily II (SF2) helicase (Gorbalenya and Koonin, 1993; Hall and Matson, 1999; Caruthers and McKay, 2002; Singleton and Wigley, 2002). Structural data from members of the SF2 helicase family suggest that nucleic acid binding occurs in a non-sequence-specific manner, via the phosphodiester backbone, rather than via interactions with the nucleic acid bases as seen in SF1 helicases (Singleton and Wigley, 2002). This would, in principle, allow SF2 helicases to bind to both ds and single-stranded (ss) nucleic acids, although, until the structure of RecG was published, all SF2 helicase complexes solved were with ss oligonucleotides. This is to be expected as the helicases concerned unwind linear ds nucleic acids, which requires translocation along a single strand. RecG is different in that it appears to translocate along a duplex, which is also not surprising as its primary function is to catalyse the movement of a branch point along a dsDNA molecule.
The crystal structure of RecG complexed with fork DNA does not reveal the exact path taken by the ‘parental’ duplex across the helicase domains, let alone identify any specific contacts with the protein or the mechanism of translocation, as this section of duplex in the substrate used was too short (Singleton et al., 2001) (Figure 2A). In this study, we identify a motif containing a helical hairpin structure in RecG that is highly conserved, and show that changes to this structure interfere with the unwinding of branched DNA molecules. The results presented indicate that the motif mediates translocation of RecG on dsDNA by coupling ATP hydrolysis to movement at the dsDNA-binding site. The primary sequence of the structural motif is very highly conserved and acts almost as a signature for RecG proteins. However, we found very similar sequences in Mfd protein, which also translocates along dsDNA (Park et al., 2002). The structure we describe may therefore define a general translocation mechanism found in other proteins that move along dsDNA (Travers, 1999; Flaus and Owen-Hughes, 2001; Saha et al., 2002).
Results
Identification of a helical hairpin motif in RecG and Mfd proteins
RecG has been identified in most species of bacteria (Sharples et al., 1999). Sequence alignments revealed a high degree of conservation, especially in the section containing the seven well-defined helicase motifs (Figure 1A). They also revealed a highly conserved sequence spanning residues 606–642 in the E.coli protein (Figure 1B). Searches of the available databases revealed a closely related sequence at a similar location in Mfd proteins (Figure 1), a transcription–repair coupling factor found in most bacteria and which acts to revive RNA polymerase stalled on the DNA or to dislodge transcription complexes blocked, for instance, by non-pairing lesions in the template strand (Selby and Sancar, 1994; Park et al., 2002). Otherwise, the sequence is unique to RecG and provides a signature motif that has enabled us to identify RecG-like proteins encoded in the nuclear genomes of Arabidopsis thalania and rice. These plant enzymes are most likely to be chloroplast proteins of bacterial ancestry. There is no homologue in the human genome. We have named the RecG signature motif TRG, for translocation in RecG.

Fig. 1. Alignments of RecG and Mfd proteins. (A) Linear representation of RecG (693 amino acids) and Mfd (1148 amino acids) aligned via helicase (blue) and TRG (yellow) motifs. (B) Alignment of sequences defining the TRG motif of RecG and Mfd proteins. The sequences shown are typical examples taken from alignments composed of 50 RecG and 56 TRCF sequences from bacterial and plant species. Residues with 100% conservation in the 50 RecG and 56 Mfd sequences are in red; those with >70% conservation are in green. The TRG motif is located 19 residues from the end of helicase motif VI in E.coli RecG. The information is summarized for RecG and Mfd in a consensus in which U represents hydrophobic residues (F, I, L, M, V, W, Y) and A and G, D and E, K and R, and S and T are considered to be functionally equivalent. Site-directed substitutions of conserved residues of E.coli RecG described in this work are indicated in blue above the alignment and numbered accordingly. The α-helices (cylinders) and loops (lines) formed by the TRG motif and shown beneath the alignments and are as described in the crystal structure of RecG from T.maritima (Singleton et al., 2001). Eco, Escherichia coli; Hin, Haemophilus influenzae; Bbu, Borrelia burgdorferi; Syn, Synechocystis species; Bsu, Bacillus subtilis; Mtu, Mycobacterium tuberculosis; Dra, Deinococcus radiodurans; Tma, Thermotoga maritima; Aae, Aquifex aeolicus; Ath, Aribidopsis thaliana.
Secondary structure analyses conducted before a crystal structure for RecG became available predicted that most of the TRG motif would form two antiparallel α-helices, from A606 to D615 and from F620 to R630, linked by a loop of four residues in RecG or seven residues in Mfd (Figure 1B). The three additional residues in Mfd provide a reliable means to distinguish between these two proteins (Figure 1B). The helical hairpin structure would place R609 and R630, as numbered in E.coli RecG, in opposing positions. The crystal structure of RecG from T.maritima confirmed the presence of a helical hairpin and the opposing positions of the arginines in the two α-helices (Figure 2B and C).
To assess the function of the TRG motif, a number of residues were selected for mutational analysis. We targeted R609, D626 and R630, which are conserved in all 50 RecG proteins and all 56 Mfd proteins analysed, F620 and Q640, which are invariant throughout the RecG sequences, and E634, which is a conserved negatively charged residue in 94% of RecG sequences. The mutant proteins made are listed in Table I.
Table I. Plasmid constructs encoding wild-type and mutant RecG proteins.
| RecG protein | Vector plasmid |
||
|---|---|---|---|
| pGEM-7Zf(–) | pT7-7 | pET-14b | |
| Wild type | pAM208 | pAM210 | pAM209 |
| E571A | pQW151 | ||
| E571R | pQW152 | ||
| R609Q | pAM260 | pAM282 | pAM309 |
| R609E | pQW110 | pQW114 | |
| F620L | pAM299 | pAM303 | pAM312 |
| D626N | pAM256 | pAM261 | pAM308 |
| R630Q | pAM297 | pAM302 | pAM310 |
| R630E | pQW123 | pQW118 | |
| R609Q E571A | pQW156 | ||
| R609E E571A | pQW157 | ||
| R609Q R630Q | pQW108 | pQW112 | |
| R609Q R630E | pQW109 | pQW113 | |
| E634Q | pAM298 | pAM306 | pAM311 |
| Q640A | pQW144 | ||
| Q640E | pQW147 | ||
Activity of mutant RecG proteins in vivo
Plasmid-encoded wild-type RecG fully complements the sensitivity of a recG null strain to UV light (Mahdi et al., 1997). To investigate the activity of the mutant proteins, we exploited a recG-null strain that also carries ruv and rpo* mutations. This strain is extremely sensitive to UV light because of the absence of both the RecG and RuvABC proteins (McGlynn and Lloyd, 2000) but, because ruv is partially suppressed by the rpo* mutation, introducing recG+ on a plasmid dramatically improves survival. It therefore provides a much more sensitive test of RecG activity than does a recG single mutant.
