

Abstract
An increasing family of neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s diseases, prion encephalopathies and cystic fibrosis is associated with aggregation of misfolded polypeptide chains which are toxic to the cell. Knowledge of the three-dimensional structure of the proteins implicated is essential for understanding why and how endogenous proteins may adopt a non-native fold. Yet, structural work has been hampered by the difficulty of handling proteins insoluble or prone to aggregation, and at the same time that is why it is interesting to study these molecules. In this review, we compare the structural knowledge accumulated for two paradigmatic misfolding disorders, Alzheimer’s disease (AD) and the family of poly-glutamine diseases (poly-Q) and discuss some of the hypotheses suggested for explaining aggregate formation. While a common mechanism between these pathologies remains to be proven, a direct comparison may help in designing new strategies for approaching their study.
Keywords: aggregation/amyloid/misfolding/poly-glutamine/structure
Misfolded proteins: the cause of a new family of diseases
We are accustomed to regarding genetic diseases as linked to the loss of function of one or more gene products. However, the ‘one gene, one protein, one function’ hypothesis has by now been contradicted more often than confirmed. One of the most interesting examples in which this concept seems to fail is represented by an increasing family of neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s diseases, prion encephalopathies and cystic fibrosis (Taylor et al., 2002). The hallmark of these otherwise unrelated diseases is the formation of aggregates containing misfolded proteins, i.e. proteins in a non-native folding state, with a conco mitant gain of function which eventually leads to neuronal death. Gain of function rather than loss of function is supported by the observation that the aggregates are toxic per se (Spillantini et al., 1998; Hardy and Selkoe, 2002). The aggregates have various supramolecular organiza tions and, in most cases, form structurally well-defined insoluble fibrillar deposits called amyloids (Glenner and Wong, 1984). In one case (prion diseases) aggregates are even believed to be responsible for disease transmission (Prusiner, 1995). What are the structural mechanisms that lead to aggregate formation of endogenous proteins? What makes the aggregates toxic? A great deal of literature has appeared in the last decade to address these questions. Since all members of the family of diseases are linked to a mechanism of aberrant protein folding, knowledge of the three-dimensional structure of the proteins implicated, both in their ‘healthy’ and in their pathological forms, is the prerequisite for understanding the mechanism of aggregate formation and, eventually, preventing it. Yet, only relatively limited structural information is currently available. Some of the structural hypotheses formulated to explain aggregate formation and toxicity have migrated from one field to the other, suggesting, at least apparently, the possibility of a somewhat similar mechanism. It is however important to question closely whether such a common mechanism exists or if the analogies are only apparent. Here we review in parallel some of the structural hypotheses currently suggested for two disease families, Alzheimer’s disease (AD) (and related tauopathies) and the family of poly-glutamine (poly-Q) diseases, selected as paradigms of misfolding disorders (Table I). A direct comparison which analyses commonalities and differences among them may be helpful for increasing our understanding of misfolding diseases.
Table I. Summary of main diseases discussed in the review.
| Disease family | Diseases | Responsible proteins | Aggregates (localization) | Reference |
|---|---|---|---|---|
| Alzheimer’s (AD) | amyloid precursor protein (APP) | plaques of fibrils (extracellular) | Hardy and Selkoe, (2002) | |
| Tauopathiesa | Amyotrophic lateral sclerosis/Parkinsonism–dementia complex | tau protein | neurofibrillary tangles (intracellular) | Lee et al., (2001) |
| Argyrophilic grain dementia | ||||
| Corticobasal degeneration | ||||
| Dementia pugilistica | ||||
| Diffuse neurofibrillary tangles with calcification | ||||
| Frontotemporal dementia with Parkinsonism linked to chromosome 17 | ||||
| Pick’s disease | ||||
| Progressive subcortical gliosis | ||||
| Progressive supranuclear palsy | ||||
| Tangle only dementia | ||||
| Poly-Q | Huntington’s (HD) | Huntingtin | amorphous or amyloid-like (intranuclear)b | Masino and Pastore, (2002) |
| Spinobulbar muscular atrophy (Kennedy disease or SBMA) | androgen receptor | |||
| Dentatorubral–pallidoluysian atrophy (Haw–River syndrome or DRPLA) | atrophin-1 | |||
| Spinocerebellar ataxia 1 (SCA1) | ataxin-1 | |||
| Spinocerebellar ataxia 2 (SCA2) | ataxin-2 | |||
| Spinocerebellar ataxia 3 (Machado–Joseph or SCA3) | ataxin-3 | |||
| Spinocerebellar ataxia 6 (SCA6) | VDCC | |||
| Spinocerebellar ataxia 7 (SCA7) | ataxin-7 |
aPathologies, other than AD, in which tau-positive neurofibrils are the predominant neuropathologic feature.
bIn SCA6 nuclear aggregates are accompanied by cytoplasmic aggregates which are predominant.
