

Abstract
In yeasts, the replication protein Cdc6/Cdc18 is required for the initiation of DNA replication and also for coupling S phase with the following mitosis. In metazoans a role for Cdc6 has only been shown in S phase entry. Here we provide evidence that human Cdc6 (HuCdc6) also regulates the onset of mitosis, as overexpression of HuCdc6 in G2 phase cells prevents entry into mitosis. This block is abolished when HuCdc6 is expressed together with a constitutively active Cyclin B/CDK1 complex or with Cdc25B or Cdc25C. An inhibitor of Chk1 kinase activity, UCN-01, overcomes the HuCdc6 mediated G2 arrest indicating that HuCdc6 blocks cells in G2 phase via a checkpoint pathway involving Chk1. When HuCdc6 is overexpressed in G2, we detected phosphorylation of Chk1. Thus, HuCdc6 can trigger a checkpoint response, which could ensure that all DNA is replicated before mitotic entry. We also present evidence that the ability of HuCdc6 to block mitosis may be regulated by its phosphorylation.
Keywords: Cdc6/HuCdc6/cell cycle/checkpoint pathway/G2 phase
Introduction
In all eukaryotic cells, DNA replication is a tightly regulated process that is coordinated with other major cell cycle events to ensure that it occurs once per cell cycle and is completed before the genome is partitioned into two daughter cells (Diffley, 1996). Central to the regulation of DNA replication are the pre-replicative complexes (pre-RCs). The core of pre-RCs is the origin recognition complex (ORC), which is composed of six polypeptides first identified in Saccharomyces cerevisiae, and later found to be conserved in all eukaryotes (Bell and Stillman, 1992; Gavin et al., 1995; Quintana et al., 1997; Tugal et al., 1998; Dhar and Dutta, 2000).
Cdc6 (Cdc18 in Schizosaccharomyces pombe) is a protein that binds to ORC and is essential for the formation and maintenance of pre-RCs in yeast (Cocker et al., 1996; Tanaka et al., 1997). ORC and Cdc6 are required for the loading of minichromosome maintenance (MCM) proteins onto chromatin and they act together with Cdt1 (Maiorano et al., 2000; Nishitani et al., 2000). Once the MCM proteins are recruited to the pre-RCs, DNA is licensed for replication, and S phase is initiated by cyclin-dependent kinases (CDKs) and the Dbf4-dependent Cdc7 kinase, which promote the loading of Cdc45, replication protein-A (RPA) and DNA polymerase α onto chromatin (Jares and Blow, 2000; Walter and Newport, 2000). After the initiation of DNA replication, the pre-RCs are disassembled and chromatin remains in a post-replicative state until the passage through the next mitosis (Hendrickson et al., 1996; Aparicio et al., 1997; Liang and Stillman, 1997). The regulated assembly and disassembly of pre-RCs ensure that DNA replication occurs only once per cell cycle (Stillman, 1996; Dutta and Bell, 1997).
The regular alternation of S phase and mitosis in eukaryotic cells is reinforced by cell cycle checkpoints (for a review see Zhou and Elledge, 2000). The checkpoints are activated following DNA damage or by incomplete DNA replication, delaying progression of the cell cycle to provide time to repair or complete the event. Therefore, mitosis can only begin after completion of DNA replication. The checkpoints that ensure the dependence of mitosis on completion of S phase are critical because of the potential for genomic instability leading to tumour formation should these controls be impaired.
The notion that cells cannot enter mitosis until DNA synthesis is complete derives from two observations. First, it was observed in a series of cell fusion experiments that when a cell in S phase was fused with a cell in G2 phase, the G2 nucleus delayed entry into mitosis until the second nucleus had completed S phase (Rao and Johnson, 1970). This indicated that cells in S phase contain a soluble inhibitor of mitosis that can block a G2 nucleus from entering mitosis. Secondly, treating cells with inhibitors of DNA replication prevents mitosis in Xenopus extracts, in yeast and in many mammalian cell lines (Schlegel and Pardee, 1986; Dasso and Newport, 1990).
Mitosis begins with the activation of Cyclin B/CDK1 by the Cdc25 phosphatases. If S phase is inhibited, Cyclin B/CDK1 is kept inactive through a cascade of checkpoint regulators, involving the Chk1 and Chk2, ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia-mutated and Rad-3 related (ATR) kinases (for a review see Zhou and Elledge, 2000). However, less is known about how a cell monitors whether S phase is complete.
In S.cerevisiae, Cdc6 is also involved in controlling mitosis. Cdc6 acts as a suppressor of mitotic catastrophe and its overexpression interferes with progression through G2 causing a dramatic delay in entry into mitosis (Bueno and Russell, 1992). High levels of Cdc18 in S.pombe also cause a cell cycle delay and block the onset of mitosis (Kelly et al., 1993; Nishitani and Nurse, 1995). The Cdc18-mediated block to mitosis is dependent on the inhibition of CDK activity and the integrity of checkpoint pathways, showing that Cdc18 acts upstream of DNA replication checkpoint genes belonging to the rad family (Greenwood et al., 1998).
In yeast, Cdc6/Cdc18 is targeted for proteolysis at the onset of S phase and degradation requires CDK-dependent phosphorylation of Cdc6 and the Skp1-Cullin-F-box-protein (SCF/CDC4) complex (Kelly et al., 1993; Baum et al., 1998; Drury et al., 2000). In human cells, phosphorylation of HuCdc6 by Cyclin A/CDK2 causes export of HuCdc6 from the nuclei after initiation of DNA replication (Jiang et al., 1999; Petersen et al., 1999) and HuCdc6 remains mainly cytoplasmic until it is targeted for proteolysis by the anaphase promoting complex/cyclosome (APC/CDH1) in early G1 (Petersen et al., 2000). We reasoned that HuCdc6 might have an additional function after S phase and it might be a good candidate for a replication checkpoint mechanism. To investigate this hypothesis, we have overexpressed HuCdc6 by microinjection into G2 phase HeLa cells and assayed the effect on mitosis. We find that HuCdc6 blocks entry into mitosis indicating that it could monitor or signal ongoing DNA replication through a checkpoint mechanism.
Results
Overexpression of HuCdc6 causes a G2 arrest in HeLa cells
To look for an additional function of HuCdc6 beyond initiation of DNA replication, we analysed the effect of HuCdc6 overexpression in G2 phase cells. We microinjected an expression plasmid encoding HuCdc6 cDNA and green fluorescent protein (GFP) as a fusion protein (pEGFP-HuCdc6) into G2-synchronized HeLa cells. As a control, we used a plasmid expressing GFP only (pEGFP) and compared cells injected with pEGFP-HuCdc6, pEGFP control and uninjected cells in each experiment. HuCdc6–GFP and GFP became detectable by epifluorescence microscopy 1 h after plasmid injection, while the majority of cells were still in G2 phase. At this point, the cells were followed by time-lapse microscopy; DIC (Differential Interference Contrast) and fluorescence images were taken every 30 min over a 10 h period and every 3 min once cells entered mitosis. The use of pEGFP as a control in each experiment allowed us to estimate the effect of injection alone on entry into and progress through mitosis. Although 60–80% of uninjected cells entered mitosis over the course of the experiment (data not shown), we routinely observed that 35–55% of GFP expressing cells entered mitosis (Figure 1A and C). The slightly lower percentage was probably due to some inevitable stress caused by the microinjection. In contrast, we found that overexpression of HuCdc6 blocked cells in G2 phase; <3% of these cells entered mitosis (Figure 1B and D). Almost all the mitotic cells (rounded and arrowed cells in Figure 1B) were uninjected, as shown by their lack of fluorescence. This result was reproducible in several independent experiments and was specific for HuCdc6 as overexpression of HuMcm5, another replication protein, had no effect on entry into mitosis (data not shown). To ensure that the G2 phase arrest was not an artefact introduced by the GFP tag, we overexpressed untagged HuCdc6 in a similar set of experiments to those described above. We were able to follow HuCdc6 injected cells by co-injecting pEGFP as a marker. Overexpression of untagged HuCdc6 also caused cells to arrest in G2 phase, in a similar fashion to pEGFP-HuCdc6 (data not shown).