The E634Q and F620L substitutions have no effect on complementation, whereas the Q640A and Q640E substitutions appear to eliminate RecG activity (Figure 3A). Changes at R609, D626 and R630 reduce complementation. Those at R609 and D626 have a rather modest effect. This is true even when the positively-charged arginine is replaced with a negatively-charged glutamate (R609E). Substitutions of R630 are much more severe. Comple mentation is reduced substantially by substituting a glutamine (R630Q), and is eliminated by substituting a glutamate (R630E). Since R609 and R630 are close together (Figure 2C), and might therefore interact, we also tested proteins in which both residues are substituted. As expected from R630E, the R609Q R630E double mutant has no activity. However, the R609Q R630Q double mutant is also inactive. The synergistic effect observed is consistent with both arginines having an important role in RecG activity. None of the proteins analysed has any effect on survival in the presence of a chromosomal recG+ allele (data not shown).

Fig. 3. Activity of wild-type RecG and TRG mutant proteins in vivo. (A) Effect of pT7-7 (vector) and recG constructs encoding the proteins indicated (wt = wild-type RecG) on survival of UV-irradiated recG-null strain N4544. (B) Agarose gel analysis of plasmid DNA extracted from the recG mutant strain showing the effect of pGEM-7Zf(–) constructs encoding wild-type or mutant RecG proteins, or no RecG (vector), on the yield of that same plasmid. Each lane shows the relative amount of plasmid recovered from equal volumes of cell culture. (C) As in (B) except that the constructs are all pT7-7 derivatives.
The data presented above have to be interpreted with caution because RecG helicase activity is known to reduce the copy number of ColE1-based plasmids by unwinding the R-loop structure used to initiate plasmid replication (Vincent et al., 1996; Fukuoh et al., 1997). When a plasmid encoding wild-type RecG is purified from a strain carrying a chromosomal recG deletion, the yield of plasmid DNA is therefore greatly reduced compared with that from the same strain carrying the vector plasmid (Figure 3B and C) (Vincent et al., 1996). Elimination of RecG helicase activity either by mutation of the ATP-binding site (K302A) or by deletion of the N-terminal branched DNA-binding site in the wedge domain (ΔN60, ΔN144) restores plasmid yields to vector levels (Figure 3B). For this reason, plasmids encoding RecG proteins with significantly reduced helicase activity might nevertheless promote DNA repair rather well because of the associated increase in plasmid copy number.
The F620L and E634Q substitutions do not restore plasmid copy number. In both cases, plasmid yield is as low as with a wild-type RecG construct (Figure 3B). Given that these proteins also promote DNA repair very effectively, we conclude that neither F620 nor E634 is essential for RecG helicase activity. In contrast, the D626N, R609Q and R630Q substitutions result in very high plasmid yields (Figure 3B), indicating that these mutations have a major effect on either the DNA binding or helicase activity of RecG. High plasmid yields consistent with inactivation of RecG are also obtained with constructs encoding RecG substitutions R609E, R630E, R609Q R630Q and R609Q R630E (data not shown). Taken together, these data indicate that both R609 and R630 are important for RecG function. They also indicate that increased plasmid copy number may be a significant factor contributing to the level of DNA repair promoted by R609E and especially R609Q (Figure 3A).
Binding and unwinding of branched DNA
Although the in vivo studies indicate that R609, D626 and R630 are important for RecG activity, they cannot differentiate defects in DNA binding from those in DNA unwinding. We therefore used bandshift assays to measure DNA binding directly. RecG forms two distinct protein–DNA complexes with junction and fork DNA substrates, a faster migrating complex 1 containing a single molecule of RecG and a slower migrating complex 2 containing two molecules (McGlynn et al., 2000). Complex 1 predominates over complex 2, which is seen only at higher concentrations of protein (Mahdi et al., 1997; McGlynn et al., 2000). The structure of RecG bound to a fork shows that only one molecule of RecG could possibly bind the DNA branch point (Figure 2). Furthermore, the bulk of the protein sits over the dsDNA arm on which it translocates. Therefore, complex 2 most probably reflects binding of a second molecule of RecG to one of the unbound arms via its dsDNA-binding site.
All the mutant proteins bind the branch point in Holliday junction and replication fork structures with an affinity similar to the wild-type protein, as indicated by the formation of complex 1 (Figure 4A; data not shown). The reduced function of the mutant proteins with substitutions at R609, D626 and R630 revealed by our in vivo analyses must be due, therefore, to an effect on helicase activity. In addition, these results confirm that the mutations have not significantly altered the overall protein fold, as the proteins still bind DNA. However, it is significant that the R609E, R630E, R609Q R630Q, and R609Q R630E proteins show more evidence of complex 2 formation with both junction and fork structures (Figure 4A; data not shown). Given the likely nature of complex 2, this indicates that the substitutions made may have increased the affinity of RecG for dsDNA. This was confirmed using a 50 bp linear dsDNA species (Figure 4B). Wild-type RecG binds this DNA, but the complexes are unstable, and significant gel retardation is seen only at very high protein concentrations. The R609E and R630E proteins form stable complexes, and at significantly lower concentrations, though still much higher than needed to bind J12 or fork DNA. Furthermore, a series of different complexes are visible. These could reflect the binding of more than one molecule of RecG or the binding of a single molecule at different positions along the DNA. The R609Q R630Q and R609Q R630E proteins give similar ladders of complexes with junction DNA (Figure 4A). This could reflect binding of additional molecules of RecG to exposed duplex arms of the structure or formation of complexes with different conformations that affect gel mobility.

Fig. 4. Band shift assays showing DNA binding activity of RecG proteins. (A) Holliday junction binding. Reactions used the His6-tagged RecG proteins indicated at 0, 0.025, 0.1, 0.4, 1.6, 6.25, 25 and 100 nM (lanes a–h and i–j) and 32P-labelled J12 DNA at 0.2 nM. (B) Linear duplex DNA binding. Reactions used the native RecG proteins indicated at 0, 50, 100, 200, 400 and 800 nM (lanes a–f, g–l and m–r) and 32P-labelled 50 bp linear dsDNA at 0.2 nM. Positions of free DNA and bound complexes are indicated on the right by horizontal and vertical lines, respectively.