A supramolecular view: fibres and aggregates
The first observation of brain aggregates was carried out by Alois Alzheimer, a German physician who, in 1906, presented a case history of a patient affected by a then unknown brain disorder (Kraepelin, 1910). The autopsy of the patient’s brain showed the presence of two distinct types of lesions: extracellular deposits forming plaques and intracellular bundles of neurofibrillary tangles (NFTs). These lesions are now considered the invariant pathological feature of AD (for a recent review see Hardy and Selkoe, 2002). The current dominant view is that the plaques deposited outside and around the neurons are intrinsically toxic (Hardy and Selkoe, 2002), although more recent findings suggest the possibility that the intraneuronal deposits are the direct cause of toxicity (Mochizuki et al., 2000; Kienlen-Campard et al., 2002). According to the so-called amyloid hypothesis, tangles are considered only a secondary event as far as the time sequence in the disease progression is concerned, being a consequence of the formation of amyloid β (Aβ) plaques (Hardy and Selkoe, 2002). When brain lesions are made up only of intracellular bundles of self-assembled tau proteins, without accumulation of Aβ in plaques, the diseases are usually distinct from AD and called tauopathies (Lee et al., 2001). NFTs are nevertheless as toxic as amyloid plaques and in tauopathies lead to clinical symptoms similar to those of AD (Spillantini et al., 1998).
The poly-Q disorders [which include Huntington’s chorea, spinobulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy and spinocerebellar ataxias (SCAs)] are also associated with the formation of neuronal nuclear inclusions (NI) (Mangiarini et al., 1996). In several cases, NI are spherical aggregates, sometimes assembled by insoluble amyloid-like fibrils (Scherzinger et al., 1999; McGowan et al., 2000). Aggregates outside the nucleus have also been observed, but it has been shown recently that nuclear localization is required for toxicity (Yang et al., 2002).
The pathologies of AD and poly-Q diseases are distinct, but both involve aggregate formation and degeneration of a specific subset of neuronal cells. The two disease families could therefore share a common mechanism of pathogenesis, in which the aggregates affect the cellular functions and eventually cause neuronal death.
A causative link between aggregation and disease is not however universally acknowledged. It has been conversely suggested that the aggregates could have a protective role for neuronal cells, being the result of the cell’s attempts to proteolytically degrade or inactivate the toxic expanded protein (Zoghbi and Orr, 2000; Hardy and Selkoe, 2002). Pathogenesis could thus be initiated by other causes and only result in amyloid fibre formation.
A molecular view: proteins or peptides?
While predisposition to AD has been linked to mutations of several genes, including those of presenilins and apolipoprotein E, mainly two protein components are present in the two types of AD aggregates (reviewed in Hardy and Selkoe, 2002). Plaques are generated by deposition of the amyloid peptides (Aβ), which are degradation products of the amyloid precursor protein (APP). APP is a transmembrane cell surface glycoprotein, expressed in five isoforms, with APP(695) being the dominant isoform in brain. APP can be cleaved by three different proteases, called α, β and γ secretases. When APP is concomitantly hydrolysed by β-secretase at the N-terminus of Aβ and by the γ-secretase within the membrane (Lichtenthaler et al., 2002), the two main products, Aβ(1–40) and Aβ(1–42), migrate outside the cell and give rise to fibrils. When APP is cut by α-secretase, the resulting soluble peptides are generally considered non-toxic, although a recent report shows that the p3 peptide derived by α- and γ-secretase cleavage of APP is also toxic (Wei et al., 2002). Aβ(1–42) readily aggregates and seeds the formation of fibrils that can then act as templates for plaque formation. Several shorter synthetic peptides corresponding to partial sequences of Aβ(1–42) are able to form fibrils in vitro and have been studied along with ‘natural’ Aβ.
NFTs are mostly composed of the tau protein (from which tauopathies are named) (Garcia and Cleveland, 2001; Hardy and Selkoe, 2002), a highly hydrophilic microtubule-associated protein whose main function is microtubule stabilization, but also covers membrane interactions and anchoring of enzymes (Brandt et al., 1995; Sontag et al., 1999). Tangle formation is accompanied by further increase of tau production, hyperphosphorylation and loss of microtubule binding in the affected neurons. Proteolysis has been proposed to be important also for tau since the C-terminus of tau, when cleaved by proteases, generates cytotoxic fragments (Canu et al., 1998). These and other fragments are thought to nucleate tangle formation from full-length tau (von Bergen et al., 2000). However, it remains much more uncertain than for the well-defined Aβ which regions of tau are responsible for tangle formation.
Poly-Q diseases result from expansion of CAG nucleotide repeats that encode poly-Q tracts within the corresponding gene product (reviewed in Paulson, 1999; Zoghbi and Orr, 2000). The resulting proteins show no sequence homology outside the poly-Q, span different lengths, have different cellular localization and, when known, different functions. The only common feature is the presence of poly-Q tracts, which are, however, located in different regions of the corresponding proteins. A direct role of poly-Q in neurotoxicity has been demonstrated (Ordway et al., 1997; Yang et al., 2002). Interestingly, pathological effects occur in patients only when the length of poly-Q exceeds a rather sharp threshold of 35–40 glutamines. The length of the poly-Q tract correlates directly with the age of onset and with the severity of the symptoms in the diseases. The molecular composition of NI is much more heterogeneous than that of the AD aggregates, since they contain the ubiquitinated affected protein, proteolytic fragments, chaperones and other poly-Q proteins. While for AD and related tauopathies the mechanism of aggregation is now widely accepted to be linked to proteolysis of APP (Hardy and Selkoe, 2002) and tau (von Bergen et al., 2000), it is still unclear whether proteolysis is a universal event also for poly-Q proteins: proteolytic fragments and not the full-length proteins have been observed in NI for at least three poly-Q diseases (huntingtin, the androgen receptor and ataxin 1) (DiFiglia et al., 1997; Wellington et al., 1998; Welch and Diamond, 2001; Lunkes et al., 2002). Huntingtin exon 1 with an expanded poly-Q is sufficient to cause a progressive neurological phenotype in transgenic mice (Mangiarini et al., 1996). However, other reports suggest that the expanded proteins are more protease resistant and that release of N-terminal fragments of huntingtin occurs also in the non-toxic non-expanded protein (Dyer and McMurray, 2001; Kim et al., 2001; Lunkes et al., 2002).