Fig. 1. In vivo analysis of HuCdc6 overexpression in G2 cells. (A) pEGFP and (B) pEGFP-HuCdc6 were microinjected into the nucleus of G2 phase HeLa cells. The behaviour of injected cells was observed by time-lapse fluorescence and DIC microscopy and images were taken every 30 min, or every 3 min after entry into mitosis over a 10 h period. Approximately 100 fluorescent cells expressing GFP or HuCdc6–GFP were classified according to their cell cycle stage: G2 phase, mitosis and after completion of mitosis (early G1 phase) during different time points starting from 1 h after microinjection. These numbers were compared with the total number of cells expressing GFP or HuCdc6–GFP (C and D). While 40–45% of cells expressing GFP went through mitosis [arrows in (A) and (B) show mitotic cells], only 3–5% of cells expressing HuCdc6–GFP did so. Representative images, mean values and standard deviations of five independent experiments are shown. (E) Comparison of expression of HuCdc6–GFP to endogenous HuCdc6 levels. A total of 1000 cells were injected with pEGFP-HuCdc6 and after 2 h were directly lysed in SDS buffer, proteins were separated on 10% SDS–PAGE and immunoblotted for HuCdc6. (F) Anti human Cdc6 antibody neutralizes HuCdc6 overexpression and abolishes the G2 phase arrest. G2 phase HeLa cells were microinjected with HuCdc6–GFP and a specific anti HuCdc6 antibody. As a control, cells were injected with the anti HuCdc6 antibody and Texas Red dextran. The percentages of cells expressing pEGFP-HuCdc6 in G2, M and G1 phases were calculated and compared with the control cells 7 h after injection. Mean values and standard deviation were calculated from at least three independent experiments.
As a further control, we microinjected a GFP-tagged version of the HuCdc6 protein purified from baculovirus-infected insect cells (Sf9) into G2 phase HeLa cells. The recombinant HuCdc6 protein blocked the cells in G2 phase (data not shown). These experiments demonstrate that upregulation of HuCdc6 protein in G2 HeLa cells blocks cell progression into mitosis.
To quantify the amount of HuCdc6 in overexpression experiments, we injected 1000 HeLa cells with pEFGP-HuCdc6 and immunoblotted for HuCdc6 (Figure 1E). After quantification by NIH Image we found that HuCdc6–GFP was expressed at ∼4-fold over the endogenous HuCdc6 in G2 cells. Moreover, HuCdc6–GFP levels were only ∼1.5-fold over these of endogenous HuCdc6 in cells blocked in S phase (data not shown).
Importantly, co-injecting an anti-HuCdc6 polyclonal antibody with pEGFP-HuCdc6 abolished the G2 phase block and cells entered mitosis in a similar fashion to control cells injected with anti-HuCdc6 antibody alone (Figure 1F). These data strengthen the conclusion that the HuCdc6-mediated G2 arrest is due to HuCdc6 overexpression. In addition, the localization of HuCdc6 (either recombinant protein or expressed from a plasmid) was mainly cytoplasmic, consistent with the localization of the endogenous HuCdc6 protein in G2 phase (Fujita, 1999). Importantly, we observed the same G2 arrest in untransformed cell lines such as NIH 3T3 mouse fibroblasts and Ptk1 kangaroo rat fibroblasts (data not shown).
We also investigated the possibility that HuCdc6 overexpression caused cells to re-replicate. Therefore we stained G2 cells overexpressing HuCdc6 with bromodeoxyuridine (BrdU). However, we did not detect DNA synthesis in these cells (data not shown). As it is difficult to determine precisely when S phase is complete and G2 phase begins, we determined whether overexpressed HuCdc6 disturbs DNA synthesis at late origins and stalls the replication forks. Overexpression of HuCdc6 in late S phase cells showed clear late patterns of BrdU incorporation, comparable with the pattern obtained from uninjected cells (data not shown).
Overexpressed HuCdc6 does not directly inhibit MPF
To enter mitosis, eukaryotic cells need to activate M-phase promoting factor (MPF), a heterodimeric complex containing a B-type cyclin and its associated serine/threonine kinase Cdc2 (CDK1 in mammalian cells; Nurse, 1990). MPF is kept inactive until the time of entry into mitosis through phosphorylation on CDK1 by the kinases Wee1 and Myt1 (Morgan, 1995). These kinases phosphorylate CDK1 at two sites, threonine residue 14 (T14) and tyrosine residue 15 (Y15) (Norbury et al., 1991; Kornbluth et al., 1994; Mueller et al., 1995). To initiate mitosis, the inhibitory phosphate groups are removed from CDK1 by the phosphatase Cdc25 and this activates MPF (Dunphy and Kumagai, 1991; Gautier et al., 1991; Millar et al., 1991). Previous work had shown that when fission yeast Cdc18 was expressed at high levels, it blocked the onset of mitosis by binding to Cdc2 and inhibiting its kinase activity (Brown et al., 1997; Greenwood et al., 1998; Lopez-Girona et al., 1998). Similarly, in budding yeast it had been shown that Cdc6 inhibited Cdc28 (CDK1) in mitosis leading to a mitotic arrest (Calzada et al., 2001; Weinreich et al., 2001). To determine whether this was also true for HuCdc6, we co-injected pEGFP-HuCdc6 in G2 HeLa cells with either a complex of Cyclin B1 and wild-type CDK1 (pCyclin B with pCDK1wt) or a constitutively active complex where the CDK1 subunit carries the mutations T14A and Y15F (CDK1AF; Hagting et al., 1998). The Cyclin B/CDK1AF complex cannot be kept inactive by the negative regulatory kinases Wee1 or Myt1 (Morgan, 1995) and does not require activation by Cdc25 (Hagting et al., 1998). We found that 40% of the cells expressing HuCdc6 and Cyclin B1/CDK1AF entered mitosis (Figure 2A and C). In agreement with a previous report (Hagting et al., 1998), we also observed that most of the cells expressing Cyclin B1/CDK1AF underwent premature mitosis (independently of HuCdc6 overexpression). This resulted in cells accumulating in mitosis and never re-entering the G1 phase (Figure 2C). In contrast, the majority of cells overexpressing HuCdc6 and wild-type Cyclin B1/CDK1 were still blocked in G2 (Figure 2B and C). These results demonstrate that HuCdc6 does not prevent mitosis simply by binding and inhibiting Cyclin B1/CDK1, indicating that HuCdc6 blocks upstream of MPF activity, possibly acting on Cdc25. There are three different Cdc25 genes in human cells; Cdc25A, Cdc25B and Cdc25C (Galaktionov and Beach, 1991; Nagata et al., 1991). To test the possibility that HuCdc6 functions at the level of Cdc25, we co-injected pEGFP-HuCdc6 together with pCdc25B or pEGFP-Cdc25C in G2 HeLa cells. Figure 3A and C shows that overexpression of Cdc25B together with HuCdc6 overcomes the G2 arrest and cells enter mitosis in exactly the same way as with Cdc25B alone (Figure 3E). High levels of Cdc25B caused the cells to round up and break down their nuclear envelope prematurely and these cells arrested in mitosis with abnormally condensed chromosomes, as previously described (Karlsson et al., 1999). Control uninjected cells, or cells expressing only Cdc25C started to enter mitosis 2 h after injection (Figure 3F), while the cells expressing HuCdc6 and Cdc25C (Figure 3B and D) entered mitosis 6 h after injection, i.e. with a 4 h delay. Thus, the HuCdc6-mediated G2 arrest could be overcome by overexpressing either Cdc25B or Cdc25C.