To confirm that R609, D626 and R630 are important to the helicase activity of RecG, we tested the ability of the mutant proteins to unwind synthetic Holliday junction and replication fork structures. The junction used, J12, has a central 12 bp core of homology within which the branch point can migrate, flanked by heterologous sequences that prevent spontaneous substrate dissociation by migration of the branch point to the DNA ends. Three types of fork were used, a full fork structure with both leading and lagging strand ends at the branch point, and two partial forks, one lacking a lagging strand and the other a leading strand. To ensure a defined substrate, the branch point in each fork was fixed by heterology.
The F620L and E634Q proteins unwind fork and Holliday junction substrates at the same rate as wild-type RecG (data not shown), confirming the in vivo data indicating that neither F620 nor E634 is important for helicase activity. In contrast, the substitutions made at R609, D626 and R630 cause a significant reduction in ATP-dependent DNA unwinding, with the loss of activity following the same general trend as that observed in the complementation assays (Figure 5A–C). It made little difference which of the three fork structures was used. The activity of each mutant relative to wild-type RecG was the same with each fork (data not shown). We therefore focused on the partial fork lacking a leading strand (Figure 5C), the preferred substrate for RecG (McGlynn and Lloyd, 2001; Gregg et al., 2002). Assays were conducted in most cases using both His6-tagged and native versions of the wild-type and mutant proteins. With the mutants, both types show very similar reductions in unwinding activity relative to wild-type RecG (Table II). R609Q retains ∼15% activity, while R609E and D626N have ∼5%. As with DNA repair, substitutions at R630 reduce unwinding quite severely; R630Q has <2% activity while R630E has <0.5%. The double mutants R609Q R630Q and R609Q R630E have lower activities than the corresponding single mutants, showing that the effects of the mutations are additive. However, neither is catalytically dead; significant unwinding can be detected using high concentrations of protein (data not shown).

Fig. 5. Unwinding of Holliday junction and forked DNA substrates by RecG proteins. (A) Dissociation of J12 DNA to flayed duplex products in 30 min reactions containing 32P-labelled J12 and the indicated RecG protein at 0, 1 and 10 nM (lanes a–c, d–f, g–i and j–l). Note that RecG can unwind J12 in either of two orientations, and therefore generates two different labelled flayed duplex products of slightly different mobility, as indicated. (B) Rates of unwinding in reactions containing J12 at an initial concentration of 0.2 nM, and the indicated proteins at 0.5 nM. Data are means of at least two independent experiments. (C) Rates of unwinding of a partial replication fork structure by RecG proteins. Reactions contained a partial fork structure, 32P labelled at the 5′ end of the lagging strand as shown in the cartoon, at an initial concentration of 0.2 nM, and the indicated proteins at 0.5 nM. Data are means of at least two independent experiments. (D) Band shift assays showing binding of native wild-type and E571R RecG proteins to a Holliday junction substrate. Reactions used the proteins indicated at 0, 0.4, 1.6, 6.25, 25 or 100 nM (lanes a–f and g–l) and 32P-labelled J12 DNA at 0.2 nM. (E) Holliday junction unwinding activity of RecG E571R. Reactions were as in (A) and used the proteins indicated at 0, 0.4, 1.6, 6.25 or 25 nM (lanes a–e and f–j) and 32P-labelled J12 DNA at 0.2 nM.
Table II. Rates of ATP hydrolysis and Holliday junction dissociation by His6-tagged and native RecG proteins.
| RecG mutation | His6-tagged protein |
Native protein |
||
|---|---|---|---|---|
| ATPasea | Helicaseb | ATPasea | Helicaseb | |
| R609Q | 18.0 ± 4.7 | 10.4 | 15.2 ± 1.1 | 19.6 |
| R609E | 10.8 ± 2.2 | 2.8 | 8.8 ± 1.2 | 6.1 |
| D626N | 8.5 | 5.9 | ||
| R630Q | 10.3 ± 1.2 | 0.8 | 11.8 ± 6.1 | 1.0 |
| R630E | 4.4 ± 2.3 | 0.2 | 2.7 ± 1.0 | 0.3 |
| R609Q R630Q | 1.0 ± 0.1 | 0.2 | 1.7 ± 0.9 | 0.9 |
aRates of ATP hydrolysis, kcat (per s), were determined using reactions containing 10 nM RecG and 250 nM cold J12, with ATP and Mg2+ both at 5 mM. Values are percenatges of the wild-type rate. The values of kcat (per s) for wild-type RecG were 184.8 (His6-tagged protein) and 123.8 (native protein). All values are the mean of at least two independent measurements.
bRates of unwinding of J12 DNA, as measured from the initial slopes of time course reactions such as those shown in Figure 5B. Values are percentages of the wild-type rate. All values are the mean of at least two independent measurements.
DNA-dependent ATP hydrolysis
Since the mutant proteins bind branched DNA with an affinity similar to that of wild-type RecG, their reduced ability to unwind such structures could be the result of a defect either at the step of ATP hydrolysis or at the coupling of this hydrolysis to movement of the DNA relative to the protein. We found that the reduced DNA unwinding activity correlates with the reduction in DNA-dependent ATP hydrolysis, although in the case of R630 substitutions the ATPase activity is slightly less affected. This was true whether we used J12 or linear dsDNA as a cofactor in the ATPase assays (Table II; data not shown). These data indicate that some stage of the ATP hydrolysis cycle is dependent on a translocation step on dsDNA, probably related to a conformational change, which is mediated via the helical hairpin and its conserved arginine residues.
Interactions between R609, R630 and E571
The crystal structure of RecG shows that R609 and R630 form a hydrogen-bonding network with E571 (see Figure 6B). We therefore tested E571A and E571R substitutions. Neither has a substantial effect on complementation (Figure 3A; data not shown). However, when E571A is combined with either R609Q or R609E, complementation activity is reduced drastically (Figure 3A). Furthermore, both E571A and E571R increase plasmid yield (Figure 3C). Analysis of the E571R protein in vitro revealed that it binds well to a Holliday junction substrate (Figure 5D). However, it has significantly reduced DNA unwinding activity (Figure 5E). The initial rate of unwinding is similar to that of the R609Q protein (Figure 5B; data not shown). Taken together, these data demonstrate that E571 is important for RecG function. The ability of the E571A and E571R proteins to promote efficient survival of a UV-irradiated recG strain therefore most probably reflects a compensatory effect of increased plasmid copy number, as for example with R609Q. Thus we conclude that E571 and its interactions with R609 and R630 are crucial for RecG function.