Clarification of the precise nature of the aggregates obviously bears important consequences for structural/functional studies of tau and poly-Q proteins.
Structural hypotheses on conformational transitions: how to solubilize the ‘insoluble’
The prevailing hypothesis for explaining aggregation and amyloid formation involves a structural transition of a polypeptide chain from a native fold to an improperly folded or misfolded conformation. Much attention has been paid to understanding the molecular mechanisms producing this transition. The starting point for these studies implies the characterization of the structure of the non-aggregated protein or protein fragments. These are often referred to as the ‘soluble form’, as opposed to aggregates, but the actual situation is more complex since the starting conformation may not necessarily be soluble, especially at concentrations suitable for structural studies, as in the case of Aβ. Structural studies are therefore strongly hampered by the low solubility in water and by the tendency of the proteins and peptides to aggregate, i.e. ironically for the very reason why it is interesting to study them. Several strategies have been devised to overcome these limitations.
In AD, Aβ are believed to exist in an α-helical conformation when it is part of the transmembrane APP (Ortega-Aznar et al., 2000), with an amphipatic character distributed along the sequence: the hydrophilic N-terminus of the Aβ is exposed to an aqueous environment whereas the highly hydrophobic C-terminus is embedded in the membrane lipids (Figure 1A). To overcome their lack of solubility and to mimic the membrane environment, most solution studies of Aβ have been performed either in mixtures of water and alcohols or in micellar solutions (Serpell, 2000; and references therein). In all these media, Aβ assume helical conformations, characterized by two helical segments interrupted by a central tract that was termed the ‘kink region’ (Coles et al., 1998; Shao et al., 1999). However, the length of the helical segments and the conformation of the kink region varies according to the media employed. In the structure of Aβ(1–40) in a trifluoroethanol (TFE)/water mixture (Sticht et al., 1995) the two helices, spanning residues 15–23 and 31–35, are separated by a disordered region. In a mixture of hexafluoroisopropanol and water the kink region of Aβ(1–42) has been found to adopt a regular type I β-turn, yielding a well-defined tertiary structure (Crescenzi et al., 2002). The striking similarity of this structure with that of the fusion domain of haemagglutinin of influenza virus hints to a possible mechanism of toxicity of Aβ (discussed later). A random coil conformation has instead been observed in aqueous solution for Met-oxidized Aβ(1–40) and smaller fragments (Jarvet et al., 2000; Zhang et al., 2000; Riek et al., 2001) and in dimethylsulfoxide and water mixtures (Crescenzi et al., 2002).

Fig. 1. Hypothetical mechanisms of conformational transitions for Aβ peptides (A), tau (B) and poly-Q containing proteins (C). (A) The peptides are proteolytically cleaved from APP, precipitate as β-structures in equilibrium with oligomers which may eventually redissolve into the membrane. The lipid double layer is shown schematically in green. (B) A schematic picture of the architecture of tau. The four repeats are indicated with R1–R4. Regions differentially spliced are coloured in green. The sites identified as calpain and caspase-3 cleavage are indicated with stars and open circles respectively (Canu et al., 1998). The proteolytic products may assemble or nucleate aggregate formation (PHFs). (C) Summary of the current models of conformational transitions involving poly-Q tracts both in the pathological and non-pathological range. For a more detailed account see Masino and Pastore (2002).
Sequence analysis of tau suggests that it is a natively unfolded protein with an even distribution of hydrophobic fragments along its sequence and up to four repeats of ∼30 residues in its central region (Ruben et al., 1991; von Bergen et al., 2000) (Figure 1B). Experimental information derived from electron microscopy (EM), small angle X-ray scattering and NMR of tau confirms that the protein is essentially unfolded with little α-helix and β-sheet content (Crowther and Wischik, 1985; Kirschner et al., 1986; Schweers et al., 1994). Peptides from tau are much more soluble than Aβ, making solution studies less problematic. Short synthetic peptides spanning tau repeat sequences are mostly random in solution but were shown to be able to aggregate as β-structures (von Bergen et al., 2000). A peptide from the C-terminus (residues 423–441), which forms an alternative nucleation region of NFTs, forms a regular helix in a TFE/water mixture with a helical stabilizing C-capping motif (Esposito et al., 2000).