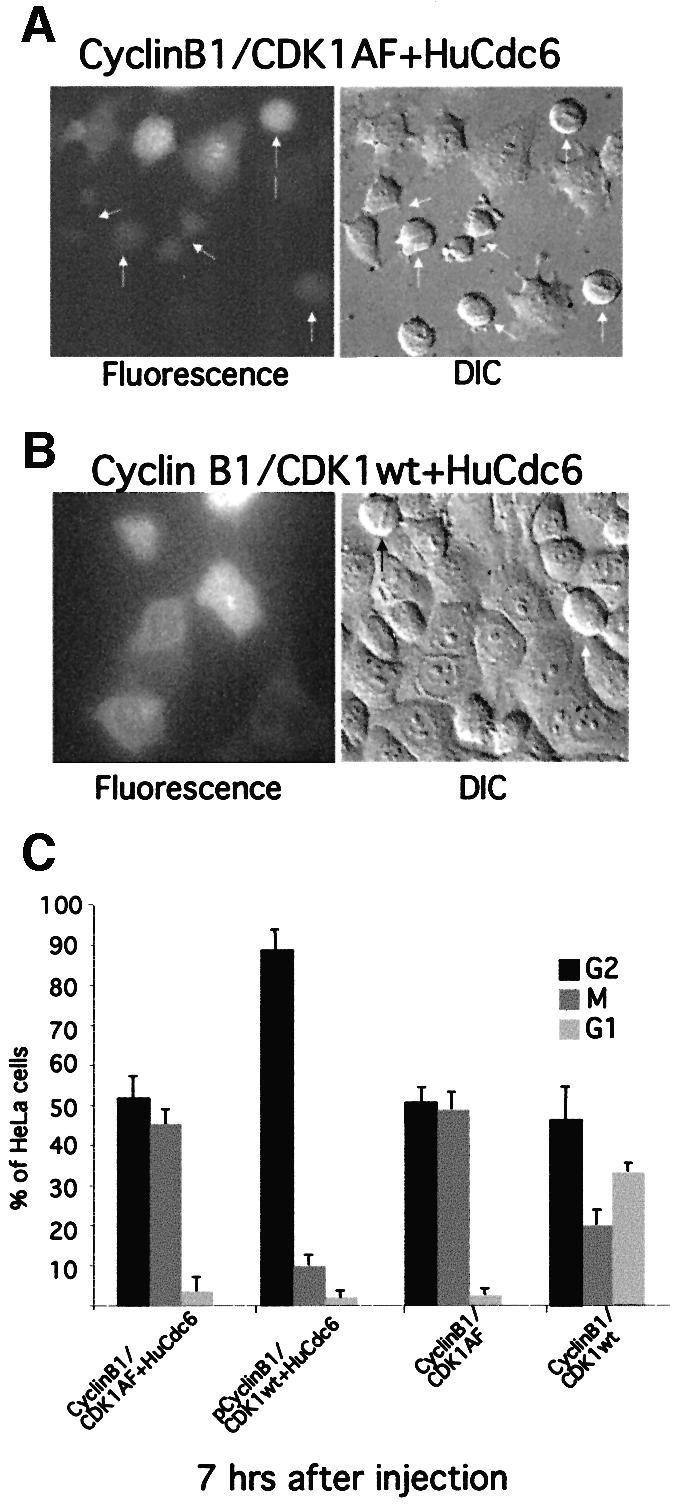
Fig. 2. Constitutively active MPF abolishes the G2 phase arrest caused by HuCdc6 overexpression. G2 phase HeLa cells were microinjected with (A) pEGFP-HuCdc6, pCyclin B1 and a constitutively active pCDK1AF or (B) with pEGFP-HuCdc6, pCyclin B1 and pCDK1wt. As controls we co-injected pCyclin B1 and pCDK1AF or pCyclin B1 and pCDK1wt were injected without HuCdc6 as a control (C). Approximately 100 fluorescent cells were counted for each sample. Fluorescence and DIC images were taken 7 h after injection. Arrows show mitotic cells. (C) The numbers of cells in G2 phase, mitosis and in early G1 phase were scored and calculated as a percentage of the total of injected cells. At least three independent experiments were performed and representative images are shown.

Fig. 3. Overexpression of Cdc25 abolishes the G2 phase arrest caused by HuCdc6–GFP. G2 phase cells were microinjected with pCdc25B and pEGFP-HuCdc6 (A) or with pEGFP-Cdc25C and pHuCdc6 (B). Arrows in (A) and (B) show mitotic cells. Cells were followed by time-lapse fluorescence and DIC microscopy as described in Figure 1, starting from 2 h after microinjection during a 7 h time course. Sixty per cent of cells entered mitosis prematurely and arrested in mitosis whether they expressed HuCdc6–GFP and Cdc25B (C) or Cdc25B alone (E). (D) Forty five per cent of cells expressing Cdc25C and HuCdc6–GFP entered and progressed through mitosis, but often with a 4 h delay compared with the 2 h delay of the control cells expressing Cdc25C only (F).
Abrogation of Chk1 activity by UCN-01 relieves Cdc6-mediated G2 arrest
The experiments described above showed that overexpression of HuCdc6 arrested cells in G2 phase, which was reminiscent of a cell cycle arrest due to DNA damage or incomplete DNA replication. Therefore, we investigated whether proteins of the replication checkpoint pathway might be involved in the Cdc6-mediated G2 arrest.
A hallmark of checkpoint activation in the presence of incomplete DNA replication is the activation of the protein kinase Chk1, which in turn phosphorylates Cdc25 on serine residue 216 creating a binding site for a 14-3-3 protein (Furnari et al., 1997; Peng et al., 1997; Sanchez et al., 1997; Zeng et al., 1998). Once the 14-3-3 protein has bound, Cdc25 activity is downregulated (Peng et al., 1997). To determine whether the HuCdc6-mediated block involved Chk1, we took advantage of the ability to inhibit Chk1 kinase activity using the inhibitor UCN-01. It had been reported that UCN-01 potently inhibited the kinase activity of Chk1, but not that of Chk2 or other kinases involved in checkpoint responses, such as ATM or ATR proteins (Graves et al., 2000).
We microinjected pEGFP-HuCdc6 in G2 HeLa cells, added 300 nM UCN-01 to the medium after microinjection and assayed the number of HuCdc6–GFP-expressing cells that entered mitosis over the next 10 h. We found that ∼21% of HuCdc6–GFP-expressing cells entered and progressed through mitosis in the presence of UCN-01 (Figure 4A and B), which was comparable to the 24% of GFP-expressing cells that entered mitosis (Figure 4B). This indicated that Chk1 was activated when HuCdc6 was overexpressed in G2 cells. To confirm this, we injected 1000 cells with pEGFP-HuCdc6 or pEGFP, lysed the cells in SDS buffer, separated the proteins by 10% SDS–PAGE and immunoblotted for Chk1. The same was done for 1000 cells treated with hydroxyurea (HU) or 1000 untreated cells (Figure 4C). No mobility shift was observed in uninjected and untreated control cells (Figure 4C lanes 2, 4 and 6). As demonstrated previously, Chk1 migrated with reduced mobility on SDS–PAGE after HU treatment (Figure 4C, lane 1), implicating phosphorylation of Chk1 (Feijoo et al., 2001). Importantly, in cells expressing HuCdc6–GFP (Figure 4C, lane 5) Chk1 showed a mobility shift comparable to that of HU-treated cells, whereas there was no change in the mobility of Chk1 in cells expressing GFP alone (Figure 4C, lane 3). These data demonstrated that overexpression of HuCdc6 in G2 HeLa cells led to phosphorylation of Chk1. HuCdc6 might have acted by binding and activating Chk1. To test this, we assayed for an interaction between HuCdc6 and Chk1 by immunoprecipitation. Extracts from G2 HeLa cells, untreated or treated with HU were immunoprecipitated and immunoblotted with anti-Chk1 or anti-HuCdc6 antibodies. Under none of the experimental conditions tested could we detect co-precipitation of the two proteins, whereas in our control experiments we found that Cyclin A co-precipitated with HuCdc6 (data not shown), in agreement with published results (Petersen et al., 1999). However, it was possible that the interaction between Chk1 and HuCdc6 is transient or does not survive our experimental conditions.