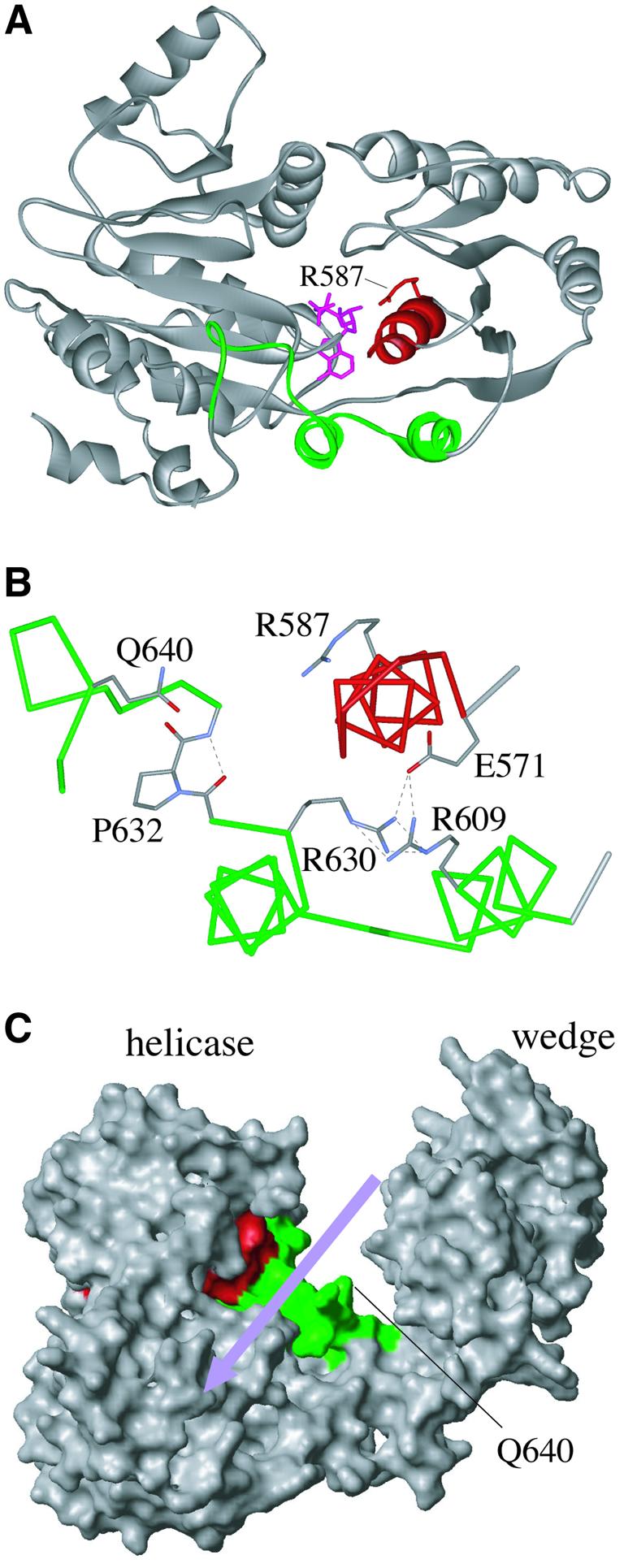
Fig. 6. Interactions between RecG helicase motif VI and the TRG motif. (A) Ribbon structure of the modelled E.coli RecG helicase domain (residues 220–660) showing how helicase motif VI (red) lies over the helical hairpin of the TRG motif (green). The bound ADP (pink) and the R587 residue extending from motif VI are also shown. (B) Hydrogen bond interactions between E571 in motif VI and both R609 and R630 in the helical hairpin structure of the TRG motif. Also shown are P632, which adds rigidity to the proposed DNA-binding loop, Q640 within the loop, which may contact the DNA phosphate backbone, and R587, which would form a salt bridge to ATP. (C) Structure of the modelled E.coli RecG showing the proposed path of dsDNA across the helicase domain (violet arrow) and projection into this channel of the loop structure of the TRG motif (green); note that the residues forming the helical hairpin are not visible in this view of RecG. Helicase motif VI is in red.
Discussion
RecG differs from all the other helicases characterized so far in that DNA unwinding is achieved via translocation on dsDNA. We have shown that a helical hairpin in RecG is critically important for DNA unwinding, and propose that this structure is a component of the motor driving translocation. Sequences forming the helical hairpin are part of a larger TRG motif that is also present in Mfd protein, a transcription–repair coupling factor that translocates on dsDNA to move or dislodge stalled RNA polymerase complexes rather than to force strand separation. The motor mechanism we propose for RecG may therefore apply more generally to proteins that utilize the energy from ATP hydrolysis to translocate on dsDNA, such as type I restriction enzymes and chromatin-remodelling factors.
The structure of RecG from T.maritima reveals that sequences forming the helical hairpin pack on the surface of the protein (Figures 2B and 6A), which would explain why substitutions within this region of E.coli RecG have no effect on protein folding or solubility. The two α-helices comprising the helical hairpin are distant from the proposed binding site for the dsDNA on which RecG translocates, and are therefore very unlikely to contact DNA directly. Nevertheless, some of the changes made greatly reduce the DNA unwinding and ATPase activities of RecG, which is correlated with reduced ability to promote repair. As binding of these proteins to the DNA branch point (complex 1) is not obviously affected, the logical conclusion is that these changes break the link between ATP hydrolysis and dsDNA translocation.