More debated is the nature of the conformation and the structural transition of poly-Q, for which experimental validation is even harder. In addition to the solubility problems, it is hard to produce this homopolymer both by conventional peptide synthesis and by recombinant methods. In 1993, Perutz proposed that poly-Q chains could form antiparallel β-strands held together by hydrogen bonds between side chain and main chain amides (Perutz et al., 1993). These ‘polar zippers’ would pair either intramolecularly (leading to a β-hairpin) or intermolecularly (leading to aggregates) when the poly-Q length exceeded a threshold. Although since then this hypothesis has strongly dominated the field, other researchers have invoked different models which imply transitions from random and α-to-β conformations (Lathrop et al., 1998; Starikov et al., 1999) (Figure 1C). In addition, an entirely different conformation, a right-handed helix with 6.2 residues/turn called µ-helix, was suggested by Monoi (1995). Such structure has a cylindrical pore along its helical axis larger than in an α-helix (∼6.6 Å) and might be favoured in poly-Q sequences owing to the unique possibility of hydrogen bonds involving the amide groups of glutamine side chains. Experimental evidence is mostly based on model peptide studies of poly-Q tracts, which have been solubilized using either hydrophilic flanking sequences or various mixtures of organic solvents, and on poly-Q sequences fused to different protein carriers (reviewed in Masino and Pastore, 2002). Attempts to characterize poly-Q structure in protein contexts different from the natural ones are justified by the observation that poly-Q is toxic independently of the specific protein (Ordway et al., 1997). Most peptide and protein studies seem to alternatively support one of these hypotheses: in one, soluble poly-Q is in random coil conformation, in the second, it forms β-structures. The apparent contradictions between these studies may be reconciled into a unifying picture by considering that the results are dominated by the strong aggregative tendency of poly-Q in water. Only when either the solvent composition, the flanking regions or the carrier protein are able to offset the insolubility of poly-Q, can this then adopt a random coil conformation. Since long poly-Q tracts have stronger tendency to aggregate, the competing effect of the environment will have a reduced effect.
Structural models of aggregates: how to study the ‘insoluble’
While the insolubility of the non-aggregated proteins hampers solution studies, a structural characterization of the aggregates is, if possible, even harder since it has to deal with their amorphous nature. These circumstances have so far prevented the determination of a consensus structure at atomic detail of any of the major aggregates. The consensus hypothesis is that amyloid fibres are constituted by β-sheet conformations. Increasing experimental evidence shows in fact that it is possible to induce β-sheet fibril formation in many proteins in vitro (reviewed in Dobson, 1999). Figure 2 shows a comparison of current models of amyloid fibres (Serpell, 2000; and references therein), NFTs (Moreno-Herrero et al., 2001) and poly-Q aggregates (Perutz et al., 2002). Direct evidence from solid state techniques, such as X-ray fibre diffraction, solid-state NMR, fourier transform-infrared and EM shows unequivocally that amyloid AD plaques are composed of long straight fibres of 6–12 nm in diameter with a basic β-sheet conformation in the fibrils (Sunde et al., 1997). The main controversy is whether in amyloid fibrils the β-strands are parallel, as indicated by work with solid-state NMR and electron paramagnetic resonance (Balbach et al., 2002; Torok et al., 2002) or antiparallel, as suggested by several X-ray diffraction studies (Serpell, 2000).
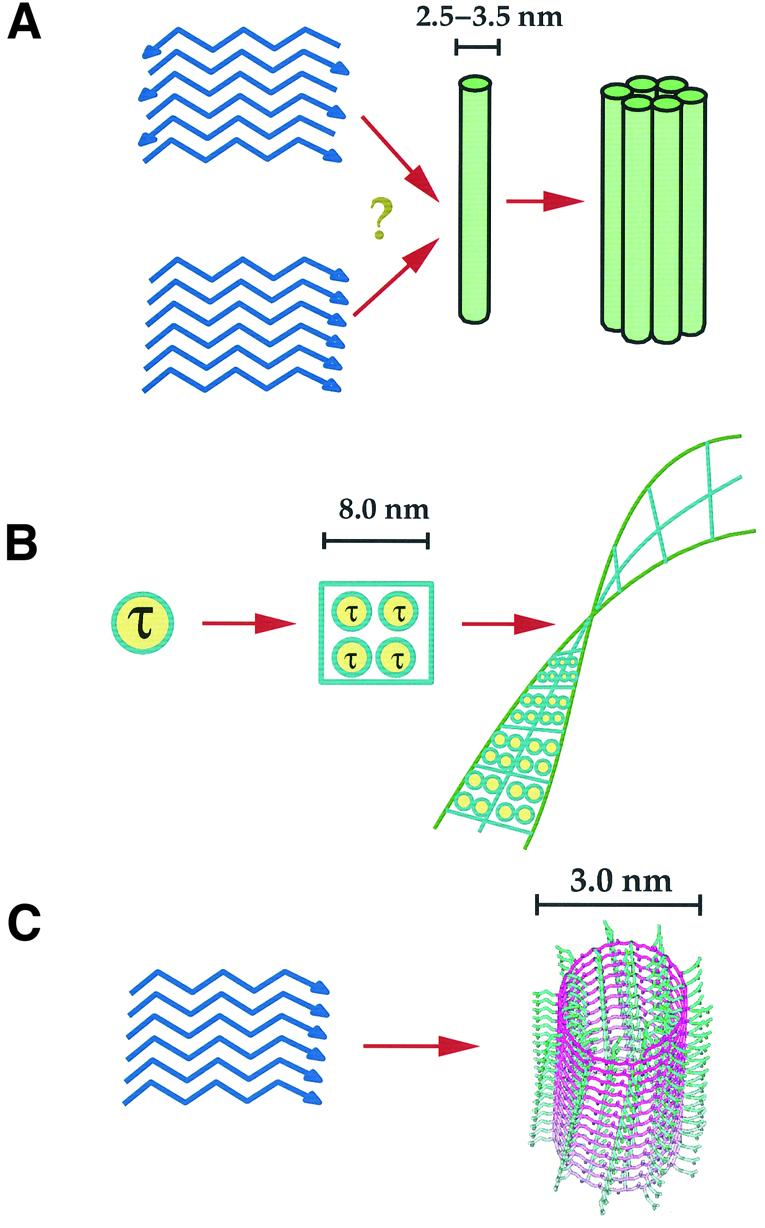
Fig. 2. Comparison of current models of amyloid fibres (A), tangles (B) and poly-Q aggregates (C). (A) The amyloid protofilament of 2.5–3.5 nm diameter would originate from β-strands running perpendicular to the fibre axes, although it is still disputed whether they run parallel or anti-parallel (Serpell, 2000). Several protofilaments are thought to further assemble to form mature fibrils. (B) Although no details are available yet about the molecular arrangement of tau in tangles, ample evidence of their morphology suggests an arrangement of paired helical filaments (Moreno-Herrero et al., 2001). (C) Model of water-filled nanotube in which the poly-Q forms a helical fibre with 20 residues per turn (Perutz et al., 2002). The main chains are shown in purple. The side chains (shown in green) protrude inside and outside the cylinder surface, according to β-sheet periodicity. Hydrogen bonds are formed both between the main chains and the side chains and would stabilize the structure.