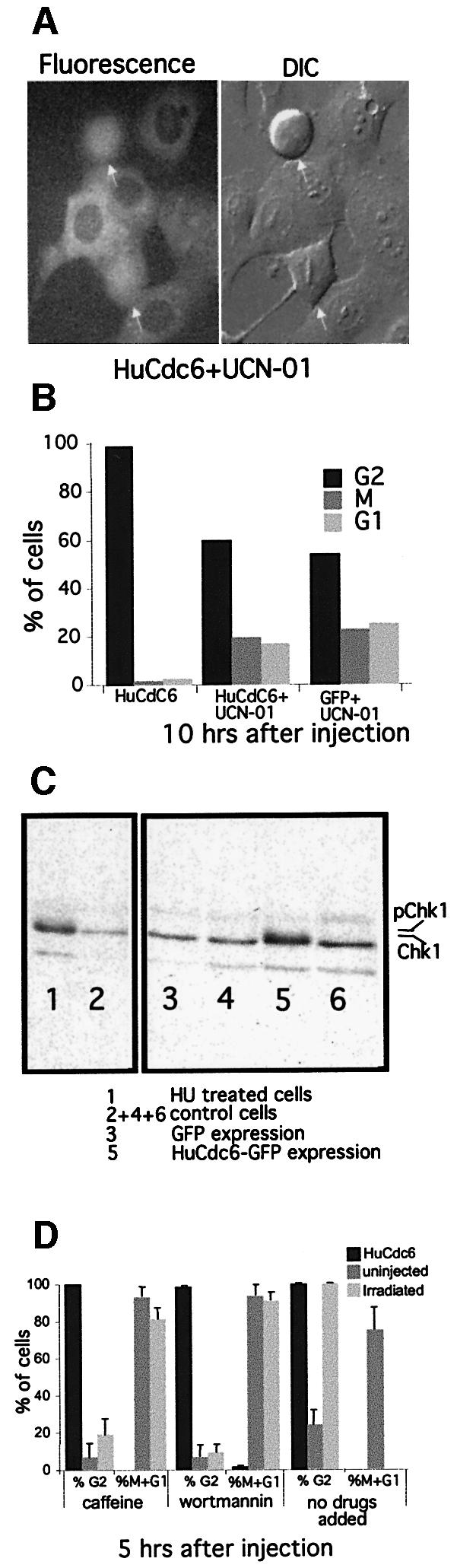
Fig. 4. An inhibitor of Chk1 kinase activity, UCN-01, abolishes HuCdc6-mediated G2 arrest. (A) G2 phase HeLa cells were microinjected with pEGFP-HuCdc6 or pEGFP as a control (not shown). UCN-01 (300 nM) was added to the medium immediately after the injections. Fluorescence and DIC images were taken during a 10 h period. G2 phase and mitotic cells (marked with an arrow) were counted from a total of ∼100 fluorescent cells over a 10 h period. (B) Percentage of cells progressing through mitosis expressing HuCdc6 or GFP. Twenty-one percent of cells expressing HuCdc6–GFP entered and progressed through mitosis comparable with the 24% of GFP expressing cells. Two experiments were performed and representative images are shown. (C) HuCdc6-mediated phosphorylation of Chk1. One thousand cells were injected with pEGFPHuCdc6 or pEGFP, uninjected but untreated or treated with HU (control cells). Proteins were separated on SDS–PAGE and immunoblotted for Chk1. Lane 1 shows a mobility shift due to phosphorylation after HU treatment, whereas control cells do not (lanes 2, 4 and 6). HuCdc6 shows a similar mobility shift as HU-treated cells (lane 1), whereas GFP alone does not (lane 3). (D) The HuCdc6-mediated G2 phase arrest is maintained in the presence of caffeine and/or wortmannin. Cells were microinjected with pEGFP-HuCdc6 in the presence or absence of 5 mM caffeine and/or 25 µM wortmannin. As a control, cells were γ-irradiated (100 kVp/min) for 15 min and caffeine and/or wortmannin were added. Cells were followed for 10 h counting cells entering and progressing through mitosis as one (%M + G1). Uninjected cells and irradiated cells entered into mitosis prematurely in the presence of caffeine and/or wortmannin. The cells expressing HuCdc6 remained arrested in G2 phase in the presence of caffeine and/or wortmannin.
We also investigated whether the two kinases upstream of Chk1, namely ATM and ATR could have been involved in the HuCdc6-mediated G2 arrest. The ATM protein kinase is a key component of the DNA damage checkpoint (Savitsky et al., 1995) and ATR is involved in the DNA replication checkpoint that is activated by unreplicated DNA (Cimprich et al., 1996; Gatei et al., 2000; Zhao and Piwnica-Worms, 2001). ATM and ATR can phosphorylate Chk1 and Chk2, respectively, to cause cell cycle arrest. We microinjected pEGFP-HuCdc6 in G2 HeLa cells and added either 5 mM caffeine or 25 µM wortmannin to the medium. Caffeine inhibits both ATM (Blasina et al., 1999; Zhou et al., 2000) and ATR function (Hall-Jackson et al., 1999), and wortmannin is believed preferentially to inhibit ATM and DNA-PK at micromolar quantities (Izzard et al., 1999). To our surprise, neither caffeine nor wortmannin, alone or in combination (data not shown) were able to abolish the G2-phase block caused by overexpressing HuCdc6 (Figure 4D). In contrast, caffeine was able to overcome a G2 arrest induced by γ-irradiation (100 kVp/min, Figure 4D). The HuCdc6-mediated block was maintained even when the drugs were added fresh to the medium twice during the experiment: before, during or straight after microinjection. Furthermore, HuCdc6 and ATR did not co-immunoprecipitate (data not shown) and ATM-deficient human fibroblasts were not able to enter mitosis in the presence of overexpressed HuCdc6 (data not shown).
Selected nonphosphorylatable mutants of HuCdc6 do not arrest HeLa cells in G2 phase
We considered the possibility that HuCdc6 might have to be modified to arrest cells in G2 phase. Although HuCdc6 was known to be phosphorylated in S phase, this was not required for the initiation of DNA replication (Petersen et al., 1999; Pelizon et al., 2000; Coverley et al., 2002). Therefore, we mutated the three CDK phosphorylation sites of HuCdc6 (serine 54, 74 and 106) to alanine. We also deleted the cyclin-binding-motif, amino acid 93–100, (Δcy-motif) as shown in Figure 5A. These mutants were injected into G2 HeLa cells as in previous experiments. HuCdc6 mutants S74A and Δcy-motif were not able to arrest cells in G2, whereas S54A and S106A both arrested cells in G2 in a similar fashion to wild-type HuCdc6 (Figure 5B). Figure 5C shows that S54A was both nuclear and cytoplasmic, S74A was completely nuclear and S106A was mainly cytoplasmic, comparable to wild-type HuCdc6 (Figure 1C). The Δcy-motif mutant was also mainly nuclear (Figure 5C). Although there was a correlation between HuCdc6 localization and its ability to arrest cells in G2, we do not believe that the localization of HuCdc6 was important because HuCdc6 bound to a functional nuclear localization signal (NLS) was constitutively nuclear but still arrested cells in G2 (data not shown). Instead it appeared more likely that the phosphorylation of HuCdc6 at specific sites was important for its function in preventing mitosis.