A closer look at the RecG structure suggests how ATP hydrolysis might be coupled to DNA translocation via the helical hairpin structure. The conserved arginines within this structure, R609 and R630, are in opposition in the two helices, with their side chains pointing towards each other rather than out into the proposed DNA-binding cleft (Figure 2C). Such juxtaposition of the positively charged residues is expected to be unfavourable, but this arrangement is stabilized by a network of hydrogen bonds involving an additional residue, E571 (Figure 6B). This residue is at the N-terminus of helicase motif VI, and is highly conserved as either a glutamate or aspartate in RecG and Mfd sequences. Motif VI forms an α-helix that sits above and parallel to the two helices of the helical hairpin motif (Figure 6B), and contains a conserved arginine residue (R587) that forms a salt bridge with the γ phosphate of ATP. In the other helicase proteins studied, motif VI is thought to move on ATP binding and hydrolysis, so linking ATPase activity to a movement of helicase domain 2 relative to domain 1 (Hall and Matson, 1999; Velankar et al., 1999; Soultanas et al., 2000; Caruthers and McKay, 2002; Dillingham et al., 2002). In addition, mutations in motif VI have been shown to uncouple ATPase and helicase activities (Hall and Matson, 1999). We propose that motif VI of RecG moves on ATP hydrolysis in a manner similar to that suggested for other helicases, which disrupts the hydrogen bonding between E571, R609 and R630 (Figure 6B), destabilizing the opposing arginine residues and causing a shift in the two helices of the motif relative to each other. This is consistent with our finding that changing any two of these three residues has a strong synergistic effect on RecG function. The role of D626 is unclear, but it is possible that the conformational change brings it into proximity with one or other of the arginine residues and forms interactions that might stabilize the new conformation. The associated movement would be transmitted to the conserved TRG sequence extending from the second helix, which forms a well-defined loop in the RecG structure (Figure 1B, residues 631–642). This loop projects into the proposed dsDNA-binding cleft (Figure 6C), and would be in the ideal position to form connections with the dsDNA, perhaps via Q640, which is present in all RecG and Mfd sequences analysed (Figure 1B, and alignments not shown). The loop is tethered at its C-terminus by an extension that hooks under the long α-helix linking the helicase and wedge domains of the protein (Figure 6A). In E.coli RecG, this loop also contains a highly conserved proline (P632, Figure 6B), which has limited flexibility and would therefore add rigidity to the loop, in addition to a number of possible hydrogen bonds.
We propose that a shift in the relative positions of the two helices in the helical hairpin, triggered on ATP hydrolysis, causes a corresponding angular movement of the loop structure, analogous to a swinging arm. This proposed mechanism has similarities to those suggested for the actin-based myosin motor (Vale and Milligan, 2000). Assuming the loop in RecG makes direct contact with the dsDNA (Figure 6C), translocation may be driven directly via a mechanical levering action of the swinging arm. Alternatively, the formation and breakage of DNA interactions by the swinging arm could provide a ratchet to trap any DNA movement achieved by other means.
Either a lever or ratchet action of the swinging arm would be consistent with previous studies demonstrating that DNA binding can be reduced dramatically by deletion of the C-terminus of RecG (Mahdi et al., 1997). It is also consistent with the higher affinity for dsDNA of proteins with substitutions at R609 or R630. These substitutions may mimic the effect of ATP hydrolysis, moving the loop structure within the dsDNA-binding cleft and freezing RecG in a conformation that enhances dsDNA contacts. The greater effect of substitutions at R630 may be explained by the fact that the affected helix is connected directly to the proposed DNA-binding loop. We have shown that Q640A and Q640E substitutions, both of which might be expected to disrupt any DNA binding via the loop structure, eliminate RecG activity in vivo. The model is also consistent with the reduced ATPase activity caused by changes to the helical hairpin motif. As mentioned, the mutant proteins might be expected to have difficulty moving through the different conformations associated with each cycle of ATP binding and hydrolysis.
RecG is one of a rapidly growing family of proteins that translocate on dsDNA using the energy derived from ATP hydrolysis. These include bacterial Mfd proteins, type I DNA restriction enzymes and eukaryotic chromatin-remodelling factors (Davies et al., 1999a; Ellis et al., 1999; Janscak et al., 1999; Firman and Szczelkun, 2000; Flaus and Owen-Hughes, 2001; Park et al., 2002; Saha et al., 2002; Szczelkun, 2002). These proteins have the seven motifs common to DNA helicases, and hydrolyse ATP only in the presence of dsDNA or chromatin. However, none shows DNA strand unwinding activity on partial duplex substrates (Richmond and Peterson, 1996; Travers, 1999; Flaus and Owen-Hughes, 2001). RecG appears to be the exception, although recent studies have shown that the DnaB ring helicase, which drives strand separation during DNA replication and is thought to encircle only the lagging strand template, can translocate on dsDNA in vitro (Kaplan and O’Donnell, 2002). RecG can be thought of as having a dsDNA translocation module connected to a strand separation module designed to operate specifically at a branch point in the bound duplex. In the case of Mfd, the translocation module is connected instead to a protein–protein interaction domain that makes specific contacts with the upstream side of transcribing RNA polymerase complexes (Park et al., 2002). Translocation shunts RNA polymerase in the direction of transcription, which can revive backtracked RNA polymerase complexes or dislodge complexes stalled by non-pairing lesions in the template strand (Selby and Sancar, 1994; Park et al., 2002). The presence in Mfd of the highly conserved TRG motif forming the helical hairpin and adjacent loop structures in RecG suggests that these two enzymes may have very similar translocation motors derived from a common ancestor.
Translocation modules of the HsdR subunits of type I restriction enzymes are coupled to nuclease domains that catalyse strand cleavage (Davies et al., 1999b). Those of chromatin-remodelling factors interact with other proteins, forming large complexes that move nucleosomes and interact with transcription factors to regulate gene expression (Cote et al., 1994; Hamiche et al., 1999; Whitehouse et al., 1999; Shen et al., 2000; Rouleau et al., 2002). Analysis of the available sequences suggests the existence in both the restriction enzymes and the remodelling factors of a conserved helical structure adjacent to the helicase domain that may serve to link ATP-triggered movement of helicase motif VI to translocation on dsDNA. However, the TRG sequences that characterize the helical hairpin and loop structures in RecG and Mfd are not evident. Structural studies of these proteins will therefore be required to test whether they have not only a RecA-like helicase motor but also a similar transmission system for driving translocation.
Materials and methods
Molecular modelling of protein structures
The structure of E.coli RecG was modelled on the co-ordinates of the T.maritima protein using Swiss-PdbViewer (Guex and Peitsch, 1997). Secondary structure predictions used PHD and Jpred (Rost, 1996; Cuff et al., 1998).