A water-filled nanotube structure formed by a single cylindrical β-sheet stabilized by backbone and side chain hydrogen bonds was suggested as a possible model for poly-Q fibres (Perutz et al., 2002). No atomic model is yet available for NFTs. Morphological studies based on EM and atomic force microscopy show tangles as formed by intra-cellular paired helical filaments (PHF), which are assembled into two twisted ribbons with a helical pitch of ∼75–80 nm (Crowther and Wischik, 1985; Pollanen et al., 1997; Moreno-Herrero et al., 2001). Conflicting evidence exists about the structural nature of PHF. A hexapeptide 306VQIVYK311 from the beginning of the third repeat of tau has been shown to assemble into thin β-filaments (although without PHF appearance) and to nucleate PHFs from full-length tau (von Bergen et al., 2000). Such results are, however, at variance with a preliminary report claiming a helical structure for unpurified PHFs isolated from brain samples of AD patients (Sadqi et al., 2002).
Why should aggregates be toxic?
Even when we understand the mechanism leading to aggregate formation, we shall still need to explain how and why these are toxic for the cell. It is hard to imagine how a common mechanism could be consistent with the very different localizations of the polypeptide chains involved in AD and poly-Q diseases. Toxicity is particularly obscure for AD where, if toxicity were due to the plaques external to the neurons, aggregates could not interfere directly with the cell metabolism. It is natural to think that plaques adjacent to the cellular membrane can directly damage it. Whereas a trivial explanation might suggest the mechanical blockage by plaques of all cellular exchanges with the outer environment, a widely invoked cause of membrane damage has been free radical oxidative stress (Varadarajan et al., 2000). On a more general level, the idea that amyloid plaques in AD act as large reservoirs of species in equilibrium with smaller neurotoxic oligomers is gaining more credit (Walsh et al., 2002).
A specific mechanism for the toxicity of oligomeric assemblies seems to be suggested by solution studies of isolated Aβ: the surprising similarity of the structure of Aβ(1–42) to the fusion domain of influenza haemag glutinin in an apolar microenvironment (Crescenzi et al., 2002) points to membrane poration as the key event for the peptide neurotoxicity. The α-helical peptide would induce formation of membrane channels, allowing the penetration of substances (such as metal ions) that can cause neuronal death (Rhee et al., 1998; Lin et al., 2001). The ability of Aβ to induce vesicle fusion (Pillot et al., 1996) and the recent observation that Aβ can enhance infection at the stage of attachment or entry into the cell of several viruses further support this hypothesis (Wojtowicz et al., 2002).
A similar mechanism has been proposed to explain the toxicity of expanded poly-Q. Poly-Q stretches can induce large membrane-depolarizing ion channels in membranes which could damage the cell by altering the ionic balance (Hirakura et al., 2000; Monoi et al., 2000). The µ-helix conformation (Monoi, 1995) would provide a justification for the puzzling threshold that separates pathological (>35–40) from non-pathological lengths of poly-Q tracts in poly-Q diseases. The minimum peptide length required for the µ-helix to span the hydrophobic core of a lipid bilayer (estimated to be ∼3 nm) is ∼37 residues. Similarly, the restraints imposed by the helical nature of the model proposed by Perutz et al. (2002) impose a minimum of 40 residues for stabilizing the nanotube structure. The poration hypothesis is however not supported by a membrane localization of most of poly-Q proteins, in conflict with the possibility of a common mechanism related to poly-Q. Alternative albeit only theoretical models relate the threshold to an intrinsic random coil instability with respect to β-conformations in chains containing >40 glutamines (Starikov et al., 1999). A structural difference could promote interactions with cellular components which do not recognize short poly-Q proteins and/or lead more readily to aggregates. In both cases, the normal cellular functions would be altered.