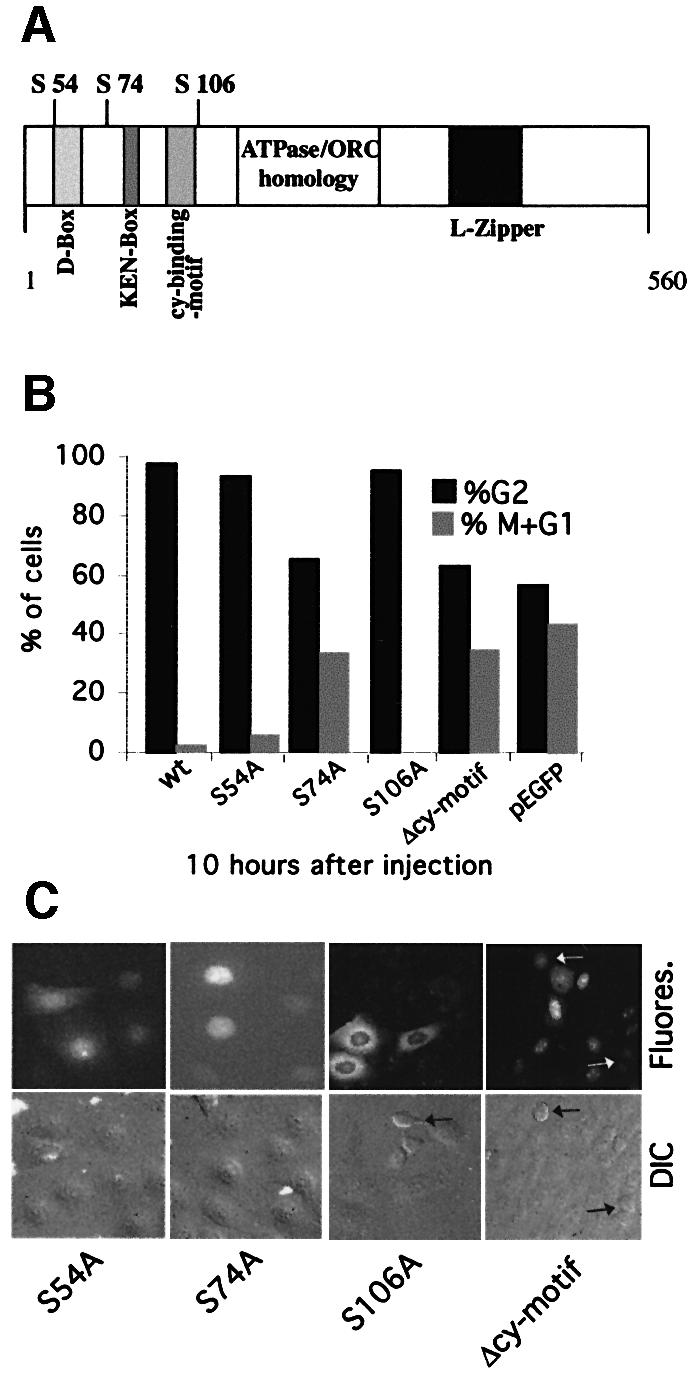
Fig. 5. Behaviour of HuCdc6 mutants in G2 cells. (A) Schematic drawing of HuCdc6 depicting features such as the CDK phosphorylation sites serines (S) 54, 74 and 106, destruction box, KEN box, the cyclin-binding-motif, the ATPase/ORC homology domain and leucine-zipper, all previously identified. (B) The pEGFP-HuCdc6 mutants S54A, S74A, S106A and Δcy-motif were injected into G2 HeLa cells and their progression into mitosis compared with GFP or wtHuCdc6–GFP-expressing cells. Cells expressing the mutants S75A and Δcy-motif cannot arrest cells in G2, whereas cells expressing S54A and S106A do arrest cells in G2 in the same fashion to wtHuCdc6. (C) Illustration of the localization of the mutants. Two independent experiments were performed and representative images are shown.
Discussion
In this work we present evidence that HuCdc6 has a function in coupling S phase with the following mitosis in human cells. We found that overexpression of HuCdc6 in G2 phase HeLa cells is sufficient to arrest the cells in G2. (i) Microinjection of pEGFP-HuCdc6, untagged pHuCdc6 or recombinant HuCdc6–GFP protein causes the cells to arrest in G2, whereas microinjection of pEGFP does not. Thus, neither the GFP tag, nor injection of plasmid DNA per se is responsible for the G2 arrest. (ii) Co-injection of a specific anti-human Cdc6 antibody with pEGFP-HuCdc6 overcomes the G2 phase block. (iii) The G2 arrest is specific for HuCdc6 and not for Mcm5 (data not shown). (iv) A specific point mutation of HuCdc6 abolishes its ability to arrest cells in G2 phase.
A previous study in HeLa cells failed to show a perturbation in cell cycle progression by FACS analysis after the transfection of HuCdc6 into asynchronous cells (Petersen et al., 2000). Although the reason for this discrepancy is not clear, we believe that it may be due to the use of different experimental approaches. For example after transfection, proliferating asynchronous cells may be able to modify exogenous HuCdc6 before they reach G2 in a way that mimics the fate of the endogenous protein making it unlikely to trigger a checkpoint response.
Previous work has shown that Cdc6/Cdc18 overexpression in yeast leads to a mitotic block in part by binding and inhibiting Cdc28 or Cdc2 (homologues of CDK1) or by inducing a checkpoint response (Bueno and Russell, 1992; Kelly et al., 1993; Nishitani and Nurse, 1995; Baum et al., 1998; Greenwood et al., 1998; Calzada et al., 2001; Weinreich et al., 2001). Our experiments suggest that HuCdc6 does not directly inhibit Cyclin B/CDK1. When HuCdc6 is co-expressed with Cyclin B and a constitutively active form of CDK1 (CDK1AF) the cells enter mitosis in similar numbers to cells co-expressing Cyclin B/CDK1AF without HuCdc6–GFP. In contrast, cells co-expressing HuCdc6–GFP with wild-type Cyclin B/CDK1 remain arrested in G2. Thus, in agreement with previous findings showing that HuCdc6 is not associated with Cyclin B/CDK1 complexes in vitro (Petersen et al., 1999), our data indicate that HuCdc6 does not directly inhibit MPF in vivo. Furthermore, overexpression of either Cdc25B or Cdc25C can overcome the G2 arrest caused by HuCdc6. The noticeable delay in time of entry into mitosis in cells co-expressing Cdc25C and HuCdc6 (4 h later than those co-expressing Cdc25B and HuCdc6) is probably due to an initial G2-phase cell arrest due to HuCdc6. This can be explained by the fact that Cdc25C needs to be phosphorylated to be activated, whereas Cdc25B is constitutively active (Lammer et al., 1998; Karlsson et al., 1999). These results strengthen our data indicating that HuCdc6 does not directly inhibit MPF, but is more likely to act upstream of MPF.