Strains and plasmids
Escherichia coli K-12 recG+ strain AB1157 and its ΔrecG263::kan derivative N3793, and wild-type strain MG1655 and its highly UV-sensitive ΔrecG263 ΔruvAC65 rpoB*35 relA1 ΔspoT207::cat eda51::Tn10 derivative N4544, have been described previously (Al-Deib et al., 1996; McGlynn and Lloyd, 2000). AM1125 is a BL21(DE3) plysS strain carrying ΔrecG263::kan (Mahdi et al., 1997). The pGEM-7Zf(–)-, pET14b- and pT7-7-derived recG+ plasmid constructs pAM208, pAM209 and pAM210, respectively, carry a cassette version of recG engineered to contain additional restriction sites that allow for convenient fragment exchange (Mahdi et al., 1997). pAM209 and pAM210 allow overexpression of RecG with or without an N-terminal His6 tag. pAM217, pAM219 and pAM220 encode RecG proteins with a mutation affecting the ATP-binding site (K320A) or N-terminal deletions of 60 (ΔN60) or 144 (ΔN144) amino acids, respectively, and were made by cloning the appropriate recG alleles excised as XbaI–HindIII fragments from constructs described previously (Mahdi et al., 1997) into pGEM-7Zf(–). pPM104 is a pBluescript II SK– (Stratagene) construct carrying the PstI–EcoRI fragment of recG from pAM208 (Mahdi et al., 1997).
Site-directed mutagenesis of recG
RecG proteins with defined amino acid substitutions in the conserved sequence motif adjacent to helicase motif VI were made by site-directed mutagenesis. The required changes to the recG sequence were made in pPM104, using the Quikchange Mutagenesis (Stratagene) procedure. The sequence changes made are available on request. Products were sequenced to confirm the presence of the required mutations, and recG mutant derivatives of pAM208, pAM209 and pAM210 were made by fragment replacement and checked by sequencing.
Media and general methods
LB broth and agar media have been cited (Al-Deib et al., 1996) and were supplemented with 100 µg/ml ampicillin for growth of strains carrying the plasmids used. Sensitivity to UV light was measured as described previously (Al-Deib et al., 1996), using strains AB1157, N3793 and N4544 transformed with either pT7-7 or derivatives carrying wild-type or mutant recG genes. Results obtained with AB1157 revealed that none of the RecG mutant proteins had a dominant-negative effect on DNA repair. To assess the effect of RecG proteins on plasmid copy number, cultures of ΔrecG263 strain N3793 carrying the required recG construct were grown overnight in LB broth supplemented with ampicillin. Plasmid DNA was extracted using a Qiagen mini-prep kit, digested with EcoRI and analysed by electrophoresis on a 1% (w/v) agarose gel, with a 1 kb DNA ladder (BRL) as a marker.
Purification of RecG proteins
All chromatography was performed at 4°C. Native RecG proteins were expressed by IPTG induction from pT7-7 constructs in strain AM1125 as described. Induced cells from 500 ml of culture were resuspended in buffer A (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 1 mM DTT), lysed by sonication and the supernatant was recovered by centrifugation. This was loaded onto a 15 ml SP-Sepharose column and eluted with a 0–1 M gradient of NaCl in buffer A. Fractions containing RecG were diluted with buffer A to a final NaCl concentration of <150 mM and loaded onto a 5 ml HiTrap heparin column. RecG was eluted with a 0–1 M NaCl gradient in buffer A. Fractions containing RecG were pooled and ammonium sulfate added to a final concentration of 0.5 M before loading onto a 5 ml phenyl–Sepharose column. Bound proteins were eluted by a stepped gradient of 0.5–0 M ammonium sulfate in buffer A. RecG was concentrated on a 5 ml HiTrap heparin column, and gel filtered in buffer A plus 100 mM NaCl on a HiPrep 16/60 Sephacryl S-200 HR column. Pure protein was dialysed against storage buffer [buffer A plus 50% (v/v) glycerol], and stored at –80°C. His6-tagged RecG proteins were expressed by IPTG induction from pET14b constructs in strain AM1125. Induced cells from 500 ml of culture were resuspended in His-tag buffer (20 mM Tris–HCl pH 7.5, 10 mM imidazole, 500 mM NaCl), lysed by sonication and the supernatant was recovered by centrifugation. The supernatant was loaded onto a 5 ml HiTrap chelating column (charged with Ni2+) and eluted with a 10–500 mM imidazole gradient. Fractions containing RecG were diluted with buffer A to a final NaCl concentration of <150 mM, and loaded onto a 5 ml HiTrap heparin column. RecG was eluted with a 0–1 M NaCl gradient in buffer A as before. Eluted protein was dialysed against storage buffer [buffer A plus 50% (v/v) glycerol], and stored at –80°C. Protein concentrations were determined using the Bradford assay with BSA as the standard, and are expressed as moles of monomeric protein.
DNA substrates
DNA substrates were made by annealing the appropriate oligonucleotides, one of which was labelled with 32P at the 5′ end, as described previously (Chan et al., 1997). DNA concentrations are in moles of the molecular structure. J12, a Holliday junction with a homologous core of 12 bp flanked by 19–20 bp heterologous arms, has been described previously (Lloyd and Sharples, 1993). The unlabelled J12 used in ATPase assays was made by annealing equal amounts of the four component oligonucleotides. Efficient production of the four-stranded structure was confirmed by gel electrophoresis through 10% polyacrylamide gels in Tris–borate–EDTA followed by ethidium bromide staining. A partial replication fork structure was made by annealing the oligonucleotides 5′-GTCGGATCCTCTAGACAGCTCCATGATCACT GGCACTGGTAGAATTCGGC-3′, 5′-CAACGTCATAGACGATTAC ATTGCTACATGGAGCTGTCTAGAGGATCCGA-3′ and 5′-TAGCA ATGTAATCGTCTATGACGTT-3′.
Linear duplex DNA was made by annealing the oligonucleotides 5′-TCGGATCCTCTAGACAGCTCCATGTAGCAATGTAATCGTCT ATGACGTTG-3′ and 5′-CAACGTCATAGACGATTACATTGCTAC ATGGAGCTGTCTAGAGGATCCGA-3′.
DNA binding assays
Binding of RecG to J12, fork and linear dsDNA substrates was measured using a band shift assay. RecG and 32P-labelled DNA were mixed in binding buffer [50 mM Tris–HCl pH 8.0, 5 mM EDTA, 1 mM DTT, 100 µg/ml BSA and 6% (v/v) glycerol] and incubated on ice for 15 min before loading on a pre-chilled 4% native polyacrylamide gel in a low ionic strength buffer (6.7 mM Tris–HCl pH 8.0, 3.3 mM sodium acetate and 2 mM EDTA). Electrophoresis was at 160 V for 90 min at 4°C. Gels were then dried and analysed by autoradiography and phosphoimaging.