Towards a unifying picture?
Although AD and poly-Q proteins were identified 10–15 years ago, still relatively few structural studies describe the conformation of these proteins both in their pathological and non-pathological forms. This is at least partially due to the specific challenges posed by this unusual class of polypeptides which respond differently to different environments. In all cases, significant information can be obtained only by using a multidisciplinary approach in which the properties in solution and in a solid-state environment are compared. While a common mechanism for misfolding diseases still remains to be proven, a unifying picture emerging from the current studies is that all peptides generated by proteolysis of the parent proteins are water insoluble and aggregate in vitro. However, protein fusion, complexation or a solvent medium can offset aggregation. It is clear that unambiguous identification of the entity prone to aggregation and a more detailed understanding of the structural bases of the conformational transition is a key step for designing possible therapies.
Acknowledgments
Acknowledgements
We are indebted to Arthur M.Lesk for providing the coordinates of the water-filled nanotube model. We thank Salvatore Adinolfi, Barbara Bardoni and Enzo Lalli, for critical reading of the manuscript.
References
- Balbach J.J., Petkova,A.T., Oyler,N.A., Antzutkin,O.N., Gordon,D.J., Meredith,S.C. and Tycko,R. (2002) Supramolecular structure in full-length Alzheimer’s β-amyloid fibrils: evidence for a parallel β-sheet organization from solid-state nuclear magnetic resonance. Biophys. J., 83, 1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R., Leger,J. and Lee,G. (1995) Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol., 13, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canu N. et al. (1998) Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J. Neurosci., 18, 7061–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles M., Bicknell,W., Watson,A.A., Fairlie,D.P. and Craik,D.J. (1998) Solution structure of amyloid β-peptide(1–40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry, 37, 11064–11077. [DOI] [PubMed] [Google Scholar]
- Crescenzi O., Tomaselli,S., Guerrini,R., Salvadori,S., D’Ursi,A., Temussi,P.A. and Picone,D. (2002) Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Eur. J. Biochem., 269, 5642–5648. [DOI] [PubMed] [Google Scholar]
- Crowther R.A. and Wischik,C.M. (1985) Image reconstruction of the Alzheimer paired helical filament. EMBO J., 4, 3661–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M., Sapp,E., Chase,K.O., Davies,S.W., Bates,G.P., Vonsattel,J.P. and Aronin,N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science, 277, 1990–1993. [DOI] [PubMed] [Google Scholar]
- Dobson C.M. (1999) Protein misfolding, evolution and disease. Trends Biochem. Sci., 24, 329–332. [DOI] [PubMed] [Google Scholar]
- Dyer R.B. and McMurray,C.T. (2001) Mutant protein in Huntington disease is resistant to proteolysis in affected brain. Nat. Genet., 29, 270–278. [DOI] [PubMed] [Google Scholar]
- Esposito G., Viglino,P., Novak,M. and Cattaneo,A. (2000) The solution structure of the C-terminal segment of tau protein. J. Pept. Sci., 6, 550–559. [DOI] [PubMed] [Google Scholar]
- Garcia M.L. and Cleveland,D.W. (2001) Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol., 13, 41–48. [DOI] [PubMed] [Google Scholar]
- Glenner G.G. and Wong,C.W. (1984) Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun., 120, 885–890. [DOI] [PubMed] [Google Scholar]
- Hardy J. and Selkoe,D.J. (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hirakura Y., Azimov,R., Azimova,R. and Kagan,B.L. (2000) Polyglutamine-induced ion channels: a possible mechanism for the neurotoxicity of Huntington and other CAG repeat diseases. J. Neurosci. Res., 60, 490–494. [DOI] [PubMed] [Google Scholar]
- Jarvet J., Damberg,P., Bodell,K., Eriksson,L.E.G. and Gräslund,A. (2000) Reversible random coil-to-sheet transition and the early stage of aggregation of the A(12–28). J. Am. Chem. Soc., 122, 4261–4268. [Google Scholar]
- Kienlen-Campard P., Miolet,S., Tasiaux,B. and Octave,J.N. (2002) Intracellular amyloid-β 1–42, but not extracellular soluble amyloid-β peptides, induces neuronal apoptosis. J. Biol. Chem., 277, 15666–15670. [DOI] [PubMed] [Google Scholar]
- Kim Y.J., Yi,Y., Sapp,E., Wang,Y., Cuiffo,B., Kegel,K.B., Qin,Z.H., Aronin,N. and DiFiglia,M. (2001) Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes and undergo calpain-dependent proteolysis. Proc. Natl Acad. Sci. USA, 98, 12784–12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner D.A., Abraham,C. and Selkoe,D.J. (1986) X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-β conformation. Proc. Natl Acad. Sci. USA, 83, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraepelin E. (1910) Psychiatrie: Ein Lehrbuch für Studierende und Artze. Barth, Leipzig, Germany, pp. 593–632.