An important finding is that Chk1 seems to be activated and phosphorylated when HuCdc6 is overexpressed in G2 phase, as seen by a mobility shift of Chk1 on western blots (Figure 4C). This corroborates the observation that UCN-01 abolishes the G2 arrest induced by HuCdc6. Although we could not see a direct interaction between Chk1 and HuCdc6 by immunoprecipitation, it is important to keep in mind that a direct interaction might not be necessary to trigger a checkpoint response. In addition, we could not demonstrate a link between HuCdc6 overexpression and the activation of Chk1 by ATM or ATR. Neither caffeine nor wortmannin abolished the G2 arrest when HuCdc6 is overexpressed whereas they did abolish a G2 arrest induced by γ-irradiation. This is slightly puzzling, since until now DNA checkpoint responses have been believed to be either ATM- or ATR-mediated. In addition we could not find a direct interaction between HuCdc6 and ATR by immunoprecipitation, but at this stage we cannot rule out an involvement of ATM and ATR in the HuCdc6 mediated arrest.
We also found that the ability of HuCdc6 to inhibit cells in G2 may be modulated by its phosphorylation. It has previously been shown that phosphorylation of HuCdc6 is not required for its function in the initiation of DNA replication, because non-phosphorylatable mutants of HuCdc6 are able to support DNA replication (Petersen et al., 1999; Pelizon et al., 2000). This raises the intriguing possibility that phosphorylated HuCdc6 is involved in the checkpoint function that prevents the cells from undergoing premature mitosis. We microinjected different phosphorylation site HuCdc6 mutants into G2-phase HeLa cells and found that mutation of serine 74 to alanine (S74A) cannot arrest cells in G2, whereas S54A and S106A mutants can. In addition, a cyclin-binding-motif deletion mutant (Δcy-motif) is also unable to arrest cells in G2, suggesting that phosphorylation of specific residues, such as S74 by cyclin/CDKs may be necessary for the checkpoint function of HuCdc6. In S.cerevisiae it has been shown that Cdc6 binding to cyclin is important for cells to exit mitosis and that a cyclin-binding motif deletion mutant arrests cells in late mitosis (Calzada et al., 2001; Weinreich et al., 2001). However, by following our Δcy-motif mutant through mitosis by live-imaging, we did not observe a delay in exit from mitosis.
Greenwood et al. (1998) showed that the Cdc18-mediated block in S.pombe is caused by the N-terminal region of the protein binding to CDK1 and also through the C-terminus operating via the DNA replication checkpoint control genes rad1, rad3, rad9, rad17, hus1 and cut5. Thus, in agreement with results from studies using yeasts (Kelly et al., 1993; Nishitani and Nurse, 1995; Baum et al., 1998; Greenwood et al., 1998), we believe that Cdc6 has a second function in human cells. Besides its role in the assembly of pre-RC during G1 phase, HuCdc6 may regulate the onset of mitosis through a checkpoint pathway. We propose that the level and/or modification of HuCdc6 protein in G2 phase might function as a signal monitoring ongoing DNA replication. Our results might also underlie some of the observations of Rao and Johnson (1970). In their cell fusion experiments, they showed that a G2 nucleus does not enter mitosis until the S phase nucleus has completed DNA replication, indicating that there are soluble inhibitors produced during S phase that are able to regulate mitosis. According to our model (Figure 6), during an unperturbed cell cycle, HuCdc6 monitors ongoing DNA replication. Once DNA replication is completed, HuCdc6 may be phosphorylated at specific sites to inactivate its ability to block mitosis. A small population of Chk1 might also be activated in late S phase, ensuring that late origins can fire without cells committing to mitosis. However, if Cdc6 is not inactivated (e.g. because HuCdc6 is overexpressed in G2 cells as in our experiments) HuCdc6 inhibitory function would impose a block to mitosis. In this case, Chk1 would be activated rapidly to ensure that cells do not enter mitosis, perhaps by being more sensitive to HuCdc6 in G2 than in S phase. In agreement with this hypothesis, we find that microinjection of recombinant protein into S phase cells does not arrest cells before mitosis, whereas the same amount of HuCdc6 protein injected into G2 cells abrogates mitotic entry (data not shown). The exact nature of the modifications that inactivate HuCdc6 is still unclear but in the light of the results presented in Figure 5, we propose that phosphorylation of specific residues in Hucdc6 is likely to play an important regulatory role.

Fig. 6. Model for potential checkpoint function of HuCdc6. Ongoing DNA replication is monitored either by proteins at the replication fork, including ATR, or by soluble factors, including HuCdc6. ATR and HuCdc6 might work in parallel pathways, both leading to the activation of Chk1 and consequent inactivation of Cdc25. The major role of the ATR pathway would be in response to stalled replication forks. Chk1 may also increase the activity of Wee1 that further prevents the activation of MPF.
In summary, our findings provide evidence that HuCdc6 may have two functions during each cell cycle. The first, well established function of HuCdc6 is in the assembly of the pre-RCs. At this point of the cell cycle HuCdc6 is unphosphorylated, localized in the nucleus and bound to chromatin. The second function of HuCdc6 after phosphorylation and export to the cytoplasm is the checkpoint-mediated coordination of S phase and mitosis. Our results indicate that a phosphorylated form of HuCdc6 might be responsible for this second function and might work as a soluble inhibitor of mitosis, providing a mechanism for coupling S phase with the following mitosis.
Materials and methods
Cell culture and synchronization
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) with 5% new born calf serum (Gibco) and 5% fetal calf serum (Gibco) at 37°C and 10% CO2. The cells were synchronized in G2 phase using a thymidine–aphidicolin regime using a well established protocol as described previously (Pines and Hunter, 1989).
UCN-01 was a gift from Dr Carl Smythe (University of Sheffield) and used at a final concentration of 300 nM. Caffeine (Sigma) was used at a final concentration of 5 mM and wortmannin (Alexis) was used at 25 µM.
Plasmid constructs and protein expression and purification
HuCdc6 PCR product nt 129–1853 inserted into a XhoI–HindIII pEGFP C2 was a gift from Dr Yoshinori Takei (Takei et al., 1999) and PCR product 210–1890 inserted into a KpnI–BamH1 pEGFP C2 produced identical data and therefore we refer in this study only to HuCdc6-pEGFP. Plasmid pHuCdc6 was a gift from Dr Magdalena Assenberg (Nuffield Department of Clinical Medicine, University of Oxford). Plasmids pEGFP-Cdc25C, pCdc25B, pCyclin B1, pCDK1 and pCDK1AF have been described previously (Hagting et al., 1998; Karlsson et al., 1999). Mutations on S54, S74 and S106 of HuCdc6, and deletion of cyclin-box 93–100 were carried out in the same way as described (Petersen et al., 1999) and were introduced into a KpnI–BamHI C2 pEGFP vector. All plasmids used in this study expressed proteins under the control of a cytomegalovirus (CMV) promoter. Plasmids were used at 100 ng/µl.
His6-tagged HuCdc6–GFP protein was expressed in baculovirus-infected Sf9 cells and purified on a Ni-agarose column as described previously (Coverley et al., 2000). Protein was used at 200 ng/µl.
Antibody production and purification
The anti-human Cdc6 rabbit polyclonal antibody was produced using a central fragment of HuCdc6 (residues 110–350) as antigen. The rabbit serum was purified according to Harlow and Lane (1988).
Immunoblotting
An estimated number of cells were plated onto a dish (Biotechs, PA). This gave us the total of 1000 cells per dish after synchronization. All of these G2 phase cells were microinjected. Cells were then directly lysed in 2× SDS buffer, subjected to 10% SDS–PAGE, transferred to nitrocellulose and detected with primary antibody against HuCdc6 (Santa-Cruz, catalogue number sc-9964) or Chk1 (Upstate, catalogue number 06-965) using HRP-conjugated anti-mouse and anti-sheep antibodies, respectively and enhanced chemiluminescence (ECL; Amersham).