Branched DNA unwinding assays
DNA unwinding assays were essentially as described previously (McGlynn and Lloyd, 1999). For standard reactions, RecG protein was mixed with 32P-labelled J12 or fork DNA in helicase buffer (20 mM Tris–HCl pH 7.5, 2 mM DTT, 100 µg/ml BSA, 5 mM ATP and 5 mM MgCl2) and incubated at 37°C for 30 min. DNA products were deproteinized by the addition of 0.2 volume of stop buffer [2.5% (w/v) SDS, 200 mM EDTA and 10 mg/ml proteinase K] and incubating for a further 10 min at 37°C. Samples were then analysed by electrophoresis using a 10% polyacrylamide gel and a Tris–borate buffer system before processing as described above. Bulk reactions were used to measure rates of unwinding. RecG at 0.5 nM was mixed in reaction buffer and kept on ice for 5 min prior to addition of labelled substrate DNA to 0.2 nM. An aliquot was removed immediately and deproteinized; this was taken as the time zero sample. The reaction was then placed at 37°C and samples subsequently removed at intervals and processed as for standard reactions.
ATPase hydrolysis
The hydrolysis of ATP was detected by measuring release of inorganic phosphate with acidic ammonium molybdate and malachite green as described previously (McGlynn et al., 2000). Typically, 50 µl reactions were established in 20 mM Tris–HCl pH 8.0, 2 mM DTT, 5 mM MgCl2, 5 mM ATP and 100 µg/ml BSA. Unlabelled J12 DNA was added to a final concentration of 250 nM. This mixture was pre-incubated at 37°C for 1 min then RecG was added to 10 nM. A 10 µl aliquot was removed immediately and added to 800 µl of the ammonium molybdate/malachite green reagent. This was the time zero sample. The RecG reaction was replaced at 37°C and further 10 µl samples removed at the indicated times. Each timed sample was incubated with the ammonium molybdate/malachite green reagent for 1 min at room temperature and then 100 µl of 34% sodium citrate solution was added. After 20 min at room temperature, the absorbance at 660 nm was measured.
Acknowledgments
Acknowledgements
We are indebted to Dale Wigley for releasing the RecG coordinates prior to publication, Peter McGlynn for advice on helicase assays and general encouragement, and Carol Buckman and Lynda Harris for excellent technical support. This work was supported by a program grant from the Medical Research Council to R.G.L. and G.J.S., and a Nottingham University Postgraduate Training Scholarship to Q.W.
References
- Al-Deib A.A., Mahdi,A.A. and Lloyd,R.G. (1996) Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J. Bacteriol., 178, 6782–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolt E.L. and Lloyd,R.G. (2002) Substrate specificity of RusA resolvase reveals the DNA structures targeted by RuvAB and RecG in vivo. Mol. Cell, 10, 187–198. [DOI] [PubMed] [Google Scholar]
- Caruthers J.M. and McKay,D.B. (2002) Helicase structure and mechanism. Curr. Opin. Struct. Biol., 12, 123–133. [DOI] [PubMed] [Google Scholar]
- Chakraverty R.K. and Hickson,I.D. (1999) Defending genome integrity during DNA replication: a proposed role for RecQ family helicases. BioEssays, 21, 286–294. [DOI] [PubMed] [Google Scholar]
- Chan S.N., Harris,L., Bolt,E.L., Whitby,M.C. and Lloyd,R.G. (1997) Sequence-specificity and biochemical characterization of the RusA Holliday junction resolvase of Escherichia coli. J. Biol. Chem., 272, 14873–14882. [DOI] [PubMed] [Google Scholar]
- Cote J., Quinn,J., Workman,J.L. and Peterson,C.L. (1994) Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science, 265, 53–60. [DOI] [PubMed] [Google Scholar]
- Cox M.M., Goodman,M.F., Kreuzer,K.N., Sherratt,D.J., Sandler,S.J. and Marians,K.J. (2000) The importance of repairing stalled replication forks. Nature, 404, 37–41. [DOI] [PubMed] [Google Scholar]
- Cuff J.A., Clamp,M.E., Siddiqui,A.S., Finlay,M. and Barton,G.J. (1998) JPred: a consensus secondary structure prediction server. Bioinformatics, 14, 892–893. [DOI] [PubMed] [Google Scholar]
- Davies G.P., Kemp,P., Molineux,I.J. and Murray,N.E. (1999a) The DNA translocation and ATPase activities of restriction-deficient mutants of EcoKI. J. Mol. Biol., 292, 787–796. [DOI] [PubMed] [Google Scholar]
- Davies G.P., Martin,I., Sturrock,S.S., Cronshaw,A., Murray,N.E. and Dryden,D.T. (1999b) On the structure and operation of type I DNA restriction enzymes. J. Mol. Biol., 290, 565–579. [DOI] [PubMed] [Google Scholar]
- Dillingham M.S., Wigley,D.B. and Webb,M.R. (2002) Direct measurement of single-stranded DNA translocation by PcrA helicase using the fluorescent base analogue 2-aminopurine. Biochemistry, 41, 643–651. [DOI] [PubMed] [Google Scholar]
- Ellis D.J., Dryden,D.T., Berge,T., Edwardson,J.M. and Henderson,R.M. (1999) Direct observation of DNA translocation and cleavage by the EcoKI endonuclease using atomic force microscopy. Nat. Struct. Biol., 6, 15–17. [DOI] [PubMed] [Google Scholar]
- Firman K. and Szczelkun,M.D. (2000) Measuring motion on DNA by the type I restriction endonuclease EcoR124I using triplex displacement. EMBO J., 19, 2094–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaus A. and Owen-Hughes,T. (2001) Mechanisms for ATP-dependent chromatin remodelling. Curr. Opin. Genet. Dev., 11, 148–154. [DOI] [PubMed] [Google Scholar]
- Fukuoh A., Iwasaki,H., Ishioka,K. and Shinagawa,H. (1997) ATP-dependent resolution of R-loops at the ColE1 replication origin by Escherichia coli RecG protein, a Holliday junction-specific helicase. EMBO J., 16, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E. and Koonin,E.V. (1993) Helicases: amino acid sequence comparisons and structure–function relationships. Curr. Opin. Struct. Biol., 3, 419–429. [Google Scholar]
- Gregg A.V., McGlynn,P., Jaktaji,R.P. and Lloyd,R.G. (2002) Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol. Cell, 9, 241–251. [DOI] [PubMed] [Google Scholar]
- Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Hall M.C. and Matson,S.W. (1999) Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol., 34, 867–877. [DOI] [PubMed] [Google Scholar]
- Hamiche A., Sandaltzopoulos,R., Gdula,D.A. and Wu,C. (1999) ATP-dependent histone octamer sliding mediated by the chromatin remodeling complex NURF. Cell, 97, 833–842. [DOI] [PubMed] [Google Scholar]