- Lathrop R.H., Casale,M., Tobias,D.J., Marsh,J.L. and Thompson,L.M. (1998) Modeling protein homopolymeric repeats: possible polyglutamine structural motifs for Huntington’s disease. Proc. Int. Conf. Intell. Syst. Mol. Biol., 6, 105–114. [PubMed] [Google Scholar]
- Lee V.M., Goedert,M. and Trojanowski,J.Q. (2001) Neurodegenerative tauopathies. Annu. Rev. Neurosci., 24, 1121–1159. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler S.F., Beher,D., Grimm,H.S., Wang,R., Shearman,M.S., Masters,C.L. and Beyreuther,K. (2002) The intramembrane cleavage site of the amyloid precursor protein depends on the length of its transmembrane domain. Proc. Natl Acad. Sci. USA, 99, 1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Bhatia,R. and Lal,R. (2001) Amyloid β protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J., 15, 2433–2444. [DOI] [PubMed] [Google Scholar]
- Lunkes A., Lindenberg,K.S., Ben-Haiem,L., Weber,C., Devys,D., Landwehrmeyer,G.B. Mandel,J.L. and Trottier,Y. (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell, 10, 259–269. [DOI] [PubMed] [Google Scholar]
- Mangiarini L. et al. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell, 87, 493–506. [DOI] [PubMed] [Google Scholar]
- Masino L. and Pastore,A. (2002) Glutamine repeats: structural hypotheses and neurodegeneration. Biochem. Soc. Trans, 30, 548–551. [DOI] [PubMed] [Google Scholar]
- McGowan D.P., van Roon-Mom,W., Holloway,H., Bates,G.P., Mangiarini,L., Cooper,G.J., Faull,R.L. and Snell,R.G. (2000) Amyloid-like inclusions in Huntington’s disease. Neuroscience, 100, 677–680. [DOI] [PubMed] [Google Scholar]
- Mochizuki A., Tamaoka,A., Shimohata,A., Komatsuzaki,Y. and Shoji,S. (2000) Aβ42-positive non-pyramidal neurons around amyloid plaques in Alzheimer’s disease. Lancet, 355, 42–43. [DOI] [PubMed] [Google Scholar]
- Monoi H. (1995) New tubular single-stranded helix of poly-l-amino acids suggested by molecular mechanics calculations: I. Homopolypeptides in isolated environments. Biophys. J., 69, 1130–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monoi H., Futaki,S., Kugimiya,S., Minakata,H. and Yoshihara,K. (2000) Poly-l-glutamine forms cation channels: relevance to the pathogenesis of the polyglutamine diseases. Biophys. J., 78, 2892–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Herrero F., Valpuesta,J.M., Pérez,M., Colchero,J., Baró,A.M., Ávila,J. and Montejo de Garcini,E. (2001) Characterization by atomic force microscopy and cryoelectron microscopy of tau polymers assembled in Alzheimer’s disease. J. Alzheimer’s Dis., 3, 443–451. [DOI] [PubMed] [Google Scholar]
- Ordway J.M. et al. (1997) Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell, 91, 753–763. [DOI] [PubMed] [Google Scholar]
- Ortega-Aznar A., de la Torre,J. and Castelvi,J. (2000) The CNS amyloid. Rev. Neurol., 30, 1175–1180. [PubMed] [Google Scholar]
- Paulson H.L. (1999) Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold. Am. J. Hum. Genet., 64, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz M.F., Staden,R., Moens,L. and DeBaere,I. (1993) Polar zippers. Curr. Biol., 3, 249–253. [DOI] [PubMed] [Google Scholar]
- Perutz M.F., Finch,J.T., Berriman,J. and Lesk,A. (2002) Amyloid fibers are water-filled nanotubes. Proc. Natl Acad. Sci. USA, 99, 5591–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillot T., Goethals,M., Vanloo,B., Talussot,C., Brasseur,R., Vandekerckhove,J., Rosseneu,M. and Lins,L. (1996) Fusogenic properties of the C-terminal domain of the Alzheimer β-amyloid peptide. J. Biol. Chem., 271, 28757–28765. [DOI] [PubMed] [Google Scholar]
- Pollanen M.S., Markiewicz,P. and Goh,M.C. (1997) Paired helical filaments are twisted ribbons composed of two parallel and aligned components: image reconstruction and modeling of filament structure using atomic force microscopy. J. Neuropathol. Exp. Neurol., 56, 79–85. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. (1995) The prion diseases. Sci. Am., 272, 48–51, 54–57. [DOI] [PubMed] [Google Scholar]
- Rhee S.K., Quist,A.P. and Lal,R. (1998) Amyloid β protein-(1–42) forms calcium-permeable, Zn2+-sensitive channel. J. Biol. Chem., 273, 13379–13382. [DOI] [PubMed] [Google Scholar]
- Riek R., Güntert,P., Döbell,H., Wipf,B. and Wüthrich,K. (2001) NMR studies in aqueous solution fail to identify significant conformational differences between the monomeric forms of two Alzheimer peptides with widely different plaque-competence, A β(1–40)(ox) and A β(1–42)(ox). Eur. J. Biochem., 268, 5930–5936. [DOI] [PubMed] [Google Scholar]