Time-lapse fluorescence imaging
For microinjection, cells were plated on a dish (Bioptechs, PA), incubated in a CO2-independent medium without phenol red (Gibco) and overlaid with mineral oil (Sigma) to prevent evaporation. Injected cells were identified by GFP or Texas Red dextran (Cambridge Bioscience) and analysed by time-lapse DIC-fluorescence microscopy as described previously (Clute and Pines, 1999; Furuno et al., 1999). For comparative analyses the parameters were kept the same: fluorescence exposure time of 200 ms, 40× oil objective with a numerical aperture of 1.0 and an image bin size of 3. Images were saved and converted into PICT format using IP lab Spectrum Software (Scanalytics Inc., VA) and exported into Adobe PhotoShop for printing.
Quantitative analysis of cells
All cells positive for GFP or Texas Red were counted as the total number of cells. Cells in G2 phase, in mitosis and cells that had progressed through mitosis (early G1) were counted separately during the course of the experiment. Their percentage was calculated from the total and compared. Cells were monitored and counted for up to 10 h after expression of the protein. In some experiments, we did not assay for cells in early G1, because we tried to keep the radiation with UV (necessary to detect GFP fluorescence) as low as possible. In all experiments we followed the cells through mitosis and after cytokinesis and counted the two daughter G1 phase cells as one pair and not separately. When using untagged plasmids, either pEGFP or Texas Red was added as a detection marker. Antibodies were co-injected with Texas Red as a marker.
Acknowledgments
Acknowledgements
The authors would like to thank Drs Yoshinori Takei for HuCdc6– pEGFP, Anja Hagting for plasmids expressing Cyclin B, CDK1 and CDK1AF, Christina Karlsson for Cdc25B and Cdc25C–GFP and Magdalena Assenberg for pHuCdc6. We thank Dr Carl Smythe for UCN-01 and helpful discussions. We thank Nicole den Elzen for introduction to the microinjection techniques, advice and comments. We thank Linda Ko-Ferrigno and Mark Madine for critically reading the manuscript. This research was supported by the Medical Research Council (MRC) and Cancer Research UK.
References
- Aparicio O.M., Weinstein,D.M. and Bell,S.P. (1997) Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell, 91, 59–69. [DOI] [PubMed] [Google Scholar]
- Baum B., Nishitani,H., Yanow,S. and Nurse,P. (1998) Cdc18 transcription and proteolysis couple S phase to passage through mitosis. EMBO J., 17, 5689–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S.P. and Stillman,B. (1992) ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature, 357, 128–134. [DOI] [PubMed] [Google Scholar]
- Blasina A., Price,B.D., Turenne,G.A. and McGowan,C.H. (1999) Caffeine inhibits the checkpoint kinase ATM. Curr. Biol., 9, 1135–1138. [DOI] [PubMed] [Google Scholar]
- Brown G.W., Jallepalli,P.V., Huneycutt,B.J. and Kelly,T.J. (1997) Interaction of the S phase regulator cdc18 with cyclin-dependent kinase in fission yeast. Proc. Natl Acad. Sci. USA, 94, 6142–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno A. and Russell,P. (1992) Dual functions of CDC6: a yeast protein required for DNA replication also inhibits nuclear division. EMBO J., 11, 2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzada A., Sacristan,M., Sanchez,E. and Bueno,A. (2001) Cdc6 cooperates with Sic1 and Hct1 to inactivate mitotic cyclin-dependent kinases. Nature, 412, 355–358. [DOI] [PubMed] [Google Scholar]
- Cimprich K.A., Shin,T.B., Keith,C.T. and Schreiber,S.L. (1996) cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl Acad. Sci. USA, 93, 2850–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P. and Pines,J. (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol., 1, 82–87. [DOI] [PubMed] [Google Scholar]
- Cocker J.H., Piatti,S., Santocanale,C., Nasmyth,K. and Diffley,J.F. (1996) An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature, 379, 180–182. [DOI] [PubMed] [Google Scholar]
- Coverley D., Pelizon,C., Trewick,S. and Laskey,R.A. (2000) Chromatin-bound Cdc6 persists in S and G2 phases in human cells, while soluble Cdc6 is destroyed in a cyclin A-cdk2 dependent process. J. Cell Sci., 113, 1929–1938. [DOI] [PubMed] [Google Scholar]
- Coverley D., Laman,H. and Laskey,R.A. (2002) Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat. Cell Biol., 4, 523–528. [DOI] [PubMed] [Google Scholar]
- Dasso M. and Newport,J.W. (1990) Completion of DNA replication is monitored by a feedback system that controls the initiation of mitosis in vitro: studies in Xenopus. Cell, 61, 811–823. [DOI] [PubMed] [Google Scholar]
- Dhar S.K. and Dutta,A. (2000) Identification and characterization of the human ORC6 homolog. J. Biol. Chem., 275, 34983–34988. [DOI] [PubMed] [Google Scholar]
- Diffley J.F. (1996) Once and only once upon a time: specifying and regulating origins of DNA replication in eukaryotic cells. Genes Dev., 10, 2819–2830. [DOI] [PubMed] [Google Scholar]
- Drury L.S., Perkins,G. and Diffley,J.F. (2000) The cyclin-dependent kinase Cdc28p regulates distinct modes of Cdc6p proteolysis during the budding yeast cell cycle. Curr. Biol., 10, 231–240. [DOI] [PubMed] [Google Scholar]
- Dunphy W.G. and Kumagai,A. (1991) The cdc25 protein contains an intrinsic phosphatase activity. Cell, 67, 189–196. [DOI] [PubMed] [Google Scholar]
- Dutta A. and Bell,S.P. (1997) Initiation of DNA replication in eukaryotic cells. Annu. Rev. Cell Dev. Biol., 13, 293–332. [DOI] [PubMed] [Google Scholar]
- Feijoo C., Hall-Jackson,C., Wu,R., Jenkins,D., Leitch,J., Gilbert,D.M. and Smythe,C. (2001) Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol., 154, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M. (1999) Cell cycle regulation of DNA replication initiation proteins in mammalian cells. Front. Biosci., 4, D816–D823. [DOI] [PubMed] [Google Scholar]
- Furnari B., Rhind,N. and Russell,P. (1997) Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science, 277, 1495–1497. [DOI] [PubMed] [Google Scholar]
- Furuno N., den Elzen,N. and Pines,J. (1999) Human cyclin A is required for mitosis until mid prophase. J. Cell Biol., 147, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaktionov K. and Beach,D. (1991) Specific activation of cdc25 tyrosine phosphatases by B-type cyclins: evidence for multiple roles of mitotic cyclins. Cell, 67, 1181–1194. [DOI] [PubMed] [Google Scholar]
- Gatei M. et al. (2000) ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet., 25, 115–119. [DOI] [PubMed] [Google Scholar]
- Gautier J., Solomon,M.J., Booher,R.N., Bazan,J.F. and Kirschner,M.W. (1991) cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell, 67, 197–211. [DOI] [PubMed] [Google Scholar]
- Gavin K.A., Hidaka,M. and Stillman,B. (1995) Conserved initiator proteins in eukaryotes. Science, 270, 1667–1671. [DOI] [PubMed] [Google Scholar]
- Graves P.R., Yu,L., Schwarz,J.K., Gales,J., Sausville,E.A., O’Connor,P.M. and Piwnica-Worms,H. (2000) The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J. Biol. Chem., 275, 5600–5605. [DOI] [PubMed] [Google Scholar]
- Greenwood E., Nishitani,H. and Nurse,P. (1998) Cdc18p can block mitosis by two independent mechanisms. J. Cell Sci., 111, 3101–3108. [DOI] [PubMed] [Google Scholar]