- Janscak P., Sandmeier,U. and Bickle,T.A. (1999) Single amino acid substitutions in the HsdR subunit of the type IB restriction enzyme EcoAI uncouple the DNA translocation and DNA cleavage activities of the enzyme. Nucleic Acids Res., 27, 2638–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan D.L. and O’Donnell,M. (2002) DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell, 10, 647–657. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. (2001) DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc. Natl Acad. Sci. USA, 98, 8461–8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd R.G. and Sharples,G.J. (1993) Dissociation of synthetic Holliday junctions by E.coli RecG protein. EMBO J., 12, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi A.A., McGlynn,P., Lovett,S.D. and Lloyd,R.G. (1997) DNA binding and helicase domains of the Escherichia coli recombination protein RecG. Nucleic Acids Res., 25, 3875–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians K.J. (2000) Replication and recombination intersect. Curr. Opin. Genet. Dev., 10, 151–156. [DOI] [PubMed] [Google Scholar]
- McGlynn P. and Lloyd,R.G. (1999) RecG helicase activity at three- and four-strand DNA structures. Nucleic Acids Res., 27, 3049–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P. and Lloyd,R.G. (2000) Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell, 101, 35–45. [DOI] [PubMed] [Google Scholar]
- McGlynn P. and Lloyd,R.G. (2001) Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl Acad. Sci. USA, 98, 8227–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P. and Lloyd,R.G. (2002) Genome stability and the processing of damaged replication forks by RecG. Trends Genet., 18, 413–419. [DOI] [PubMed] [Google Scholar]
- McGlynn P., Al-Deib,A.A., Liu,J., Marians,K.J. and Lloyd,R.G. (1997) The DNA replication protein PriA and the recombination protein RecG bind D-loops. J. Mol. Biol., 270, 212–221. [DOI] [PubMed] [Google Scholar]
- McGlynn P., Mahdi,A.A. and Lloyd,R.G. (2000) Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res., 28, 2324–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P., Lloyd,R.G. and Marians,K.J. (2001) Formation of Holliday junctions by regression of stalled replication forks: RecG stimulates fork regression even when the DNA is negatively supercoiled. Proc. Natl Acad. Sci. USA, 98, 8235–8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.S., Marr,M.T. and Roberts,J.W. (2002) E.coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell, 109, 757–767. [DOI] [PubMed] [Google Scholar]
- Richmond E. and Peterson,C.L. (1996) Functional analysis of the DNA-stimulated ATPase domain of yeast SWI2/SNF2. Nucleic Acids Res., 24, 3685–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. (1996) PHD: predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol., 266, 525–539. [DOI] [PubMed] [Google Scholar]
- Rouleau N., Domans’kyi,A., Reeben,M., Moilanen,A.M., Havas,K., Kang,Z., Owen-Hughes,T., Palvimo,J.J. and Janne,O.A. (2002) Novel ATPase of SNF2-like protein family interacts with androgen receptor and modulates androgen-dependent transcription. Mol. Biol. Cell, 13, 2106–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A., Wittmeyer,J. and Cairns,B.R. (2002) Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev., 16, 2120–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby C.P. and Sancar,A. (1994) Mechanisms of transcription–repair coupling and mutation frequency decline. Microbiol. Rev., 58, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples G.J., Ingleston,S.M. and Lloyd,R.G. (1999) Holliday junction processing in bacteria: insights from the evolutionary conservation of RuvABC, RecG and RusA. J. Bacteriol., 181, 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Mizuguchi,G., Hamiche,A. and Wu,C. (2000) A chromatin remodelling complex involved in transcription and DNA processing. Nature, 406, 541–544. [DOI] [PubMed] [Google Scholar]
- Singleton M.R. and Wigley,D.B. (2002) Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol., 184, 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton M.R., Scaife,S. and Wigley,D.B. (2001) Structural analysis of DNA replication fork reversal by RecG. Cell, 107, 79–89. [DOI] [PubMed] [Google Scholar]
- Sogo J.M., Lopes,M. and Foiani,M. (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science, 297, 599–602. [DOI] [PubMed] [Google Scholar]
- Soultanas P., Dillingham,M.S., Wiley,P., Webb,M.R. and Wigley,D.B. (2000) Uncoupling DNA translocation and helicase activity in PcrA: direct evidence for an active mechanism. EMBO J., 19, 3799–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczelkun M.D. (2002) Kinetic models of translocation, head-on collision and DNA cleavage by type I restriction endonucleases. Biochemistry, 41, 2067–2074. [DOI] [PubMed] [Google Scholar]
- Tercero J.A. and Diffley,J.F. (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature, 412, 553–557. [DOI] [PubMed] [Google Scholar]
- Travers A. (1999) An engine for nucleosome remodeling. Cell, 96, 311–314. [DOI] [PubMed] [Google Scholar]
- Vale R.D. and Milligan,R.A. (2000) The way things move: looking under the hood of molecular motor proteins. Science, 288, 88–95. [DOI] [PubMed] [Google Scholar]
- Velankar S.S., Soultanas,P., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]
- Vincent S.D., Mahdi,A.A. and Lloyd,R.G. (1996) The RecG branch migration protein of Escherichia coli dissociates R-loops. J. Mol. Biol., 264, 713–721. [DOI] [PubMed] [Google Scholar]
- Whitby M.C. and Lloyd,R.G. (1998) Targeting Holliday junctions by the RecG branch migration protein of Escherichia coli. J. Biol. Chem., 273, 19729–19739. [DOI] [PubMed] [Google Scholar]
- Whitby M.C., Vincent,S. and Lloyd,R.G. (1994) Branch migration of Holliday junctions: identification of RecG protein as a junction specific DNA helicase. EMBO J., 13, 5220–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I., Flaus,A., Cairns,B.R., White,M.F., Workman,J.L. and Owen-Hughes,T. (1999) Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature, 400, 784–787. [DOI] [PubMed] [Google Scholar]