- Ruben G.C., Iqbal,K., Grundke-Iqbal,I., Wisniewski,H.M., Ciardelli,T.L. and Johnson,J.E.,Jr (1991) The microtubule-associated protein tau forms a triple-stranded left-hand helical polymer. J. Biol. Chem., 266, 22019–22027. [PubMed] [Google Scholar]
- Sadqi M., Hernandez,F., Pan,U., Perez,M., Schaeberle,M.D., Avila,J. and Munoz,V. (2002) α-helix structure in Alzheimer’s disease aggregates of tau-protein. Biochemistry, 41, 7150–7155. [DOI] [PubMed] [Google Scholar]
- Scherzinger E., Sittler,A., Schweiger,K., Heiser,V., Lurz,R., Hasenbank,R., Bates,G.P., Lehrach,H. and Wanker,E.E. (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc. Natl Acad. Sci. USA, 96, 4604–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers O., Schonbrunn-Hanebeck,E., Marx,A. and Mandelkow,E. (1994) Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for β-structure. J. Biol. Chem., 269, 24290–24297. [PubMed] [Google Scholar]
- Serpell L.C. (2000) Alzheimer’s amyloid fibrils: structure and assembly. Biochim. Biophys. Acta, 1502, 16–30. [DOI] [PubMed] [Google Scholar]
- Shao H., Jao,S., Ma,K. and Zagorski,M.G. (1999) Solution structures of micelle-bound amyloid β-(1–40) and β-(1–42) peptides of Alzheimer’s disease. J. Mol. Biol., 285, 755–773. [DOI] [PubMed] [Google Scholar]
- Sontag E., Nunbhakdi-Craig,V., Lee,G., Brandt,R., Kamibayashi,C., Kuret,J., White,C.L.,III, Mumby,M.C. and Bloom,G.S. (1999) Molecular interactions among protein phosphatase 2A, tau and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem., 274, 25490–25498. [DOI] [PubMed] [Google Scholar]
- Spillantini M.G., Bird,T.D. and Ghetti,B. (1998) Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain. Pathol., 8, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starikov E.B., Lehrach,H. and Wanker,E.E. (1999) Folding of oligoglutamines: a theoretical approach based upon thermodynamics and molecular mechanics. J. Biomol. Struct. Dyn., 17, 409–427. [DOI] [PubMed] [Google Scholar]
- Sticht H., Bayer,P., Willbold,D., Dames,S., Hilbich,C., Beyreuther,K., Frank,R.W. and Rosch,P. (1995) Structure of amyloid A4-(1–40)-peptide of Alzheimer’s disease. Eur. J. Biochem., 233, 293–298. [DOI] [PubMed] [Google Scholar]
- Sunde M., Serpell,L.C., Bartlam,M., Fraser,P.E., Pepys,M.B. and Blake,C.C. (1997) Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol., 273, 729–739. [DOI] [PubMed] [Google Scholar]
- Taylor J.P., Hardy,J. and Fischbeck,K.H. (2002) Toxic proteins in neurodegenerative disease. Science, 296, 1991–1995. [DOI] [PubMed] [Google Scholar]
- Torok M., Milton,S., Kayed,R., Wu,P., McIntire,T., Glabe,C.C. and Langen,R. (2002) Structural and dynamic features of Alzheimer’s Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J. Biol. Chem., 277, 40810–40815. [DOI] [PubMed] [Google Scholar]
- Varadarajan S., Yatin,S., Aksenova,M. and Butterfield,D.A. (2000) Review: Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol., 130, 184–208. [DOI] [PubMed] [Google Scholar]
- von Bergen M., Friedhoff,P., Biernat,J., Heberle,J., Mandelkow,E.M. and Mandelkow,E. (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming β structure. Proc. Natl Acad. Sci. USA, 97, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D.M., Klyubin,I., Fadeeva,J.V., Cullen,W.K., Anwyl,R., Wolfe,M.S., Rowan,M.J. and Selkoe,D.J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature, 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Wei W., Norton,D.D., Wang,X. and Kusiak,J.W. (2002) Aβ 17–42 in Alzheimer’s disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain, 125, 2036–2043. [DOI] [PubMed] [Google Scholar]
- Welch W.J. and Diamond,M.I. (2001) Glucocorticoid modulation of androgen receptor nuclear aggregation and cellular toxicity is associated with distinct forms of soluble expanded polyglutamine protein. Hum. Mol. Genet., 10, 3063–3074. [DOI] [PubMed] [Google Scholar]
- Wellington C.L. et al. (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem., 273, 9158–9167. [DOI] [PubMed] [Google Scholar]
- Wojtowicz W.M., Farzan,M., Joyal,J.L., Carter,K., Babcock,G.J., Israel,D.I., Sodroski,J. and Mirzabekov,T. (2002) Stimulation of enveloped virus infection by β-amyloid fibrils. J. Biol. Chem., 277, 35019–35024. [DOI] [PubMed] [Google Scholar]
- Yang W., Dunlap,J.R., Andrews,R.B. and Wetzel,R. (2002) Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet., 11, 2905–2917. [DOI] [PubMed] [Google Scholar]
- Zhang S. et al. (2000) The Alzheimer’s peptide a β adopts a collapsed coil structure in water. J. Struct. Biol., 130, 130–141. [DOI] [PubMed] [Google Scholar]
- Zoghbi H.Y. and Orr,H.T. (2000) Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci., 23, 217–247. [DOI] [PubMed] [Google Scholar]