- Hagting A., Karlsson,C., Clute,P., Jackman,M. and Pines,J. (1998) MPF localization is controlled by nuclear export. EMBO J., 17, 4127–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Jackson C.A., Cross,D.A., Morrice,N. and Smythe,C. (1999) ATR is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene, 18, 6707–6713. [DOI] [PubMed] [Google Scholar]
- Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Hendrickson M., Madine,M., Dalton,S. and Gautier,J. (1996) Phosphorylation of MCM4 by cdc2 protein kinase inhibits the activity of the minichromosome maintenance complex. Proc. Natl Acad. Sci. USA, 93, 12223–12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzard R.A., Jackson,S.P. and Smith,G.C. (1999) Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res., 59, 2581–2586. [PubMed] [Google Scholar]
- Jares P. and Blow,J.J. (2000) Xenopus cdc7 function is dependent on licensing but not on XORC, XCdc6, or CDK activity and is required for XCdc45 loading. Genes Dev., 14, 1528–1540. [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Wells,N.J. and Hunter,T. (1999) Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc. Natl Acad. Sci. USA, 96, 6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson C., Katich,S., Hagting,A., Hoffmann,I. and Pines,J. (1999) Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J. Cell Biol., 146, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly T.J., Martin,G.S., Forsburg,S.L., Stephen,R.J., Russo,A. and Nurse,P. (1993) The fission yeast cdc18+ gene product couples S phase to START and mitosis. Cell, 74, 371–382. [DOI] [PubMed] [Google Scholar]
- Kornbluth S., Sebastian,B., Hunter,T. and Newport,J. (1994) Membrane localization of the kinase which phosphorylates p34cdc2 on threonine 14. Mol. Biol. Cell, 5, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammer C., Wagerer,S., Saffrich,R., Mertens,D., Ansorge,W. and Hoffmann,I. (1998) The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J. Cell Sci., 111, 2445–2453. [DOI] [PubMed] [Google Scholar]
- Liang C. and Stillman,B. (1997) Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev., 11, 3375–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A., Mondesert,O., Leatherwood,J. and Russell,P. (1998) Negative regulation of Cdc18 DNA replication protein by Cdc2. Mol. Biol. Cell, 9, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorano D., Moreau,J. and Mechali,M. (2000) XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature, 404, 622–625. [DOI] [PubMed] [Google Scholar]
- Millar J.B., Blevitt,J., Gerace,L., Sadhu,K., Featherstone,C. and Russell,P. (1991) p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc. Natl Acad. Sci. USA, 88, 10500–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D.O. (1995) Principles of CDK regulation. Nature, 374, 131–134. [DOI] [PubMed] [Google Scholar]
- Mueller P.R., Coleman,T.R., Kumagai,A. and Dunphy,W.G. (1995) Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science, 270, 86–90. [DOI] [PubMed] [Google Scholar]
- Nagata A., Igarashi,M., Jinno,S., Suto,K. and Okayama,H. (1991) An additional homolog of the fission yeast cdc25+ gene occurs in humans and is highly expressed in some cancer cells. New Biol., 3, 959–968. [PubMed] [Google Scholar]
- Nishitani H. and Nurse,P. (1995) p65cdc18 plays a major role controlling the initiation of DNA replication in fission yeast. Cell, 83, 397–405. [DOI] [PubMed] [Google Scholar]
- Nishitani H., Lygerou,Z., Nishimoto,T. and Nurse,P. (2000) The Cdt1 protein is required to license DNA for replication in fission yeast. Nature, 404, 625–628. [DOI] [PubMed] [Google Scholar]
- Norbury C., Blow,J. and Nurse,P. (1991) Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J., 10, 3321–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P. (1990) Universal control mechanism regulating onset of M-phase. Nature, 344, 503–508. [DOI] [PubMed] [Google Scholar]
- Pelizon C., Madine,M.A., Romanowski,P. and Laskey,R.A. (2000) Unphosphorylatable mutants of Cdc6 disrupt its nuclear export but still support DNA replication once per cell cycle. Genes Dev., 14, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.Y., Graves,P.R., Thoma,R.S., Wu,Z., Shaw,A.S. and Piwnica-Worms,H. (1997) Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science, 277, 1501–1505. [DOI] [PubMed] [Google Scholar]
- Petersen B.O., Lukas,J., Sorensen,C.S., Bartek,J. and Helin,K. (1999) Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J., 18, 396–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen B.O. et al. (2000) Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev., 14, 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. and Hunter,T. (1989) Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell, 58, 833–846. [DOI] [PubMed] [Google Scholar]
- Quintana D.G., Hou,Z., Thome,K.C., Hendricks,M., Saha,P. and Dutta,A. (1997) Identification of HsORC4, a member of the human origin of replication recognition complex. J. Biol. Chem., 272, 28247–28251. [DOI] [PubMed] [Google Scholar]
- Rao P.N. and Johnson,R.T. (1970) Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature, 225, 159–164. [DOI] [PubMed] [Google Scholar]
- Sanchez Y., Wong,C., Thoma,R.S., Richman,R., Wu,Z., Piwnica-Worms,H. and Elledge,S.J. (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science, 277, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Savitsky K., Sfez,S., Tagle,D.A., Ziv,Y., Sartiel,A., Collins,F.S., Shiloh,Y. and Rotman,G. (1995) The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet., 4, 2025–2032. [DOI] [PubMed] [Google Scholar]
- Schlegel R. and Pardee,A.B. (1986) Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science, 232, 1264–1266. [DOI] [PubMed] [Google Scholar]
- Stillman B. (1996) Cell cycle control of DNA replication. Science, 274, 1659–1664. [DOI] [PubMed] [Google Scholar]
- Takei Y., Yamamoto,K. and Tsujimoto,G. (1999) Identification of the sequence responsible for the nuclear localization of human Cdc6. FEBS Lett., 447, 292–296. [DOI] [PubMed] [Google Scholar]
- Tanaka T., Knapp,D. and Nasmyth,K. (1997) Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell, 90, 649–660. [DOI] [PubMed] [Google Scholar]
- Tugal T., Zou-Yang,X.H., Gavin,K., Pappin,D., Canas,B., Kobayashi,R., Hunt,T. and Stillman,B. (1998) The Orc4p and Orc5p subunits of the Xenopus and human origin recognition complex are related to Orc1p and Cdc6p. J. Biol. Chem., 273, 32421–32429. [DOI] [PubMed] [Google Scholar]
- Walter J. and Newport,J. (2000) Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase α. Mol. Cell, 5, 617–627. [DOI] [PubMed] [Google Scholar]
- Weinreich M., Liang,C., Chen,H.H. and Stillman,B. (2001) Binding of cyclin-dependent kinases to ORC and Cdc6p regulates the chromosome replication cycle. Proc. Natl Acad. Sci. USA, 98, 11211–11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y., Forbes,K.C., Wu,Z., Moreno,S., Piwnica-Worms,H. and Enoch,T. (1998) Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature, 395, 507–510. [DOI] [PubMed] [Google Scholar]
- Zhao H. and Piwnica-Worms,H. (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol., 21, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.B. and Elledge,S.J. (2000) The DNA damage response: putting checkpoints in perspective. Nature, 408, 433–439. [DOI] [PubMed] [Google Scholar]
- Zhou B.B., Chaturvedi,P., Spring,K., Scott,S.P., Johanson,R.A., Mishra,R., Mattern,M.R., Winkler,J.D. and Khanna,K.K. (2000) Caffeine abolishes the mammalian G2/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J. Biol. Chem., 275, 10342–10348. [DOI] [PubMed] [Google Scholar]